

Summary
Detailed genomic contact maps have revealed that chromosomes are structurally organized in megabase-sized topologically associated domains (TADs) that encompass smaller subTADs. These domains segregate in the nuclear space to form active and inactive nuclear compartments, but cause and consequence of compartmentalization are largely unknown. Here, we combined lacO/lacR binding platforms with allele-specific 4C technologies to track their precise position in the three-dimensional genome upon recruitment of NANOG, SUV39H1, or EZH2. We observed locked genomic loci resistant to spatial repositioning and unlocked loci that could be repositioned to different nuclear subcompartments with distinct chromatin signatures. Focal protein recruitment caused the entire subTAD, but not surrounding regions, to engage in new genomic contacts. Compartment switching was found uncoupled from transcription changes, and the enzymatic modification of histones per se was insufficient for repositioning. Collectively, this suggests that trans-associated factors influence three-dimensional compartmentalization independent of their cis effect on local chromatin composition and activity.
Graphical Abstract
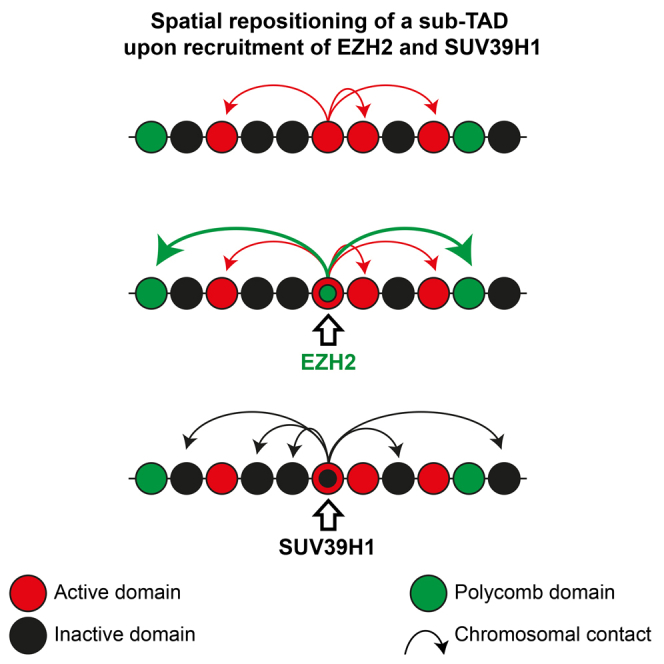
Highlights
-
•
4C technology reveals nuclear repositioning of genomic loci upon protein recruitment
-
•
Given subTAD can adopt multiple nuclear positions depending on the recruited factor
-
•
Switching between nuclear compartments can be uncoupled from transcriptional changes
-
•
Histone-modifying SUV39H1 relies on its chromodomain for locus repositioning
Wijchers et al. combine 4C technology with lacO/lacR binding platforms to show that a given genomic locus can adopt multiple nuclear positions depending on the recruited protein. Focal protein recruitment can move an entire subTAD, but repositioning appeared largely unrelated to changes in its chromatin composition and activity.
Introduction
Nuclear organization of interphase chromosomes is considered to be an important regulator of genome function. Chromosomes occupy nonrandom positions in the nucleus, and the position of a gene relative to nuclear compartments is thought to influence the probability of expression. Microscopy studies and high-throughput chromosome conformation capture (3C) techniques (de Wit and de Laat, 2012) have greatly increased our knowledge of the three-dimensional configuration of the genome in the nucleus (Bickmore and van Steensel, 2013). However, the mechanisms that govern the spatial arrangement of chromosomes and gene positioning in the nucleus are still poorly understood.
Individual chromosomes occupy visually discrete volumes within the nucleus (chromosome territories) (Bolzer et al., 2005). Hi-C and 5C studies revealed that chromosomes are partitioned into hundreds of distinct chromatin interaction domains called topologically associating domains (TADs) (Dixon et al., 2012, Nora et al., 2012). Most physical contacts between gene promoters and their regulatory elements are thought to take place within such domains (Dixon et al., 2015, Shen et al., 2012). TADs vary in their transcriptional activity, DNA sequence, and chromatin composition (Tanay and Cavalli, 2013). In Hi-C data, this segregation of different chromatin types in the nucleus is apparent as a biphasic state with an A and B compartment (Lieberman-Aiden et al., 2009), which have recently been subdivided into A1–2 and B1–4, each with their own chromatin signature (Rao et al., 2014). The A compartment represents colocalization of transcriptionally active or permissive chromatin and B the spatial crowding of inactive chromatin at the nuclear periphery (Guelen et al., 2008), nucleoli (Németh et al., 2010, van Koningsbruggen et al., 2010), and chromocenters (Wijchers et al., 2015). Since this spatial segregation is linked to gene expression, the composition of the A and B compartments is cell-type specific (Dixon et al., 2015). Although this suggests a role for nuclear compartmentalization in gene regulation, to what extent cell-type-specific compartment switches are a cause or consequence of gene activity is largely unclear.
Cell-type-specific nuclear arrangements imply that genomic loci must possess an intrinsic flexibility to allow repositioning in response to differentiation signals. Live-cell microscopy studies demonstrated that gene dynamics are generally constrained during interphase (Chubb et al., 2002), and that with a few exceptions (Chuang et al., 2006), gene repositioning generally requires mitosis before new locations can be adopted in daughter cells (Kumaran and Spector, 2008). Genes are physical parts of much larger chromosomes and will therefore rely on neighboring chromosomal segments for their freedom to move. Neuronal differentiation-induced activation of Mash1 is accompanied by visual relocation toward the nuclear center, dragging along immediately flanking genes that nevertheless remain transcriptionally silent (Williams et al., 2006). Repositioning of genomic regions relative to the nuclear lamina occurs on both single-gene and multi-gene scales (Peric-Hupkes et al., 2010). The factors that influence the scale at which nuclear repositioning is regulated, and the consequences of locus repositioning for surrounding regions, are still unknown.
There are numerous examples of endogenous genes that change nuclear positions during lineage commitment, as judged from fluorescence in situ hybridization (FISH) studies. When silenced, they tend to locate to more peripheral nuclear locations in the nucleus (Peric-Hupkes et al., 2010) or closer to chromocenters (Brown et al., 1997), whereas they adopt a more internal nuclear position when activated (Peric-Hupkes et al., 2010). Repositioning of genes to the nuclear periphery through physical tethering leads to repression of associated genes in many, but not all, cases (Kumaran and Spector, 2008). Peripheral positioning therefore correlates with, but does not strictly imply, gene silencing.
So far, studies designed to experimentally alter the protein composition of a given gene locus and follow its nuclear positioning in a given cell type relied on microscopic techniques that measure location relative to visibly distinct nuclear structures such as the nuclear periphery, chromocenters, nucleoli, Polycomb bodies, speckles, transcription factories, or other gene loci. These nuclear landmarks all contain different DNA- and chromatin-binding proteins. The general perception from these studies is that tethering of compartment-specific proteins leads to locus recruitment to the corresponding nuclear entity. But how do we reconcile this with the compartmentalized genome conformation appreciated from Hi-C studies?
Few studies used 3C-based methods to monitor conformational changes that follow upon binding of regulatory proteins to a locus. 4C technology is highly suited for this, as it unbiasedly assays the genome-wide contacts of a locus of choice (Simonis et al., 2006). Blocking transcription and depleting RNA polymerase II from active gene loci did not cause any major changes in their genome-wide 4C contact maps (Palstra et al., 2008), nor did the recruitment of the glucocorticoid receptor to its target genes (Hakim et al., 2011). The introduction of the β-globin locus control region (a “super enhancer”) into an already active chromosomal region did not induce new contacts but specifically strengthened those already formed with regions targeted by shared transcription factors (Noordermeer et al., 2011). A similarly subtle change in preferred contacts with cognate chromosomal regions was seen when the pluripotency factor NANOG was recruited to an artificial lac operator cassette (de Wit et al., 2013). These latter observations suggest that genes can reside in more than just two (A and B) genomic environments, a notion that receives support from data suggesting that various types of chromosomal regions with similar chromatin signatures preferentially cluster in the nuclear space (Bantignies et al., 2011, Rao et al., 2014, Tolhuis et al., 2011). However, these findings are generally based on genome-wide correlations, but little evidence from experimental locus repositioning is available to support these concepts.
To start exploring the flexibility of different genomic regions in response to the recruitment of various chromatin regulators, we followed chromosomally integrated lacO/lacR platforms and surrounding sequences by 4C technology, and we assessed how repositioning relates to transcriptional changes and epigenetic features.
Results
Experimental System to Manipulate Locus Positioning in the Nucleus
To manipulate the nuclear positioning of defined genomic loci, we sequentially integrated a recruitment platform into two genomic regions of the same embryonic stem cells (ESCs) (Figure 1A). These ESCs were F1s derived from 129/Sv and C57BL/6 mouse strains, with only the alleles from the latter carrying the recruitment platform. This platform consisted of a kanamycin resistance gene flanked on either side by an array of 120 lac operator (lacO) repeats that serve as high-affinity binding sites for the Escherichia coli Lac repressor (lacR) (Figure 1A). After integration of the first array on chromosome 8 (previously used in de Wit et al., 2013), the additional floxed Neomycin resistance gene was removed to allow a second integration on chromosome 11 in the same cells. Correct and single integrations were validated by Southern blot (Figure S1A, available online).
Figure 1.

A lacO/lacR Recruitment Platform to Induce Nuclear Repositioning
(A) Schematic view of lacO integration sites on chr8 and chr11. trans-acting factors are recruited to the lacO array when fused to EGFP-lacR. See also Figure S1.
(B) Western blots confirming fusion proteins are of the correct size. Protein markers (left) and expected size of fusion protein (below) are indicated.
(C) Nuclear distributions of the fusion proteins used.
(D) ChIP with GFP antibody to confirm binding of each fusion protein to the lacO cassette.
We then expressed three different transcription and chromatin factors fused to an enhanced green fluorescent protein (EGFP)-lacR moiety to direct these proteins to the lacO cassettes. These factors included EZH2, the component of the Polycomb Repressive Complex 2 (PRC2) that is responsible for H3K27 trimethylation; SUV39H1, the histone methyltransferase factor that deposits H3K9me3 in constitutive heterochromatin; and NANOG, a pluripotency transcription factor that can have an activating as well as repressive effect on transcription (Liang et al., 2008). Viral transduction, followed by 10 days of cellular selection and expansion to obtain sufficient cells with a large enough (>70%) proportion of GFP-positive cells for subsequent analysis, led to expression of fusion proteins of the expected size but at varying levels (Figure 1B). EGFP-lacR showed a uniform nuclear distribution, except for two bright spots marking the lacO arrays (Figure 1C). These two bright foci were also visible in EGFP-lacR-NANOG cells, which otherwise displayed a more grainy pattern than EGFP-lacR alone, consistent with the thousands of NANOG binding sites across the genome. EGFP-lacR-EZH2 was found throughout the nucleus with multiple bright foci, presumably highlighting the presence of Polycomb bodies (Cheutin and Cavalli, 2014). EGFP-lacR-SUV39H1 showed the typical enrichment in DAPI-dense pericentromeric heterochromatin foci (PCH) (Aagaard et al., 1999). We checked the expression of a number of endogenous target genes in cells ectopically expressing EGFP-lacR-EZH2 and EGFP-lacR-NANOG and found this essentially unaltered or slightly reduced (in case of EZH2 expression) (Figure S1B). Binding of the fusion proteins to the lacO arrays was further verified by chromatin immunoprecipitation (ChIP) using antibodies against GFP, confirming again that all proteins bound to the array, albeit with different efficiencies (Figure 1D). Thus, all fusion proteins bound to the lacO arrays and showed the expected nuclear distribution.
Locus Susceptibility to Spatial Repositioning Depends on Genomic Location and Associated Factors
To explore the ability of the regulatory proteins to change the nuclear position of the lacO loci, we applied 4C-seq (Splinter et al., 2012), a 3C-based technology which probes for chromosomal regions spatially juxtaposed to a genomic site of interest. For the lacO locus on chromosome 11 (chr11), we used a 4C viewpoint in the Neomycin resistance (Neo) gene. To follow the lacO cassette on chromosome 8 (chr8), which no longer had a Neo gene, we employed an allele-specific 4C-seq (Splinter et al., 2011), taking advantage of a SNP in the same gene (Nfix) but 30 kilobases (kb) upstream of the lacO array. The SNP allowed distinguishing contacts made by the lacO-containing BL/6 allele and the wild-type 129/Sv allele, and we confirmed that it produced almost identical profiles to those of the Neo viewpoint in single-targeted cells (Figure S2A). Interchromosomal contacts are not nearly as abundant as contacts within a given chromosome, and their robust detection by 4C would require deeper sequencing of more complex 4C libraries than those analyzed here. Therefore, as in most Hi-C studies, we limit ourselves to the analysis of intrachromosomal contacts, which, in contrast, can readily be identified.
We found that binding of each chromatin protein had little impact on the genomic contacts made by the lacO locus on chr8 (Figure S2B). Very few, mostly quantitative, contact changes occurred within the otherwise unaltered genomic environment. Only upon EZH2 recruitment was one prominent new contact seen, with an H3K27me3-rich chromosomal region located at ∼127 Mb of chr8 (Figure S2B; data not shown). In these same cells, all three chromatin proteins, but not EGFP-lacR alone (Figure 2A), had a much more pronounced impact on the contact profiles of the lacO locus on chr11 (Figures 2B–2D). NANOG and EZH2 recruitment each led to new contacts with regions not seen by the untargeted, or LacR-bound, array (Figures 2B and 2C); these changes were relatively subtle in the case of NANOG, whereas EZH2 recruitment induced prominent new contacts across a number of loci. Even more dramatic changes in contacts were seen after binding of SUV39H1, which led to massive loss of 4C signals across nearly all commonly contacted loci and a concomitant gain in 4C signals at normally ignored intervening chromosomal parts (Figure 2D). Thus, it appears that the susceptibility of a locus to change its 3D genomic neighborhood depends on genomic location. Since newly juxtaposed regions were different between NANOG-, EZH2-, and SUV39H1-transduced cells, the direction and degree of repositioning appears to be determined by associated trans-acting factors.
Figure 2.

Susceptibility to Spatial Repositioning Depends on Genomic Location and trans-Acting Factors
(A–D) 4C profiles comparing chromosome-wide contacts of the lacO cassette on chr11 in (A) untransduced and EGFP-lacR transduced cells, (B) EGFP-lacR and EGFP-lacR-NANOG transduced cells, (C) EGFP-lacR and EGFP-lacR-EZH2 transduced cells, and (D) EGFP-lacR and EGFP-lacR-SUV39H1 transduced cells. The position of the lacO array is indicated. Arrowheads point at clear examples of differentially contacted regions. See also Figure S2.
Factor-Dependent Switching between Subcompartments with Distinct Chromatin Signatures
To further explore factor-dependent repositioning, we quantitatively compared normalized 4C profiles of transduced and untransduced cells and characterized the differentially contacted chromosomal segments. The targeting of NANOG induced only small shifts in the LacO-contacted regions, but the chromosomal parts that gained contacts were enriched in binding sites for NANOG (Figure S3), as seen before (de Wit et al., 2013). EGFP-lacR-Ezh2 profiles also globally matched that of EGFP-lacR, except at two regions that showed a very prominent accumulation of contacts, one being the HoxB cluster and the other being a cluster of Cbx genes (Figure 3A). These loci efficiently recruit both the Polycomb PRC2 complex protein EZH2 and the PRC1 subunit Ring1B (Ku et al., 2008) and are highly enriched for H3K27me3 (Figure 3A), a modification for which EZH2 is responsible (Simon and Kingston, 2009). Systematic analysis indeed showed that EZH2 recruitment to LacO promoted contacts with other chromosomal regions enriched for the H3K27me3 mark (Figure 3B). This shows that EZH2 promotes spatial crowding of genomic regions with a Polycomb chromatin signature.
Figure 3.

Local Chromatin Signature Influences Direction of Nuclear Repositioning
(A) 4C profiles of the lacO array viewpoint from EGFP-lacR (blue) and EGFP-lacR-EZH2 (red) transduced cells. Density of H3K27me3 sites is indicated below profiles. Red and blue bars above profiles indicate windows where 4C signal was significantly enriched in EGFP-lacR-EZH2 versus EGFP-lacR. See also Figure S4.
(B) Enrichment values of indicated histone modifications in regions significantly enriched upon EZH2 binding to the lacO (red bars in A). See also Figure S3.
(C) Same as (A), but for EGFP-lacR (blue) and EGFP-lacR-SUV39H1 (red). Density of H3K9me3 sites is indicated. Bottom panel displays Hi-C principle component values, corresponding to A and B compartments.
(D) As in (B), but for regions enriched upon SUV39H1 binding to the lacO.
EGFP-lacR-SUV39H1 binding to the lacO led to massive changes in contact frequencies across large chromosomal intervals (Figure 3C). This time, the newly contacted domains were depleted for the H3K27me3 chromatin mark and lacked H3K4me3, but were strongly enriched for H3K9me3, the histone mark deposited by SUV39H1 (Figure 3D). Thus, like EZH2 and NANOG, SUV39H1 binding causes the locus to change its preferred genomic neighbors in nuclear space. The three factors, however, induce different topological changes, each promoting locus movement to distinct nuclear environments occupied by genomic regions with the corresponding associated factors and chromatin signatures. To validate the 4C results, we performed DNA FISH using BAC probes against the integration site, two SUV39H1-contacted regions, and one EZH2-contacted region (the HoxB locus), plus a probe against the LacO repeats. Whereas the LacO-targeted site and its corresponding untargeted site on the homologous chromosome showed very similar contact frequencies with each of the three tested regions, we found significantly increased contact frequencies (p value < 0.001; one-sided test) with the HoxB locus exclusively upon EZH2 recruitment to the LacO-targeted site. Similarly, upon SUV39H1 recruitment the LacO site showed significantly increased FISH contacts (p value < 0.01 for both) with the two 4C-predicted SUV39H1 target regions (Figure S4). Thus, as extensively shown before (see, e.g., Simonis et al., 2006), FISH confirms that our 4C approach faithfully identifies long-range chromosomal contacts.
Since the locus on chr11 can be forced to move to at least three different genomic environments, our data confirm recent evidence for the existence of multiple subcompartments (Rao et al., 2014) within the “active” A and “inactive” B compartments (Lieberman-Aiden et al., 2009). We intersected the 4C contact profiles with Hi-C data generated from ESCs to investigate how the gained and lost contacts compared to regions belonging to the originally defined A and B compartments (Lieberman-Aiden et al., 2009). The untargeted locus on chr11 resides in the active A compartment (Figure 3C). This is true also for the HoxB and the Cbx gene clusters that are nevertheless ignored by the locus unless EZH2 is recruited. The same is true for NANOG-dense regions: they belong to the A compartment but are only selected by lacO as preferred genomic neighbors upon NANOG recruitment. As shown before, NANOG-dense chromosomal regions and Polycomb-associated regions each preferentially cluster among themselves (de Wit et al., 2013, Denholtz et al., 2013, Vieux-Rochas et al., 2015). Thus, we independently corroborate recent high-resolution Hi-C data showing that the A compartment can be further subdivided into subcompartments in which similarly typed chromosomal regions specifically accumulate (Rao et al., 2014). In ESCs, the cell-type-specific pluripotency factors establish one such subcompartment, and Polycomb group proteins establish another A-type subcompartment. Depending on its associated proteins, an integrated LacO platform can switch between these subcompartments.
Interestingly, SUV39H1 recruitment caused the locus to switch from the A to the B compartment. Regions originally surrounding the untargeted lacO locus that massively lost contacts upon SUV39H1 binding all belonged to the A compartment, whereas the chromosomal parts that accumulated 4C signals showed a striking correlation with those belonging to the B compartment. In such instances, new contacts were observed across the entire B compartment domains extending to their boundaries, beyond which a loss in contacts was seen. This shows that in the same cell type a locus can switch environments not only within a compartment but also from one compartment to another.
Spatial Repositioning Is Regulated at the SubTAD Level
To validate these results, we performed reciprocal 4C-seq experiments with viewpoints located in newly identified EZH2- or SUV39H1-specific contact domains. 4C profiles from the HoxB locus (∼6 Mb away) confirmed prominent new contacts around the lacO integration site in EGFP-lacR-Ezh2 transgenic cells that were absent in untransduced cells (Figure 4A). Similarly, looking from a locus containing the Asic2 gene (>20 Mb away), i.e., one of the regions contacted specifically upon SUV39H1 recruitment to LacO, revealed a clear SUV39H1-specific peak at the position of the lacO transgene (Figure 4B). This confirms the chromosome conformational changes induced by the local recruitment of these chromatin factors. Looking more closely at the reciprocal 4C contact data, we noticed that the segment around the lacO integration site with the highest differential signal spanned a similar-sized region for both EGFP-lacR-Ezh2- and EGFP-lacR-Suv39h1-transduced cells (Figures 4C and 4D). The size and the borders of this region corresponded to a structural domain appreciable from a Hi-C contact matrix (Figure 4E) that we produced for mouse ESCs (based on the analysis of 26 million valid Hi-C tags with a cis/total enrichment of 74%; Geeven et al., 2015). This region measures approximately 200 kb, which is much smaller than the originally defined TAD at this chromosomal location (spanning ∼840 kb, from chr11:101480001–102320000) (Dixon et al., 2012) and would therefore better qualify as a subTAD (Phillips-Cremins et al., 2013). It therefore appears that focal recruitment of trans-acting factors can cause an entire subTAD to get engaged in new contacts.
Figure 4.

A Repositioned Locus Drags along Its Associated SubTAD
(A) 4C profiles looking from the EZH2-induced contact at the HoxB cluster (indicated by arrowhead) in EGFP-lacR (blue) and EGFP-lacR-EZH2 (red) transgenic cells. Location of the lacO integration site is indicated by dashed line. See also Figure S4.
(B) Same as (A), but for EGFP-lacR (blue) and EGFP-lacR-SUV39H1 (red), looking from a SUV39H1-induced contact in the Asic2 gene. Note that the 4C is not allele specific, implying that contacts come from both alleles, which effectively dilutes the signal at the lacO integration site.
(C) Same as (A), zoomed in on the lacO locus. The gray area highlights the region with the highest differential 4C signal.
(D) Same as (B), zoomed in on the lacO locus. The gray area highlights the region with the highest differential 4C signal.
(E) Hi-C contact matrix of region surrounding the lacO locus on chr11 (data from ESCs). Dashed lines correspond to the borders of the gray area in (C) and (D). Note that the borders coincide with edges of a block of locally increased contact frequencies (lacO subTAD). Red arrowheads indicate positions of viewpoints used in (F).
(F) Allele-specific 4C profiles comparing the lacO and wild-type alleles upon binding of EZH2 for a viewpoint 335 kb upstream of the lacO in a neighboring subTAD, 80 kb upstream within the same subTAD, and 209 kb and 418kb downstream of the LacO in neighboring subTADs.
(G) Boxplots showing the percentage of reads coming from the lacO-transgenic allele (versus wild-type allele) across regions mapping to the B compartment upon EGFP-lacR-SUV39H1 (red) or EGFP-lacR only (blue) binding. See also Figure S5.
To further investigate the extent to which the 3D positioning of flanking sequences is influenced by protein recruitment to LacO, we performed allele-specific 4C from viewpoints that lie up- and downstream of the lacO array and compared profiles between the lacO-transgenic and untargeted allele. Upon EZH2 binding to the lacO locus, a viewpoint in the same subTAD but 80 kb upstream of LacO showed the same switch in chromosomal contacting partners as seen for the lacO array itself (Figure 4F), making many new distal contacts with the upstream HoxB locus and the downstream Cbx cluster. Viewpoints further upstream (−335 kb) and downstream (+209 and +418 kb) of lacO, both located in directly neighboring subTADs (Figure 4E), hardly showed these differential contacts upon EZH2 and SUV39H1 binding (Figure 4F). Similarly, only the viewpoint located at −80 kb in the same subTAD as lacO followed lacO in its A → B compartment switch upon SUV39H1 binding, while viewpoints in neighboring subTADs appeared resistant to this repositioning (Figures 4G and S5). We conclude that the 3D positioning of chromosomal segments can be controlled at the level of subTADs, i.e., at the submegabase level involving chromosomal regions that measure only a few hundreds of kilobases in size. Sequences belonging to the same subTAD experience the same genomic environment, while those belonging to flanking subTADs may occupy another nuclear subcompartment.
Nuclear Compartment Switching and Gene Expression Are Not Causally Related
Having established a system where we can manipulate the spatial genomic neighborhood of a subTAD, we then wished to understand the relationship with gene expression. We first asked whether transcriptional changes could be a driving force behind repositioning, focusing on the expression of the Neo gene that was cointegrated with and immediately flanking the lacO array on chr11. Neo expression was reduced by the binding of the trans-acting factors (Figure 5A), consistent with their roles as transcriptional repressors (Liang et al., 2008). EGFP-LacR binding by itself was also sufficient to repress Neo expression (Figure 5A), a phenomenon seen before and attributed to the tight binding of lacR to DNA (Dubarry et al., 2011). In the case of EGFP-lacR alone, repression occurred without nuclear repositioning of the locus; for NANOG and EZH2 this was accompanied by engagement with a few new but mutually exclusive contacting regions, and for SUV39H1 this occurred concomitant with a seemingly complete switch toward the B compartment. We conclude therefore that a change in transcriptional output is insufficient for nuclear repositioning of the subTAD.
Figure 5.

Spatial Repositioning Does Not Drive Gene Expression Changes
(A) Quantitative RT-PCR analysis of Neomycin (two primer sets) and control gene (Hprt). Data was normalized to Actb and expressed as fold-change over untransduced (UT) cells. Error bars represent SD.
(B) Schematic overview of genes surrounding the lacO integration site. Genes that are transcriptionally inactive in ES cells are indicated by “off.” The gray area corresponds to the subTAD that is repositioned upon SUV39H1 binding (see also Figure 4).
(C) Quantitative RT-PCR on the two genes most nearby the lacO integration site. Note that the downregulated Rundc3a gene is only 36 bp downstream of the lacO transgene. Error bars represent SD.
(D) SNP-based expression analysis of the more distal Atxn713 and Ubtf genes. Data plotted are the fold-changes in the fraction of reads coming from the lacO-targeted allele. Values of all replicates were normalized to the first untransduced (UT) replicate. Error bars represent SD.
Vice versa, repositioning may induce expression changes of the genes that comigrate with lacO to new compartments upon factor tethering. To investigate whether and to what extent this occurs, we focused on the genes immediately surrounding LacO in the same subTAD of chr11 (Figure 5B). In SUV39H1-transduced cells these genes follow LacO (Figure 4G) and migrate from the A to the B compartment in a large proportion of the cells (Figure 3C). We analyzed their expression by quantitative RT-PCR (Figure 5C) or by next-generation sequencing of PCR-amplified cDNA products (Figure 5D), using SNPs that allowed discrimination of the lacO-transgenic allele. From this, we found that only the immediately flanking Rundc3a gene, with its promoter 36 bp downstream of the integration site, displayed decreased expression levels. It did so, however, not only upon SUV39H1 binding but also upon EGFP-lacR binding (Figure 5C), which does not induce spatial repositioning of the domain. Rundc3a expression was unaffected (or even slightly upregulated) upon EZH2 recruitment, despite this factor causing spatial repositioning to Polycomb bodies. Altered Rundc3 gene expression therefore cannot be attributed to nuclear repositioning. The expression of other genes across the repositioned subTAD was unaffected upon SUV39H1 and EZH2 recruitment (Figures 5C and 5D). We therefore conclude that nuclear repositioning of the subTAD does not drive changes in its gene expression, and vice versa. In summary, genome-wide datasets clearly show that transcriptional control and genomic compartmentalization correlate, but consistent with other findings (Dixon et al., 2015, Palstra et al., 2008, Therizols et al., 2014) our results emphasize that they are often not causally related.
Local Modification of Histones Is Not Sufficient for Nuclear Repositioning
We next wished to investigate the role of chromatin composition in the spatial repositioning. Consistent with their enzymatic activity, EZH2 and SUV39H1 recruitment led to local deposition of H3K27me3 and H3K9me3, respectively, at the lacO locus on chr11 (Figures 6A–6C). Although we did not extensively analyze the corecruitment of other PcG proteins, we did notice that EZH2 (a PRC2 complex member) attracted Ring1B, a PRC1 component (Figure S6A). The H3K27me3 on chr11 did not spread far into adjacent regions: relative levels increased somewhat across the subTAD but remained low in absolute terms, with the exception of a site at +60 kb that is naturally rich in H3K27me3 (Figures 6A and 6B). This is consistent with recent findings that EZH2 spreading upon induced recruitment of Polycomb group proteins to a tetO transgene is usually limited to <10 kb (Blackledge et al., 2014). Similarly, upon SUV39H1 recruitment to lacO, there was a modest increase in H3K9me3 levels at the surrounding sequences, but their absolute levels remained marginal compared to the array itself (Figure 6C).
Figure 6.

The Role of Chromatin in Spatial Repositioning
(A) Schematic overview of lacO locus on chr11, with respect to CTCF binding sites (gray), shared CTCF-cohesin sites (blocks), H3K27me3 (blue), and H3K9me3 (red) histone modifications. Locations of ChIP primers are indicated. Grey area corresponds to the subTAD that is repositioned upon EZH2 or SUV39H1 binding (Figure 4).
(B) ChIP for H3K27me3 in untransduced (light blue) and EGFP-lacR-EZH2 (dark blue) cells for the genomic sites indicated in (A). Input-normalized data are expressed as enrichment over the (H3K27me3-rich) Pax2 promoter. See also Figure S6.
(C) As in (B) but for H3K9me3 in untransduced (light red) and EGFP-lacR-SUV39H1 (dark red) transduced cells. Input-normalized data is expressed as enrichment over the (H3K9me3-rich) Mfap3 gene.
(D) Schematic display of the wild-type and mutant SUV39H1 proteins. See also Figure S6.
(E) Nuclear distributions of the fusion proteins used.
(F) ChIP data confirming H3K9me3 deposition upon binding of both wild-type and mutant SUV39H1. Error bars represent SD.
(G) Quantitative RT-PCR analysis of Neomycin (two primer sets). Data were normalized to Actb and expressed as fold-change over EGFP-lacR cells. Error bars represent SD.
(H) 4C profiles of the lacO array viewpoint (arrowhead) from EGFP-lacR (blue) and EGFP-lacR-SUV39H1ΔCD (red) cells. Bottom panel displays Hi-C principle component values, with positive and negative corresponding to A and B compartments, respectively.
The deposition of these chromatin marks and the concomitant repositioning of the locus to nuclear subcompartments of similar chromatin signature (Figure 3) raise the possibility that histone modifications play an active role in the engagement of specific long-range contacts and establishment of nuclear subcompartments. To further investigate this, we constructed a mutant SUV39H1 that lacked the chromodomain (CD) but retained the histone methyltransferase domains (Figure 6D). The CD is required for binding to H3K9me3, and, correspondingly, this SUV39H1ΔCD mutant no longer localized to PCH (Figure 6E). It still bound to the lacO array and efficiently deposited H3K9me3 (Figures 6E,6F, and S6B). The SUV39H1ΔCD mutant was also fully capable of repressing Neo gene expression to levels lower than that already induced by EGFP-LacR alone and even slightly lower than that induced by wild-type SUV39H1(Figure 6G). The latter seems to correspond to the somewhat higher levels of H3K9me3 deposited by the SUV39H1ΔCD mutant (Figure 6F). Despite this evident activity, however, SUV39H1ΔCD binding, in contrast to SUV39H1, did not result in locus repositioning. We no longer observed distinct interactions with genomic regions in the B compartment (Figure 6H), but instead, SUV39H1ΔCD gave similar 4C profiles to those of EGFP-LacR transduced cells (Figure 6H). This implies that local deposition of H3K9 methylation may control gene silencing but is insufficient to direct spatial repositioning.
Discussion
The link between nuclear localization, chromatin composition, and gene expression is primarily based on correlations extracted from genome-wide datasets. Microscopy studies tracing the location of individual loci in general confirm established relationships but also reveal exceptions (Kumaran and Spector, 2008) and show that there is large cell-to-cell variability in nuclear positioning of given loci (Kind et al., 2013). Tracing a locus by microscopy relies on measuring distances relative to visible landmarks such as the nuclear periphery, chromocenters, or selected other loci, but how such movement relates to the TADs, compartments, and other topological features appreciable from high-throughput 3C data is unclear. With the aim to bridge this gap and establish cause and consequence in the three-way relationship between nuclear organization, gene expression, and chromatin composition, we combined lacO/LacR recruitment platforms with allele-specific 4C technology. We recruited a selection of trans-acting proteins that could modulate these parameters at defined genomic locations in the same cell type. Although endogenous genes are usually not targeted by so many copies of a given protein, repressive H3K27me3 and H3K9me3 marks can span genomic regions of tens of kb (Hawkins et al., 2010), not unlike the ∼10 kb lacO array integrated here.
Susceptibility to Spatial Repositioning Depends on Genomic Location
All three trans-acting factors were able to change the spatial positioning of the lacO locus when integrated upstream of Rundc3a on chr11, albeit to different extents. However, the same lacO array integrated in the Nfix gene on chr8 seemed almost impervious to these large-scale repositioning influences, showing only minor small-scale changes (Figure S2; de Wit et al., 2013). As data from both integration sites comes from the same cell with identical amounts of lacO repeats in each transgene, these differential responses cannot be explained by technical differences. This proves, therefore, that genomic context is an important determinant of the flexibility of a locus to roam the nucleus (Krijger and de Laat, 2013). What elements in the genomic habitat surrounding these lacO arrays are responsible for the differential capacity to relocalize is still unclear. Differences in gene density, transcriptional activity, and distance to telomere and centromere may play a role (Brown et al., 2008), but most of these parameters are not markedly different between the lacO loci on chr8 and chr11. For example, both loci localize to the A (active) compartment in ESCs. The integration site at 102.2 Mb on chr11 (122 Mb in size) is considerably closer to the telomere, though, and therefore possibly more flexible than the integration site at 87.5 Mb on chr8 (129 Mb in size). Studies using more systematic approaches such as TRIP (Akhtar et al., 2014) could perhaps help to further dissect the parameters dictating locus mobility in the future.
Changes in Nuclear Position Are Controlled by Topological Constraints
As each locus is physically connected to the rest of its chromosome, its repositioning will by default carry along adjacent chromosomal regions. Differentiation-induced changes in association with the nuclear lamina and A/B compartments usually correspond to one or multiple neighboring TADs (Dixon et al., 2015, Peric-Hupkes et al., 2010). However, the causes and consequences of these movements are unclear; do these represent the coordinated movement of the complete TADs, or are they mostly the result of passive movement following the regulated repositioning of a single gene?
Here, we establish that localized chromatin modification of a relatively small chromosomal segment induces new contacts with chromosomal regions elsewhere on the same chromosome. Our reciprocal 4C analysis reveals that the remainder of the associated subTAD, rather than the megabase-scaled TAD, also engages in the same contacts. Thus, the nuclear position of a locus can be controlled at the level of subTADs, which hints at a hitherto unknown flexibility of subTADs to adopt a different nuclear position from the remainder of its associated TAD. Overall, we expect that spatial contact partners of a given subTAD will be selected based on their chromatin composition and linear proximity on the chromosome, but that with additional constraints imposed by the remaining segments of the chromosome, each will have their own preference for a spatial neighborhood.
trans-Acting Factors Form Nuclear Subcompartments
What determines where a locus is repositioned to? Although binding of all three trans-acting factors had a repressive effect on gene expression, they guided their target locus in different directions or kept it at the same location. This suggests that loci can have a remarkable flexibility in their nuclear localization and that loci with the same transcriptional status can occupy different subcompartments in the nucleus. It also shows that the repositioning to different subcompartments cannot be the mere consequence of transcriptional changes.
Our data suggest that specific nuclear subcompartments form as a consequence of affinities between proteins and/or modifications associated with the participating chromosomal regions. All three trans-acting factors showed a strong preference to move the target locus to genomic neighborhoods with a chromatin signature corresponding to that deposited by these factors at the lacO array. How these proteins impose the observed repositioning is not entirely clear. Given the limited freedom of chromatin to roam the interphase nucleus, the substantial repositioning that we measure probably depends on one or multiple rounds of mitosis, as also seen in other systems (Kumaran and Spector, 2008). Polycomb group proteins have the intrinsic property to aggregate to establish a compact chromatin structure when bound to nearby nucleosomes (Francis et al., 2004). Likewise, the deposition of H3K9me3 by SUV39H1 serves as a docking platform for a whole range of heterochromatic proteins, including heterochromatin protein 1, which is thought to oligomerize to promote chromatin compaction (Grewal and Jia, 2007). Similar to experimentally induced chromatin loops through spatial interaction between two dimerization domains at an enhancer and promoter (Deng et al., 2012), the inclination of such protein complexes to self-associate may therefore increase the probability of the lacO locus to stabilize contacts with genomic sites that are bound by the same factors.
Despite the strong association of chromatin modifications with topological organization of the genome, the failure of our mutant SUV39H1 to reposition the lacO locus despite its normal methyltransferase activity implies that focal histone modifications are not sufficient to drive spatial repositioning. The mutant SUV39H1 version lacked the chromodomain, a protein module responsible for binding to H3K9me3. We previously demonstrated that recruitment of only the chromodomain of CBX1/HP1beta was sufficient to relocate and tether a lacO array to PCH, presumably through the simultaneous binding of lacR to the lacO repeats and the chromodomain to the H3K9me3 decorations abundant in PCH (Wijchers et al., 2015). Our current finding that SUV39H1 requires its chromodomain for repositioning seems fully consistent with this. Then why is the H3K9me3 deposition on LacO by itself not sufficient for PCH-associated chromodomains to grab and reposition the locus? A possible explanation may be that there are not sufficient H3K9me3 molecules per lacO allele to efficiently compete for stable associations with chromodomain-containing proteins that are accumulated at PCH, or that the presence of a mutant SUV39H1 acts as steric hindrance and shields H3K9me3 from chromodomains. Alternatively, the N-terminal part of SUV39H1 that was removed is also responsible for interactions with HP1 and required for recruitment of the complete set of classical heterochromatin proteins (Muramatsu et al., 2013). The heterochromatin established at the lacO array by the mutant SUV39H1 may therefore lack particular modules that are involved in nuclear repositioning.
Altogether, our data show that subTADs differ in their susceptibility to adopt new nuclear positions. We identify nuclear subcompartments where subTADs with similar epigenetic features can be recruited. Associated trans-acting factors drive this nuclear compartmentalization independent of their effect on local chromatin composition and activity. While there seems to be no causal relationship between compartmentalization and gene expression, this 3D organization will cause spatial crowding of transcription and chromatin factors, which is likely to contribute to maintenance of transcriptional states.
Experimental Procedures
lacO Targeting and LacR-Fusion Transduction
The lacO array was inserted in chr8 and chr11 via site-specific homologous recombination, as described (de Wit et al., 2013). LacO-transgenic cells were transduced with EGFP-lacR fusions using lentivirus, selected with puromycin for approximately 10 days when cells had reached sufficient numbers for collection, and tested for purity by flow cytometry (minimum 70% GFP positive). Further details can be found in the Supplemental Information.
4C-Seq Analysis
4C sample preparation was performed as described (Splinter et al., 2012); data mapping and analysis was as described (de Wit et al., 2013). To allow direct comparisons of 4C profiles of different samples, mapped reads are normalized for sequencing depth by multiplying by a factor such that the total sum of mapped reads in cis (i.e., on the bait chromosome), after discarding the 4C fragend with the highest multiplicity, equals one million. For visualization, we compute the average 4C signal across windows of 51 consecutive fragends (centered on the 25th) per experiment. Per condition we plot the average of this signal as taken from two independent biological replicates.
FISH
FISH was performed as described previously and detailed in the Supplemental Information. ImageJ (Image5D plugin) software was used to score touching (no unstained pixel in between FISH signals) and nontouching (with unstained pixel(s) in between) alleles.
Author Contributions
P.J.W., P.H.L.K., and Y.Z. performed all experiments with help from A.D., M.J.A.M.V., M.J., C.V.-Q., C.V., and H.T. All computational and statistical analyses were performed by G.G. L.C.M.A.-G. and P.J.V. provided reagents. P.J.W., P.H.L.K., and W.d.L. designed and supervised the experiments, and the manuscript was written by P.J.W. and W.d.L.
Acknowledgments
We thank Daan Noordermeer, Erik Splinter, Petra Klous, and Harmen van de Werken for technical support; Niels Geijsen for sharing phage2 constructs; Stefan van der Elst for FACS sorting; Stieneke van den Brink for ESC culture; and Anko de Graaff and the Hubrecht Imaging Center for supporting the imaging. This work was supported by a Long-Term Postdoctoral Fellowship of the Human Frontier Science Program (LT000709/2013) to A.D., an NWO/CW TOP grant (714.012.002), an NWO VICI grant 724.012.003, a NanoNextNL grant, an EU grant 2010-259743 (MODHEP), and a European Research Council Starting Grant (209700, “4C”) to W.d.L.
Published: January 28, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.01.001.
Accession Numbers
The accession number for the 4C data reported in this paper is GEO: GSE76174.
Supplemental Information
References
- Aagaard L., Laible G., Selenko P., Schmid M., Dorn R., Schotta G., Kuhfittig S., Wolf A., Lebersorger A., Singh P.B. Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3-9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J. 1999;18:1923–1938. doi: 10.1093/emboj/18.7.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar W., Pindyurin A.V., de Jong J., Pagie L., Ten Hoeve J., Berns A., Wessels L.F., van Steensel B., van Lohuizen M. Using TRIP for genome-wide position effect analysis in cultured cells. Nat. Protoc. 2014;9:1255–1281. doi: 10.1038/nprot.2014.072. [DOI] [PubMed] [Google Scholar]
- Bantignies F., Roure V., Comet I., Leblanc B., Schuettengruber B., Bonnet J., Tixier V., Mas A., Cavalli G. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 2011;144:214–226. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- Bickmore W.A., van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152:1270–1284. doi: 10.1016/j.cell.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Blackledge N.P., Farcas A.M., Kondo T., King H.W., McGouran J.F., Hanssen L.L., Ito S., Cooper S., Kondo K., Koseki Y. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 2014;157:1445–1459. doi: 10.1016/j.cell.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolzer A., Kreth G., Solovei I., Koehler D., Saracoglu K., Fauth C., Müller S., Eils R., Cremer C., Speicher M.R., Cremer T. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.E., Guest S.S., Smale S.T., Hahm K., Merkenschlager M., Fisher A.G. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell. 1997;91:845–854. doi: 10.1016/s0092-8674(00)80472-9. [DOI] [PubMed] [Google Scholar]
- Brown J.M., Green J., das Neves R.P., Wallace H.A., Smith A.J., Hughes J., Gray N., Taylor S., Wood W.G., Higgs D.R. Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J. Cell Biol. 2008;182:1083–1097. doi: 10.1083/jcb.200803174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheutin T., Cavalli G. Polycomb silencing: from linear chromatin domains to 3D chromosome folding. Curr. Opin. Genet. Dev. 2014;25:30–37. doi: 10.1016/j.gde.2013.11.016. [DOI] [PubMed] [Google Scholar]
- Chuang C.H., Carpenter A.E., Fuchsova B., Johnson T., de Lanerolle P., Belmont A.S. Long-range directional movement of an interphase chromosome site. Curr. Biol. 2006;16:825–831. doi: 10.1016/j.cub.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Chubb J.R., Boyle S., Perry P., Bickmore W.A. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. 2002;12:439–445. doi: 10.1016/s0960-9822(02)00695-4. [DOI] [PubMed] [Google Scholar]
- de Wit E., de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E., Bouwman B.A., Zhu Y., Klous P., Splinter E., Verstegen M.J., Krijger P.H., Festuccia N., Nora E.P., Welling M. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013;501:227–231. doi: 10.1038/nature12420. [DOI] [PubMed] [Google Scholar]
- Deng W., Lee J., Wang H., Miller J., Reik A., Gregory P.D., Dean A., Blobel G.A. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. doi: 10.1016/j.cell.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denholtz M., Bonora G., Chronis C., Splinter E., de Laat W., Ernst J., Pellegrini M., Plath K. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 2013;13:602–616. doi: 10.1016/j.stem.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubarry M., Loïodice I., Chen C.L., Thermes C., Taddei A. Tight protein-DNA interactions favor gene silencing. Genes Dev. 2011;25:1365–1370. doi: 10.1101/gad.611011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis N.J., Kingston R.E., Woodcock C.L. Chromatin compaction by a polycomb group protein complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- Geeven G., Zhu Y., Kim B.J., Bartholdy B.A., Yang S.M., Macfarlan T.S., Gifford W.D., Pfaff S.L., Verstegen M.J., Pinto H. Local compartment changes and regulatory landscape alterations in histone H1-depleted cells. Genome Biol. 2015;16:289. doi: 10.1186/s13059-015-0857-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal S.I., Jia S. Heterochromatin revisited. Nat. Rev. Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W., van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Hakim O., Sung M.H., Voss T.C., Splinter E., John S., Sabo P.J., Thurman R.E., Stamatoyannopoulos J.A., de Laat W., Hager G.L. Diverse gene reprogramming events occur in the same spatial clusters of distal regulatory elements. Genome Res. 2011;21:697–706. doi: 10.1101/gr.111153.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins R.D., Hon G.C., Lee L.K., Ngo Q., Lister R., Pelizzola M., Edsall L.E., Kuan S., Luu Y., Klugman S. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J., Pagie L., Ortabozkoyun H., Boyle S., de Vries S.S., Janssen H., Amendola M., Nolen L.D., Bickmore W.A., van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–192. doi: 10.1016/j.cell.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Krijger P.H., de Laat W. Identical cells with different 3D genomes; cause and consequences? Curr. Opin. Genet. Dev. 2013;23:191–196. doi: 10.1016/j.gde.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Ku M., Koche R.P., Rheinbay E., Mendenhall E.M., Endoh M., Mikkelsen T.S., Presser A., Nusbaum C., Xie X., Chi A.S. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran R.I., Spector D.L. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 2008;180:51–65. doi: 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Wan M., Zhang Y., Gu P., Xin H., Jung S.Y., Qin J., Wong J., Cooney A.J., Liu D., Songyang Z. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat. Cell Biol. 2008;10:731–739. doi: 10.1038/ncb1736. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu D., Singh P.B., Kimura H., Tachibana M., Shinkai Y. Pericentric heterochromatin generated by HP1 protein interaction-defective histone methyltransferase Suv39h1. J. Biol. Chem. 2013;288:25285–25296. doi: 10.1074/jbc.M113.470724. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Németh A., Conesa A., Santoyo-Lopez J., Medina I., Montaner D., Péterfia B., Solovei I., Cremer T., Dopazo J., Längst G. Initial genomics of the human nucleolus. PLoS Genet. 2010;6:e1000889. doi: 10.1371/journal.pgen.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer D., de Wit E., Klous P., van de Werken H., Simonis M., Lopez-Jones M., Eussen B., de Klein A., Singer R.H., de Laat W. Variegated gene expression caused by cell-specific long-range DNA interactions. Nat. Cell Biol. 2011;13:944–951. doi: 10.1038/ncb2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palstra R.J., Simonis M., Klous P., Brasset E., Eijkelkamp B., de Laat W. Maintenance of long-range DNA interactions after inhibition of ongoing RNA polymerase II transcription. PLoS One. 2008;3:e1661. doi: 10.1371/journal.pone.0001661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D., Meuleman W., Pagie L., Bruggeman S.W., Solovei I., Brugman W., Gräf S., Flicek P., Kerkhoven R.M., van Lohuizen M. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.T., Hookway T.A., Guo C., Sun Y. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., Aiden E.L. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V.V., Ren B. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J.A., Kingston R.E. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Simonis M., Klous P., Splinter E., Moshkin Y., Willemsen R., de Wit E., van Steensel B., de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat. Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- Splinter E., de Wit E., Nora E.P., Klous P., van de Werken H.J., Zhu Y., Kaaij L.J., van Ijcken W., Gribnau J., Heard E., de Laat W. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev. 2011;25:1371–1383. doi: 10.1101/gad.633311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E., de Wit E., van de Werken H.J., Klous P., de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 2012;58:221–230. doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Tanay A., Cavalli G. Chromosomal domains: epigenetic contexts and functional implications of genomic compartmentalization. Curr. Opin. Genet. Dev. 2013;23:197–203. doi: 10.1016/j.gde.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Therizols P., Illingworth R.S., Courilleau C., Boyle S., Wood A.J., Bickmore W.A. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science. 2014;346:1238–1242. doi: 10.1126/science.1259587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B., Blom M., Kerkhoven R.M., Pagie L., Teunissen H., Nieuwland M., Simonis M., de Laat W., van Lohuizen M., van Steensel B. Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet. 2011;7:e1001343. doi: 10.1371/journal.pgen.1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Koningsbruggen S., Gierlinski M., Schofield P., Martin D., Barton G.J., Ariyurek Y., den Dunnen J.T., Lamond A.I. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell. 2010;21:3735–3748. doi: 10.1091/mbc.E10-06-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieux-Rochas M., Fabre P.J., Leleu M., Duboule D., Noordermeer D. Clustering of mammalian Hox genes with other H3K27me3 targets within an active nuclear domain. Proc. Natl. Acad. Sci. USA. 2015;112:4672–4677. doi: 10.1073/pnas.1504783112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijchers P.J., Geeven G., Eyres M., Bergsma A.J., Janssen M., Verstegen M., Zhu Y., Schell Y., Vermeulen C., de Wit E., de Laat W. Characterization and dynamics of pericentromere-associated domains in mice. Genome Res. 2015;25:958–969. doi: 10.1101/gr.186643.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.R., Azuara V., Perry P., Sauer S., Dvorkina M., Jørgensen H., Roix J., McQueen P., Misteli T., Merkenschlager M., Fisher A.G. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J. Cell Sci. 2006;119:132–140. doi: 10.1242/jcs.02727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.