

Abstract
Adenosine A2A receptors and ATP-activated K+ (KATP) channels contribute to part of the cerebral vasodilatory response to systemic hypoxia, but other mediators are likely involved. Epoxyeicosatrienoic acids (EETs) are cerebral vasodilators and are released from astrocytes exposed to hypoxia. Moreover, stimulation of metabotropic glutamate receptors (mGluR) produces vasodilation by an EET-dependent mechanism. Here, we tested the hypothesis that EET signaling and mGluR activation contribute to hypoxic vasodilation. Laser-Doppler flow was measured over cerebral cortex of anesthetized rats subjected to stepwise reductions in arterial oxygen saturation to 50-70%. Hypoxic reactivity was calculated as the slope of the change in laser-Doppler flow vs. the reciprocal of arterial oxygen content. Hypoxic reactivity significantly decreased from 9.2 ± 1.9 (±95% confidence interval) in controls with vehicle treatment to 2.6 ± 1.4 with the EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid, to 3.0 ± 1.5 with the EET synthesis inhibitor MS-PPOH, to 1.9 ± 2.3 with the combined mGluR subtype 1 and 5 antagonists 2-methyl-6-(phenylethynyl)pyridine and LY367385, to 5.6 ± 1.2 with the KATP channel inhibitor glibenclamide, and to 5.8 ± 2.3 with the A2A receptor antagonist SCH58261. However, reactivity was not significantly altered by the A2B receptor antagonist MRS1754 (6.7 ± 1.8; P = 0.28 Dunnett's test) or by the 20-hydroxyeicosatetraenoic acid synthesis inhibitor HET0016 (7.5 ± 2.3; P = 0.6). These data indicate that, in addition to the known contributions of A2A receptors and KATP channels to the increase in cerebral blood flow during hypoxia, EETs and mGluRs make a major contribution, possibly by mGluR stimulation and hypoxia-induced release of EETs. In contrast, A2B receptors do not make a major contribution, and 20-hydroxyeicosatetraenoic acid does not significantly limit hypoxic vasodilation.
Keywords: cerebral circulation, cytochrome P-450, EET, hypoxia, vasodilation
hypoxic hypoxia, defined as a decrease in arterial partial pressure of O2 (PaO2), produces an increase in cerebral blood flow (CBF) that helps maintain bulk delivery of O2 to the brain's microcirculation (20, 45, 48). However, the mechanisms of hypoxic vasodilation are incompletely understood. Some early studies with nonspecific adenosine receptor antagonists theophylline and 8-phenyltheophylline showed a reduced CBF response to hypoxia (16, 23, 31), and a more recent study with adenosine A2A receptor gene knockout mice and the A2A receptor antagonist ZM241385 showed an attenuated CBF response during a brief, 30-s exposure to severe hypoxia (30). Moreover, brain tissue concentration of adenosine increases in severe hypoxia (36, 50), and cultured astrocytes release adenosine in a graded fashion during hypoxia (21). However, in contrast to previous reports (16, 31), theophylline failed to block pial arterial dilation to moderate hypoxia (14), suggesting that adenosine's contribution to vasodilation may require more severe hypoxia. In addition to adenosine, ATP-activated K+ (KATP) channels have been implicated in cerebral hypoxic vasodilation. The sulfonylurea receptor antagonist glibenclamide attenuated pial artery dilation and the CBF response to hypoxia (40, 44, 46). However, neither adenosine nor sulfonylurea receptor antagonists completely block the CBF response to hypoxia, indicating that other mechanisms are also likely to be involved.
Oxygen-dependent enzymes may act as oxygen sensors, and oxygenases have been postulated to participate in the cerebrovascular response to hypoxia. In 1981, Traystman et al. (47) reported that the broad spectrum oxygenase inhibitors imipramine and metyrapone markedly blunt the CBF response to hypoxia. The epoxygenase inhibitor miconazole has also been reported to decrease hypoxia-induced pial artery dilation in newborn piglets (24). The epoxygenase enzyme, cytochrome P-450 (CYP) 2C11, is expressed in rat astrocytes (3, 38), and exposure of astrocytes to a hypoxic environment increases formation and release of epoxyeicosatrienoic acids (EETs) from arachidonic acid by the catalytic action of CYP 2C11 (51). Hypoxic astrocytes also exhibit an increase in calcium-activated K+ (KCa) channel activity and superoxide production that is suppressed by the epoxygenase inhibitor N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide (MS-PPOH). EETs are capable of opening KCa channels in astrocytes and in cerebral vascular smooth muscle (10, 15, 51), possibly mediated by opening of transient receptor potential vanilloid 4 channels (4, 5). Thus EETs may serve as an astrocyte-based signaling mechanism that contributes to hypoxia-induced smooth muscle hyperpolarization and vasodilation. On the other hand, a hypoxic environment reduces production of the vasoconstrictor 20-hydroxyeicosatetraenoic acid (20-HETE) in cerebral artery muscle cells; such a reduction can increase activity of arterial KCa channel current and produce hyperpolarization and cerebral arterial vasodilation (12).
The evidence supporting the possible role of EETs in hypoxic vasodilation is somewhat analogous to the evidence supporting the role of EETs in neurovascular coupling. Glutamate released from neurons acts on astrocyte metabotropic glutamate receptors (mGluR). Neuronal activation or pharmacological activation of mGluR produces an increase in CBF that is inhibited by MS-PPOH, the EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE), and mGluR group I antagonists (28, 41). Exposure of astrocytes to glutamate causes release of EETs (1, 33) and opens astrocyte KCa channels by an epoxygenase-dependent mechanism (13).
Based on the aforementioned evidence that both hypoxia and glutamate cause release of EETs and EET-dependent signaling in astrocytes, we formulated the hypothesis that mGluR and EETs contribute to the increase in CBF during systemic hypoxemia, as outlined schematically in Fig. 1. Here, we tested the effect of mGluR and EET inhibitors on the CBF response to graded levels of hypoxemia. These effects were contrasted to those produced by glibenclamide and the selective A2A receptor antagonist SCH58261 (35). The effect of the A2B antagonist MRS1754 (18), which is known to attenuate the CBF response to neuronal activation (41), was also examined. Furthermore, the synthesis of the vasoconstrictor 20-HETE is O2 dependent (7, 12), and a reduction in 20-HETE production under hypoxic conditions could promote vasodilation. Thus we also tested whether an inhibitor of 20-HETE synthesis augments the CBF response to hypoxia.
Fig. 1.
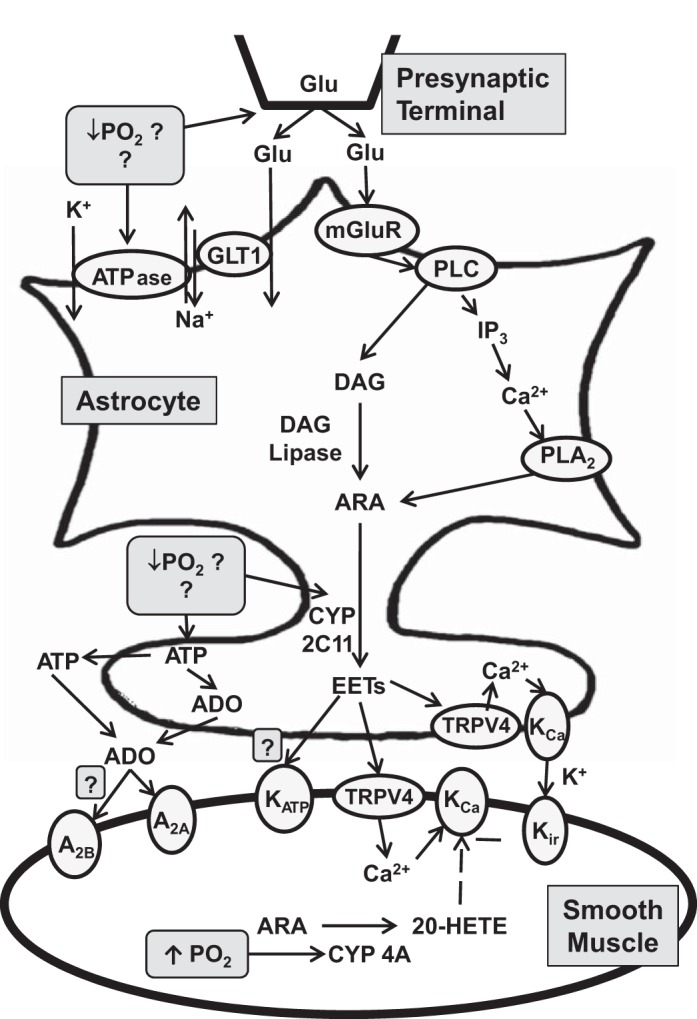
Schematic diagram of possible signaling mechanisms in astrocytes and vascular smooth muscle that result in cerebrovascular vasodilation during hypoxemia. ADO, adenosine; ARA, arachidonic acid; DAG, diacylglycerol; GLT1, primary astrocytic glutamate transporter; Glu, glutamate; IP3, inositol triphosphate; mGluR, metabotropic glutamate receptor; PLA2, phospholipase A2; PLC, phospholipase C; CYP, cytochrome P-450; EET, epoxyeicosatrienoic acids; TRPV4, transient receptor potential vanilloid 4; KCa, calcium-activated K+; Kir, inwardly rectifying K+ channel; 20-HETE, 20-hydroxyeicosatetraenoic acid. Dashed line indicates negative effect.
METHODS
All procedures were approved by the Johns Hopkins University Animal Care and Use Committee and conformed to the guidelines of the National Institutes of Health. Male Wistar rats were anesthetized initially with 5% isoflurane. Anesthesia was maintained by ventilation with 2% isoflurane in O2-enriched air during surgery. The lungs were mechanically ventilated through a tracheostomy, and a femoral artery and vein were catheterized. Rectal temperature was maintained at ∼37°C throughout the experiment.
To measure the CBF response to hypoxia, we positioned a laser-Doppler flow (LDF) probe (Perimed, Stockholm, Sweden) over an area in which the skull was thinned to translucency and corresponded to whisker barrel cortex (7 mm lateral from bregma). The LDF probe provides a measure of relative changes in red blood cell flux in an underlying volume of ∼1 mm3.
To administer drugs locally to the cerebral cortex where LDF was monitored, we drilled two small holes in the skull at ∼3 mm dorsal and ventral to the LDF recording site, as previously described (41). In the dorsal craniotomy, a 120-μm tapered catheter was secured subdurally for fluid infusion, and the ventral craniotomy was used for passive fluid drainage through a small hole in the dura mater. Artificial cerebrospinal fluid containing 156 mM Na+, 3 mM K+, 1.25 mM Ca2+, 0.66 mM Mg2+, 133 mM Cl−, 25 mM HCO3−, 6.7 mM urea, and 3.7 mM dextrose was superfused over cortex at a rate of 5 μl/min. Vehicle or inhibitors were superfused for 1 h before hypoxia induction and continued throughout the period of hypoxia.
Eight groups were subjected to hypoxia after a single treatment with vehicle or inhibitors. These groups included superfusion with vehicle (n = 20), the EET antagonist 14,15-EEZE (30 μM; n = 13), the EET synthesis inhibitor MS-PPOH (20 μM; n = 13), the A2A antagonist SCH58261 (1 μM; n = 18), the A2B antagonist MRS1754 (1 μM; n = 13), the sulfonylurea receptor antagonist glibenclamide (10 μM; n = 13), and the 20-HETE synthesis inhibitor HET0016 (1 μM; n = 15). The concentrations of these drugs were based on those previously shown to inhibit cerebrovascular reactivity (27, 37, 41, 46). To inhibit mGluR, we administered the group I mGluR subtype 1 antagonist (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385; 0.5 mg/kg iv) and the mGluR subtype 5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP; 0.5 mg/kg iv) 20 min before hypoxia (n = 15). The doses and routes of administration of the mGluR antagonists were chosen based on their ability to inhibit the increase in LDF during neuronal activation (52).
After the surgical procedures were completed and before administration of inhibitors, anesthesia was switched from isoflurane to α-chlorolose (33 mg/kg plus 12 mg·kg−1·h−1). In addition, pancuronium bromide (0.3 mg/kg iv) was administered before induction of hypoxia to prevent hypoxia-induced ventilatory efforts. Hypoxic hypoxia was produced by stepwise reductions in inspired O2 over the range of 21% to ∼10%. Ventilation at each decrement in inspired O2 lasted 10 min before the next reduction in inspired O2. Supplemental CO2 was added to the inspired gas mixture as needed to prevent a decrease in end-tidal CO2. Samples of arterial blood were obtained before induction of hypoxia and at 7 min after each change in inspired O2. Samples were analyzed for PaO2, arterial partial pressure of CO2 (PaCO2), and pH (ABL80, Radiometer, Copenhagen, Denmark) and for arterial hemoglobin concentration, O2 saturation, and O2 content (OSM3 hemoximeter, Radiometer). Mean arterial blood pressure (MABP) and LDF were averaged over the last 2 min of each level of hypoxia. If MABP decreased by >20% from the normoxic baseline, the experiment was terminated.
Data were obtained at four to seven different levels of oxygenation in each rat. The increase in CBF during hypoxia is characterized by an inverse relationship with arterial O2 content (19). For each rat, hypoxic responsivity was calculated as the slope of the regression line of LDF, expressed as a fraction of the normoxic baseline, vs. the reciprocal of arterial O2 content (ml O2/dl). Hypoxic responses were compared among the eight groups by ANOVA. Post hoc comparisons with the vehicle group were made by the two-tailed Dunnett's test at the 0.05 significance level. For ease of presentation, data also were pooled into incremental bins of arterial O2 saturation and expressed as means ± 95% confidence intervals. Within each bin, MABP, PaCO2, pH, and hemoglobin concentration were compared with those of the vehicle-treated group by ANOVA and the Dunnett's test at the 0.05 significance level. Cerebrovascular resistance (CVR) was calculated from the MABP/LDF data and expressed as a ratio of baseline CVR. Because normality test failed, CVR data in each group were compared with that in the vehicle group with the Mann-Whitney test at the 0.05 significance level.
RESULTS
Graded decreases in inspired O2 in the mechanically ventilated rats produced decreases in PaO2 from 110 ± 5 Torr to as low as 55 ± 6 Torr. More severe levels of hypoxia were not studied because the decrease in MABP that occurs in anesthetized rats can confound interpretation of the data. As expected, we observed graded increases in LDF with increasing severity of hypoxia in the control group treated with vehicle (Fig. 2A). The increase in LDF was markedly less in groups treated with the EET antagonist 14,15-EEZE and the EET synthesis inhibitor MS-PPOH. Although a small decrease in MABP occurred with 50–70% arterial O2 saturation, MABP was similar among groups. Moreover, PaCO2 was also similar and well maintained in these groups.
Fig. 2.
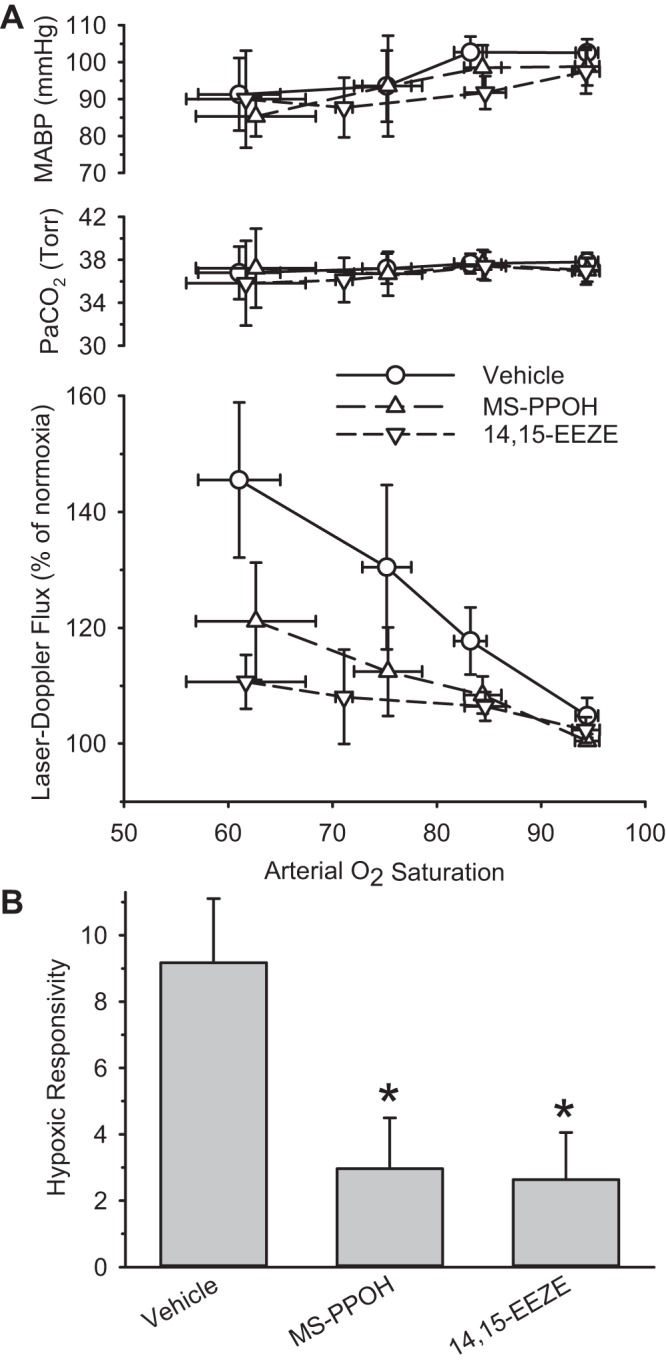
A: arterial Pco2 (PaCO2), mean arterial blood pressure (MABP), and cortical laser-Doppler flow (LDF) are sorted by range of arterial O2 saturation and are plotted as means and 95% confidence intervals for groups treated with vehicle (n = 20), the EET synthesis inhibitor N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide (MS-PPOH; n = 13), or the EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE; n = 13). B: hypoxic responsivity, derived from the regression slope of the relative change of LDF vs. the reciprocal of arterial O2 content for each rat, is plotted for groups treated with vehicle, MS-PPOH, and 14,15-EEZE. *P < 0.05 from vehicle group.
A single ANOVA on all eight groups of hypoxic responsivity, calculated from the slope of the LDF response and the reciprocal of arterial O2 content, passed the normality and equal variance tests, and an effect of treatment was significant (F = 8.11; degrees of freedom = 7,112). Hypoxic responsivity was significantly reduced by 71% in the 14,15-EEZE-treated group (Dunnett's test) and by 68% in the MS-PPOH-treated group compared with that of the vehicle-treated group (Fig. 2B).
Concurrent treatment with the mGluR antagonists MPEP and LY367385 also markedly reduced the LDF response to hypoxia without producing significant differences in MABP or PaCO2 between groups (Fig. 3). Hypoxic responsivity was significantly reduced by 79%, which was comparable in magnitude to the reduction observed in the 14,15-EEZE- and MS-PPOH-treated groups.
Fig. 3.
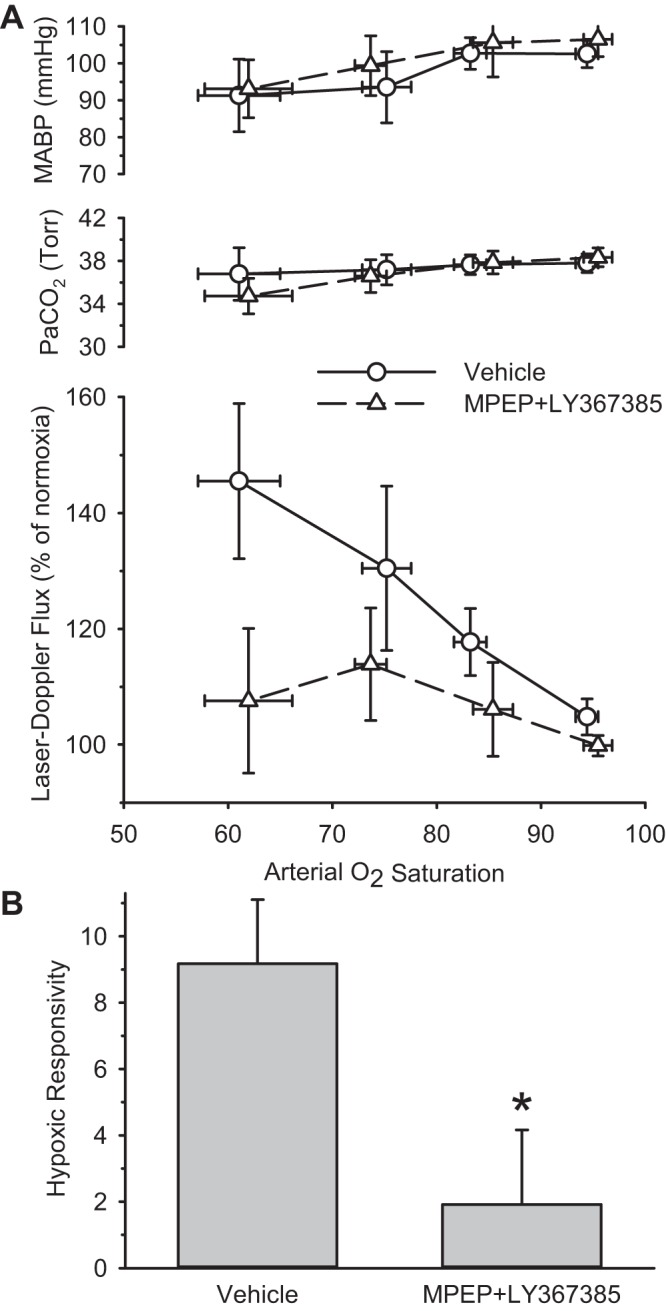
A: PaCO2, MABP, and cortical LDF are sorted by range of arterial O2 saturation and are plotted as means and 95% confidence intervals for groups treated with vehicle (n = 20) or the metabotropic glutamate receptor antagonists 2-methyl-6-(phenylethynyl)pyridine (MPEP) and (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385; n = 15). B: hypoxic responsivity is plotted for groups treated with vehicle and MPEP plus LY-367385. *P < 0.05 from vehicle group.
Treatment with the adenosine A2A antagonist SCH58261 attenuated the hypoxic responsivity to a moderate extent (37%; Fig. 4). In the case of treatment with the A2B antagonist MRS1754, the 27% reduction in hypoxic responsivity did not reach statistical significance (P = 0.28).
Fig. 4.
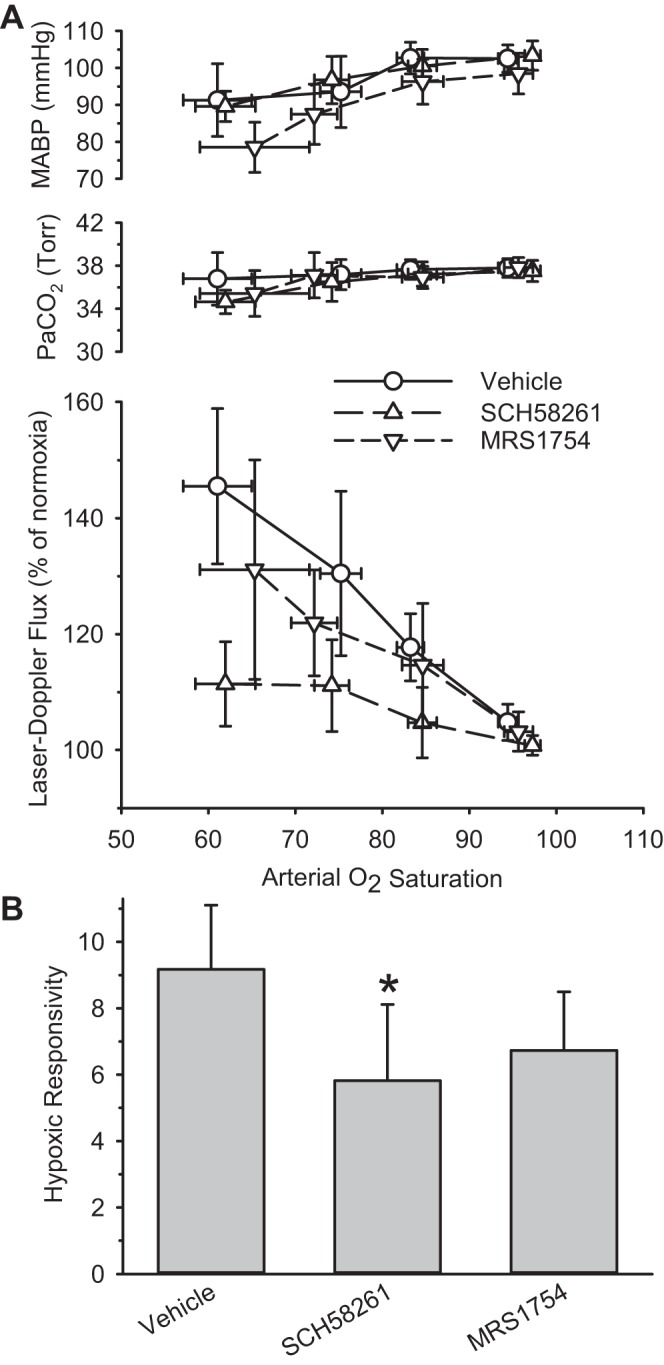
A: PaCO2, MABP, and cortical LDF are sorted by range of arterial O2 saturation and are plotted as means and 95% confidence intervals for groups treated with vehicle (n = 20), the A2A antagonist SCH58261 (n = 18), or the A2B antagonist MRS1754 (n = 13). B: hypoxic responsivity is plotted for groups treated with vehicle, SCH58261, and MRS1754. *P < 0.05 from vehicle group.
Treatment with glibenclamide moderately attenuated the LDF response to hypoxia (39%; P = 0.038) compared with that of the vehicle-treated group (Fig. 5). Treatment with the 20-HETE synthesis inhibitor HET0016 did not significantly alter the LDF response to hypoxia (P = 0.6).
Fig. 5.
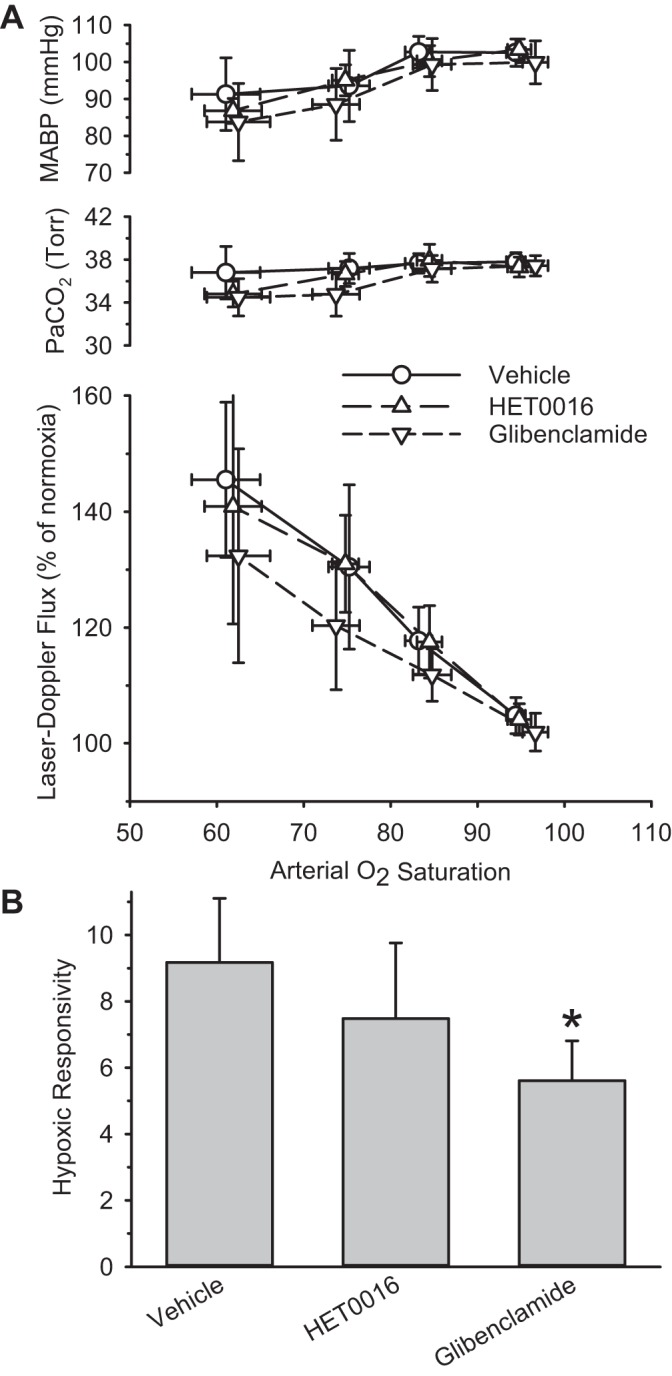
A: PaCO2, MABP, and cortical LDF are sorted by range of arterial O2 saturation and are plotted as means and 95% confidence intervals for groups treated with vehicle (n = 20), the 20-HETE synthesis inhibitor HET0016 (n = 15), or the ATP-activated K+ channel inhibitor glibenclamide (n = 13). B: hypoxic responsivity is plotted for groups treated with vehicle, HET0016, and glibenclamide. *P < 0.05 from vehicle group.
ANOVA on the eight groups indicated no significant effect of treatment on MABP (P = 0.15) or on PaCO2 (P = 0.31) for data in the 50–70% arterial O2 saturation range. In addition, arterial hemoglobin concentration remained unchanged and was similar among groups at each level of hypoxia (Table 1). However, groups treated with MS-PPOH, MPEP plus LY367385, and HET0016 did display mild metabolic acidosis compared with the vehicle-treated group at the most severe range of hypoxia (Table 1).
Table 1.
Arterial hemoglobin concentration and pH at different ranges of arterial O2 saturation
| Arterial O2 Saturation |
||||
|---|---|---|---|---|
| Treatment | 90–100% | 80–90% | 70–80% | 50–70% |
| Hemoglobin, g/l | ||||
| Vehicle | 106 ± 5 | 105 ± 4 | 106 ± 8 | 108 ± 6 |
| MS-PPOH | 104 ± 4 | 106 ± 7 | 106 ± 12 | 109 ± 7 |
| 14,15-EEZE | 104 ± 4 | 102 ± 6 | 104 ± 10 | 115 ± 10 |
| MPEP + LY367385 | 108 ± 6 | 109 ± 7 | 108 ± 5 | 108 ± 5 |
| SCH58261 | 113 ± 3 | 113 ± 5 | 110 ± 6 | 111 ± 5 |
| MRS1754 | 103 ± 4 | 104 ± 4 | 101 ± 5 | 104 ± 6 |
| Glibenclamide | 106 ± 3 | 104 ± 4 | 105 ± 5 | 105 ± 6 |
| HET0016 | 107 ± 8 | 112 ± 5 | 111 ± 4 | 106 ± 4 |
| pH | ||||
| Vehicle | 7.38 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.02 | 7.39 ± 0.02 |
| MS-PPOH | 7.36 ± 0.01 | 7.37 ± 0.01 | 7.36 ± 0.02 | 7.31 ± 0.04* |
| 14,15-EEZE | 7.37 ± 0.02 | 7.37 ± 0.01 | 7.35 ± 0.03 | 7.34 ± 0.03 |
| MPEP + LY367385 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.36 ± 0.02 | 7.34 ± 0.02* |
| SCH58261 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.36 ± 0.02 | 7.35 ± 0.03 |
| MRS1754 | 7.37 ± 0.01 | 7.36 ± 0.01 | 7.35 ± 0.02 | 7.34 ± 0.03 |
| Glibenclamide | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.34 ± 0.02 | 7.35 ± 0.02 |
| HET0016 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.35 ± 0.01 | 7.34 ± 0.01* |
Values are means ± 95% confidence intervals.
MS-PPOH, N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide; 14,15-EEZE, 14,15-epoxyeicosa-5(Z)-enoic acid; LY367385, (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid; MPEP, 2-methyl-6-(phenylethynyl)pyridine.
P < 0.05 from vehicle group by Dunnett's test.
Because the slope of the LDF response to hypoxia can be disproportionately influenced by the response at the most severe level of LDF, we examined whether vasodilation was affected by drug treatment at moderate and severe hypoxia. CVR was used as a measure of vasodilation. To compare CVR with the vehicle group, values in each rat were interpolated at 80% arterial O2 saturation to represent moderate hypoxia and at 65% arterial O2 saturation to represent severe hypoxia. The reduction CVR during hypoxia was significantly attenuated in groups treated with MS-PPOH, 14,15-EEZE, MPEP plus LY367385, and SCH58261 (Fig. 6). The attenuation occurred at both 80% and 65% arterial O2 saturation.
Fig. 6.

Cerebrovascular resistance (CVR; calculated as a ratio of baseline) is displayed as box-whisker plots (5, 25, 50, 75, and 95 percentiles) at 80% arterial O2 saturation (A) and 65% arterial O2 saturation (B) for the 8 treatment groups. *P < 0.05 from vehicle group.
DISCUSSION
The major new findings of this study are that EETs and mGluR contribute substantially to the increase in CBF and the decrease in CVR during acute hypoxic hypoxia, and that the roles of EETs and mGluR are at least as prominent as the contributions of A2A receptors and KATP channels. In addition, the CVR data indicate that EETs, mGluR, and A2A receptors contribute to the vasodilation seen at both moderate and severe levels of hypoxia, thereby indicating that recruitment of these mechanisms does not require a large threshold of hypoxemia. In contrast, our results indicate that A2B receptors do not play a major role in hypoxic vasodilation, and that the vasoconstrictor 20-HETE does not constrain the increase in CBF or decrease in CVR.
Rat astrocytes express the epoxygenase enzyme CYP 2C11 (3, 38), and cultured astrocytes release EETs in response to hypoxia (51). Trigeminal and sphenopalatine ganglia express the epoxygenase enzymes CYP 2J3 and 2J4 (17). Stimulation of these ganglia produces cerebral hyperemia that is blocked by topical application of the EET antagonist 14,15-EEZE to cortex, but not by the CYP epoxygenase inhibitor MS-PPOH, possibly because the EETs are synthesized in the ganglia and released on the cortex. The fact that we can inhibit hypoxic vasodilation by similar amounts with topical application of either MS-PPOH or 14,15-EEZE suggests that the EETs that mediate hypoxic vasodilation are synthesized and active locally. The similar effect of MS-PPOH and 14,15-EEZE also suggests that hypoxia likely increased de novo EET synthesis, rather than solely increasing the release of stored EETs, at least in the 10-min steady-state conditions for each stepwise change in inspired O2. Although we did not monitor LDF during the 1-h period of drug infusion before hypoxia, we previously did not observe effects of these drugs on baseline LDF that would account for a decrease in hypoxic vasodilation (28, 37).
EETs are known to cause KCa channels to open in cerebrovascular smooth muscle cells, producing hyperpolarization and vasodilation (10, 17). Thus EETs that are released from other cells could act on arteriolar smooth muscle to produce vasodilation. Furthermore, EETs are involved in opening of KCa channels in cultured astrocytes exposed to hypoxia or glutamate (13, 51). Release of K+ from astrocytes via KCa channels has been postulated to open inward-rectifier K+ channels on adjacent vascular smooth muscle and lead to vasorelaxation during activation of astrocytes (15). Lastly, EETs have been implicated in opening KATP channels in myocardial cells and mesenteric arteries (29); a similar mechanism could operate in cerebrovascular smooth muscle where glibenclamide has been shown to limit hypoxic vasodilation (40, 44, 46). The ability of glibenclamide to attenuate the LDF response in the present study is consistent with this possibility, although the effect on the LDF response slope was moderate, and an effect on CVR did not attain statistical significance.
The generation of EETs or 20-HETE from arachidonic acid by CYP epoxygenases or CYP 4A ω-hydroxylase requires oxygen. The hypoxic insult also generates production of reactive oxygen species, which enhances degradation of 20-HETE without affecting the bioavailability of the EETs (9, 48). Thus the lack of effect of HET0016 on hypoxic vascular reactivity could be related to reduced endogenous 20-HETE synthesis and/or increased degradation during hypoxia.
Exposure of astrocytes to glutamate is known to increase release of EETs (1, 33) and to open astrocyte KCa channels (13). The effect of glutamate on astrocyte signaling is largely attributed to stimulation of astrocyte mGluR receptors (13, 52), although astrocyte mGluR5 expression appears to decline during development (43). In the present study, mGluR antagonists were as effective as EET antagonists in reducing the LDF response to hypoxia. This finding is consistent with the possibility that stimulation of mGluR is responsible for an increase in EET signaling during hypoxia. We did not investigate the mechanism for increased mGluR stimulation during hypoxia in this study. Increased glutamate release arising from anoxic depolarization of neurons requires severe reductions in O2 consumption, but cerebral O2 consumption is generally maintained at arterial O2 saturations above 50% (20). In our study, the mGluR antagonist-induced attenuation of the LDF response was already evident with moderate hypoxia. A more likely explanation is a graded release of glutamate from neurons or astrocytes, or possibly a reduced glutamate uptake by astrocytes. However, our data do not reveal the upstream O2-sensing mechanism responsible for activated mGluR and EETs production.
Although our study was limited to the effects of acute hypoxia, EETs may also be important in chronic hypoxia. The gene for CYP 2C11 appears to have a hypoxia-inducible factor-1α binding element (26), exposure of cultured astrocytes to hypoxia for 24 h increases expression of CYP 2C11 (51), and ischemic preconditioning is able to increase CYP 2C11 expression in brain (2). In mesenteric arteries, the epoxygenase CYP 2C9 is upregulated at 48 h of hypoxia and results in vascular smooth muscle hyperpolarization (6). Thus the role of EETs in the vascular response to hypoxia may not be limited to the acute stage.
Exposure of mice to 30 s of hypoxia reveals an impaired LDF response in A2A null mice and in wild-type mice treated with the A2A antagonist ZM241385 (30). In the present study, we chose SCH58261 as an A2A antagonist because of its high selectivity and potency (35). The 37% attenuation of the LDF response that we observed with a different inhibitor in a different species further supports a role for A2A receptors in cerebral vasodilation during hypoxia. With arterial hypotension, A2A receptors have been found to contribute to autoregulatory vasodilation (22, 42), but MABP was not severely decreased with the level of hypoxia achieved in our experiments, and the MABP decrease in the SCH58261-treated group was similar to that in the vehicle-treated group.
We are not aware of studies that have examined effects of A2B antagonists on CBF during hypoxia. A2B receptors are expressed in cerebral artery muscle cells (11), but they require a higher concentration of adenosine for activation than do A2A receptors (32). The lack of a significant effect of MRS1754 on the increase in LDF response with arterial O2 saturation above 50% suggests that the increase in adenosine is not sufficient to activate these receptors. A higher adenosine concentration could be achieved if A2B receptors are localized near ecto-endonucleases that break down ATP released from astrocytes, as is thought to occur with neuronal activation (49). Interestingly, A2B antagonists, including MRS1754, attenuate the increase in LDF during whisker stimulation (41), thereby indicating some differences in vasodilatory signaling mechanisms between neuronal activation and hypoxemia. Consistent with the concept of nonidentical signaling mechanisms, the LDF response to whisker stimulation is not affected by manipulation of O2 availability during neuronal activation (25).
The large effect of 14,15-EEZE, MS-PPOH, and mGluR antagonists on the LDF response to hypoxia was not attributable to a lower MABP or PaCO2. Most of the inhibitors were administered by superfusion over cerebral cortex and should have had minimal effect on systemic hemodynamics. The exception was the mGluR antagonists, which were given systemically to match the effective dose and route of administration that are known to inhibit the LDF response to whisker stimulation (52). Although systemic administration of the mGluR antagonists did not affect the MABP response to hypoxia, we cannot exclude a systemic neurohumoral effect that might modulate the local LDF response to hypoxia. However, such an effect is unlikely to fully account for the large suppression of the LDF response to hypoxia.
Hypoxia opens KCa channels in cerebral artery smooth muscle (9). The open-state probability of these channels is augmented by inhibition of 20-HETE and counteracted by exogenous 20-HETE (12). However, decreasing the Po2 of isolated cerebral microvascular smooth muscle cells decreases endogenous 20-HETE concentration (12) and could thereby permit other vasodilators to act unencumbered. The lack of effect of HET0016 on the increase in LDF during hypoxia supports the concept that 20-HETE synthesis is already reduced during hypoxia and not constraining the increase in flow. In contrast to the lack of effect of the 20-HETE synthesis inhibitor in hypoxic vasodilation, 20-HETE appears to be involved with the vasoconstrictor response to increased oxygenation in brain and skeletal muscle (7). For example, pial arteriole constriction in response to cell-free hemoglobin (39) or to the onset of pulmonary ventilation in fetal sheep (34) is blocked by inhibitors of 20-HETE synthesis. Therefore, the vasoactive effects of CYP metabolites of arachidonic acid depend on the directional changes in oxygenation: 20-HETE plays a more prominent role with increases in oxygenation, whereas EETs play a prominent role with decreases in brain oxygenation. These results differ somewhat from those obtained in gracilis muscle, where MS-PPOH has no effect on hypoxic vasodilation and 20-HETE inhibitors attenuate hypoxic vasodilation and hyperoxic vasoconstriction (7, 8).
In conclusion, we present pharmacological evidence that the increase in CBF during hypoxic hypoxia depends largely on activation of EETs and mGluR group I. We also confirm a role for the A2A receptor and KATP channels, but their contribution appears to be less than those of EETs and mGluR. In contrast, A2B and 20-HETE do not play a significant role in hypoxic vasodilation. Because applying an mGluR group I agonist produces an increase in LDF that is blocked by MS-PPOH and 14,15-EEZE (28), mGluR likely acts upstream in the signaling cascade. These results imply that hypoxia without ischemia increases metabotropic glutamate signaling, leading to synthesis and release of EETs.
GRANTS
This study was supported by National Institute of Neurological Disorders and Stroke Grant R01 NS38684 (R. C. Koehler), by National Heart, Lung, and Blood Institute Grants R01 HL033833, R01 HL092105, and R01 HL105997 (D. R. Harder), and by a grant from the Veterans Administration Research Career Scientist Award (D. R. Harder).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: X.L., D.G., D.R.H., and R.C.K. conception and design of research; X.L. performed experiments; X.L. and R.C.K. analyzed data; X.L., D.G., D.R.H., and R.C.K. interpreted results of experiments; X.L. and R.C.K. drafted manuscript; X.L., D.G., D.R.H., and R.C.K. approved final version of manuscript; D.G., D.R.H., and R.C.K. edited and revised manuscript; R.C.K. prepared figures.
ACKNOWLEDGMENTS
The authors thank Claire F. Levine for editorial assistance.
REFERENCES
- 1.Alkayed NJ, Birks EK, Narayanan J, Petrie KA, Kohler-Cabot AE, Harder DR. Role of P-450 arachidonic acid expoygenase in the response of cerebral blood flow to glutamate in rats. Stroke 28: 1066–1072, 1997. [DOI] [PubMed] [Google Scholar]
- 2.Alkayed NJ, Goyagi T, Joh HD, Klaus J, Harder DR, Traystman RJ, Hurn PD. Neuroprotection and P450 2C11 upregulation after experimental transient ischemic attack. Stroke 33: 1677–1684, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke 27: 971–979, 1996. [DOI] [PubMed] [Google Scholar]
- 4.Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A 110: 6157–6162, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97: 1270–1279, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Earley S, Pastuszyn A, Walker BR. Cytochrome P-450 epoxygenase products contribute to attenuated vasoconstriction after chronic hypoxia. Am J Physiol Heart Circ Physiol 285: H127–H136, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Frisbee JC, Krishna UM, Falck JR, Lombard JH. Role of prostanoids and 20-HETE in mediating oxygen-induced constriction of skeletal muscle resistance arteries. Microvasc Res 62: 271–283, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Frisbee JC, Roman RJ, Krishna UM, Falck JR, Lombard JH. Relative contributions of cyclooxygenase- and cytochrome P450 ω-hydroxylase-dependent pathways to hypoxic dilation of skeletal muscle resistance arteries. J Vasc Res 38: 305–314, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Gebremedhin D, Bonnet P, Greene AS, England SK, Rusch NJ, Lombard JH, Harder DR. Hypoxia increases the activity of Ca2+-sensitive K+ channels in cat cerebral arterial muscle cell membranes. Pflügers Arch 428: 621–630, 1994. [DOI] [PubMed] [Google Scholar]
- 10.Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol Heart Circ Physiol 263: H519–H525, 1992. [DOI] [PubMed] [Google Scholar]
- 11.Gebremedhin D, Weinberger B, Lourim D, Harder DR. Adenosine can mediate its actions through generation of reactive oxygen species. J Cereb Blood Flow Metab 30: 1777–1790, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gebremedhin D, Yamaura K, Harder DR. Role of 20-HETE in the hypoxia-induced activation of Ca2+-activated K+ channel currents in rat cerebral arterial muscle cells. Am J Physiol Heart Circ Physiol 294: H107–H120, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Gebremedhin D, Yamaura K, Zhang C, Bylund J, Koehler RC, Harder DR. Metabotropic glutamate receptor activation enhances the activities of two types of Ca2+-activated K+ channels in rat hippocampal astrocytes. J Neurosci 23: 1678–1687, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haller C, Kuschinsky W. Moderate hypoxia: reactivity of pial arteries and local effect of theophylline. J Appl Physiol 63: 2208–2215, 1987. [DOI] [PubMed] [Google Scholar]
- 15.Higashimori H, Blanco VM, Tuniki VR, Falck JR, Filosa JA. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. Am J Physiol Cell Physiol 299: C1068–C1078, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman WE, Albrecht RF, Miletich DJ. The role of adenosine in CBF increases during hypoxia in young vs aged rats. Stroke 15: 124–129, 1984. [DOI] [PubMed] [Google Scholar]
- 17.Iliff JJ, Wang R, Zeldin DC, Alkayed NJ. Epoxyeicosanoids as mediators of neurogenic vasodilation in cerebral vessels. Am J Physiol Heart Circ Physiol 296: H1352–H1363, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji X, Kim YC, Ahern DG, Linden J, Jacobson KA. [3H]MRS 1754, a selective antagonist radioligand for A2B adenosine receptors. Biochem Pharmacol 61: 657–663, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones MD Jr, Traystman RJ, Simmons MA, Molteni RA. Effects of changes in arterial O2 content on cerebral blood flow in the lamb. Am J Physiol Heart Circ Physiol 240: H209–H215, 1981. [DOI] [PubMed] [Google Scholar]
- 20.Koehler RC, Traystman RJ, Zeger S, Rogers MC, Jones MD Jr. Comparison of cerebrovascular response to hypoxic and carbon monoxide hypoxia in newborn and adult sheep. J Cereb Blood Flow Metab 4: 115–122, 1984. [DOI] [PubMed] [Google Scholar]
- 21.Kulik TB, Aronhime SN, Echeverry G, Beylin A, Winn HR. The relationship between oxygen and adenosine in astrocytic cultures. Glia 58: 1335–1344, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Kusano Y, Echeverry G, Miekisiak G, Kulik TB, Aronhime SN, Chen JF, Winn HR. Role of adenosine A2 receptors in regulation of cerebral blood flow during induced hypotension. J Cereb Blood Flow Metab 30: 808–815, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laudignon N, Farri E, Beharry K, Rex J, Aranda JV. Influence of adenosine on cerebral blood flow during hypoxic hypoxia in the newborn piglet. J Appl Physiol 68: 1534–1541, 1990. [DOI] [PubMed] [Google Scholar]
- 24.Leffler CW, Smith JS, Edrington JL, Zuckerman SL, Parfenova H. Mechanisms of hypoxia-induced cerebrovascular dilation in the newborn pig. Am J Physiol Heart Circ Physiol 272: H1323–H1332, 1997. [DOI] [PubMed] [Google Scholar]
- 25.Lindauer U, Leithner C, Kaasch H, Rohrer B, Foddis M, Fuchtemeier M, Offenhauser N, Steinbrink J, Royl G, Kohl-Bareis M, Dirnagl U. Neurovascular coupling in rat brain operates independent of hemoglobin deoxygenation. J Cereb Blood Flow Metab 30: 757–768, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu M, Alkayed NJ. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) 1alpha-linked induction of P450 2C11 epoxygenase in astrocytes. J Cereb Blood Flow Metab 25: 939–948, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Li C, Falck JR, Roman RJ, Harder DR, Koehler RC. Interaction of nitric oxide, 20-HETE, and EETs during functional hyperemia in whisker barrel cortex. Am J Physiol Heart Circ Physiol 295: H619–H631, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Li C, Gebremedhin D, Hwang SH, Hammock BD, Falck JR, Roman RJ, Harder DR, Koehler RC. Epoxyeicosatrienoic acid-dependent cerebral vasodilation evoked by metabotropic glutamate receptor activation in vivo. Am J Physiol Heart Circ Physiol 301: H373–H381, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu T, Ye D, Wang X, Seubert JM, Graves JP, Bradbury JA, Zeldin DC, Lee HC. Cardiac and vascular KATP channels in rats are activated by endogenous epoxyeicosatrienoic acids through different mechanisms. J Physiol 575: 627–644, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miekisiak G, Kulik T, Kusano Y, Kung D, Chen JF, Winn HR. Cerebral blood flow response in adenosine 2a receptor knockout mice during transient hypoxic hypoxia. J Cereb Blood Flow Metab 28: 1656–1664, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Morii S, Ngai AC, Ko KR, Winn HR. Role of adenosine in regulation of cerebral blood flow: effects of theophylline during normoxia and hypoxia. Am J Physiol Heart Circ Physiol 253: H165–H175, 1987. [DOI] [PubMed] [Google Scholar]
- 32.Ngai AC, Coyne EF, Meno JR, West GA, Winn HR. Receptor subtypes mediating adenosine-induced dilation of cerebral arterioles. Am J Physiol Heart Circ Physiol 280: H2329–H2335, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Nithipatikom K, Grall AJ, Holmes BB, Harder DR, Falck JR, Campbell WB. Liquid chromatographic-electrospray ionization-mass spectrometric analysis of cytochrome P450 metabolites of arachidonic acid. Anal Biochem 298: 327–336, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Ohata H, Gebremedhin D, Narayanan J, Harder DR, Koehler RC. Onset of pulmonary ventilation in fetal sheep produces pial arteriolar constriction dependent on cytochrome P450 ω-hydroxylase activity. J Appl Physiol 109: 412–417, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ongini E, Dionisotti S, Gessi S, Irenius E, Fredholm BB. Comparison of CGS 15943, ZM 241385 and SCH 58261 as antagonists at human adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol 359: 7–10, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Park TS, Van Wylen DG, Rubio R, Berne RM. Increased brain interstitial fluid adenosine concentration during hypoxia in newborn piglet. J Cereb Blood Flow Metab 7: 178–183, 1987. [DOI] [PubMed] [Google Scholar]
- 37.Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, Traystman RJ, Koehler RC. Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. Am J Physiol Heart Circ Physiol 283: H2029–H2037, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Peng X, Zhang C, Alkayed NJ, Harder DR, Koehler RC. Dependency of cortical functional hyperemia to forepaw stimulation on epoxygenase and nitric oxide synthase activities in rats. J Cereb Blood Flow Metab 24: 509–517, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Qin X, Kwansa H, Bucci E, Roman RJ, Koehler RC. Role of 20-HETE in the pial arteriolar constrictor response to decreased hematocrit after exchange transfusion of cell-free polymeric hemoglobin. J Appl Physiol 100: 336–342, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shankar V, Armstead WM. Opioids contribute to hypoxia-induced pial artery dilation through activation of ATP-sensitive K+ channels. Am J Physiol Heart Circ Physiol 269: H997–H1002, 1995. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Liu X, Gebremedhin D, Falck JR, Harder DR, Koehler RC. Interaction of mechanisms involving epoxyeicosatrienoic acids, adenosine receptors, and metabotropic glutamate receptors in neurovascular coupling in rat whisker barrel cortex. J Cereb Blood Flow Metab 28: 111–125, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin HK, Shin YW, Hong KW. Role of adenosine A2B receptors in vasodilation of rat pial artery and cerebral blood flow autoregulation. Am J Physiol Heart Circ Physiol 278: H339–H344, 2000. [DOI] [PubMed] [Google Scholar]
- 43.Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 339: 197–200, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taguchi H, Heistad DD, Kitazono T, Faraci FM. ATP-sensitive K+ channels mediate dilatation of cerebral arterioles during hypoxia. Circ Res 74: 1005–1008, 1994. [DOI] [PubMed] [Google Scholar]
- 45.Todd MM, Wu B, Maktabi M, Hindman BJ, Warner DS. Cerebral blood flow and oxygen delivery during hypoxemia and hemodilution: role of arterial oxygen content. Am J Physiol Heart Circ Physiol 267: H2025–H2031, 1994. [DOI] [PubMed] [Google Scholar]
- 46.Tomiyama Y, Brian JE Jr, Todd MM. Cerebral blood flow during hemodilution and hypoxia in rats: role of ATP-sensitive potassium channels. Stroke 30: 1942–1947, 1999. [DOI] [PubMed] [Google Scholar]
- 47.Traystman RJ, Gurtner GH, Rogers MC, Jones MD Jr, Koehler RC. A possible role of oxygenases in the regulation of cerebral blood flow. In: Cardiovascular Physiology. Neural Control Mechanisms, edited by Kovach AGB, Sandor P, Kollai P. Budapest: Pergammon, 1981, p. 167–177. [Google Scholar]
- 48.Ulatowski JA, Bucci E, Razynska A, Traystman RJ, Koehler RC. Cerebral blood flow during hypoxic hypoxia with plasma-based hemoglobin at reduced hematocrit. Am J Physiol Heart Circ Physiol 274: H1933–H1942, 1998. [DOI] [PubMed] [Google Scholar]
- 49.Vetri F, Xu H, Mao L, Paisansathan C, Pelligrino DA. ATP hydrolysis pathways and their contributions to pial arteriolar dilation in rats. Am J Physiol Heart Circ Physiol 301: H1369–H1377, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. Am J Physiol Heart Circ Physiol 241: H235–H242, 1981. [DOI] [PubMed] [Google Scholar]
- 51.Yamaura K, Gebremedhin D, Zhang C, Narayanan J, Hoefert K, Jacobs ER, Koehler RC, Harder DR. Contribution of epoxyeicosatrienoic acids to the hypoxia-induced activation of Ca2+-activated K+ channel current in cultured rat hippocampal astrocytes. Neuroscience 143: 703–716, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6: 43–50, 2003. [DOI] [PubMed] [Google Scholar]