

Summary
Antisense long non-coding (aslnc)RNAs represent a substantial part of eukaryotic transcriptomes that are, in yeast, controlled by the Xrn1 exonuclease. Nonsense-Mediated Decay (NMD) destabilizes the Xrn1-sensitive aslncRNAs (XUT), but what determines their sensitivity remains unclear. We report that 3′ single-stranded (3′-ss) extension mediates XUTs degradation by NMD, assisted by the Mtr4 and Dbp2 helicases. Single-gene investigation, genome-wide RNA analyses, and double-stranded (ds)RNA mapping revealed that 3′-ss extensions discriminate the NMD-targeted XUTs from stable lncRNAs. Ribosome profiling showed that XUT are translated, locking them for NMD activity. Interestingly, mutants of the Mtr4 and Dbp2 helicases accumulated XUTs, suggesting that dsRNA unwinding is a critical step for degradation. Indeed, expression of anticomplementary transcripts protects cryptic intergenic lncRNAs from NMD. Our results indicate that aslncRNAs form dsRNA that are only translated and targeted to NMD if dissociated by Mtr4 and Dbp2. We propose that NMD buffers genome expression by discarding pervasive regulatory transcripts.
Keywords: lncRNA, Xrn1, NMD, dsRNA, RNA helicases
Graphical Abstract
Highlights
-
•
Xrn1-sensitive Unstable Transcripts (XUTs) are 3′-extended isoforms of stable lncRNAs
-
•
Nonsense-Mediated Decay preferentially targets long XUTs with single-stranded 3′ end
-
•
Antisense XUTs form double-stranded RNA in vivo
-
•
Formation of double-stranded RNA protects XUTs from Nonsense-Mediated Decay
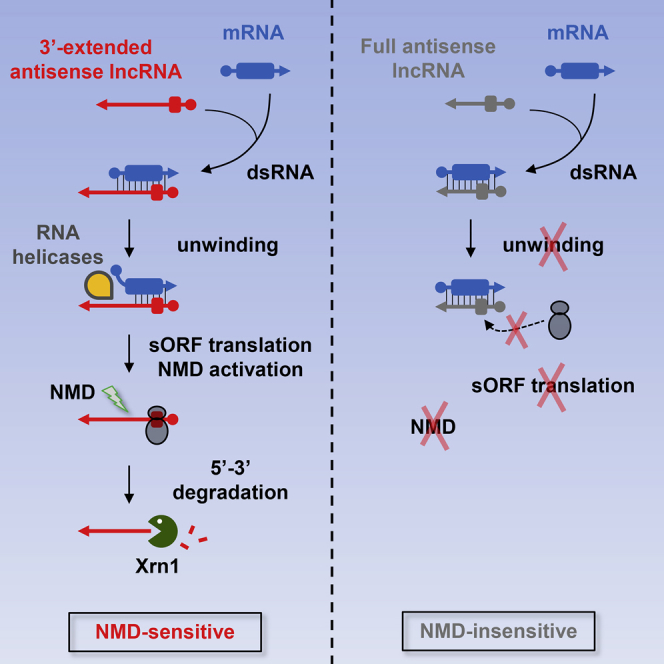
Wery et al. used single-gene investigation, genome-wide RNA analyses, and double-stranded (ds)RNA in vivo mapping to show that antisense Xrn1-sensitive Unstable Transcripts (XUTs) form double-stranded RNA in yeast and that 3′ single-stranded extension mediates XUTs degradation by the Nonsense-Mediated Decay (NMD) pathway, assisted by the Mtr4 and Dbp2 RNA helicases.
Introduction
Emergence of high-density tilling arrays and high-throughput sequencing technologies have led to the major discovery that transcription in Eukaryotes is not limited to protein-coding genes and transcription units of the non-coding (nc) rRNAs, tRNAs, and small nucle(ol)ar (sn)(o)RNAs. Rather, eukaryotic genomes are pervasively transcribed, i.e. most if not the whole genome can be virtually transcribed in at least one condition depending on cell type, developmental stage, or environment. For instance, while protein-coding genes represent only 2% of the human genome (Taft et al., 2007), up to 75% of it was shown to be transcribed in at least one of the cell lines analyzed in the ENCODE project (Djebali et al., 2012). This pervasive transcription produces a plethora of ncRNAs species (Berretta and Morillon, 2009), distinct from rRNAs, tRNAs, and sn(o)RNAs, that are commonly classified according to their size into small (<200 nt) and long (≥200 nt) ncRNAs. Both classes co-exist in most Eukaryotes and function as co-regulators of many cellular processes, including gene expression (Mercer et al., 2009, Rinn and Guttman, 2014, Rinn and Chang, 2012, Wery et al., 2011). Small and long ncRNAs have broad impacts on development and dysregulation of several of them has been associated to various diseases including cancer and neurological disorders (Batista and Chang, 2013, Taft et al., 2010).
The budding yeast S. cerevisiae constitutes a notable exception among Eukaryotes, since it has lost the RNAi system during evolution and lacks small ncRNAs (Drinnenberg et al., 2009). Hence, S. cerevisiae has become a prominent model to specifically study the effects of long (l)ncRNAs, which might be partially hidden by the effects of small ncRNAs in other eukaryotic models. Several classes of lncRNAs have been described in S. cerevisiae (Tisseur et al., 2011, Tudek et al., 2015). Strikingly, the majority of them are unstable as the consequence of their extensive degradation in the nucleus or in the cytoplasm. Consequently, they are not detectable in wild-type (WT) cells, but accumulate upon inactivation of the machineries responsible for their degradation. These unstable lncRNAs include the Cryptic Unstable Transcripts (CUTs) that are sensitive to the nuclear exosome-dependent 3′-5′ RNA decay pathway (Neil et al., 2009, Wyers et al., 2005, Xu et al., 2009), the Xrn1-sensitive (X)UTs that we identified as a class of antisense regulatory lncRNAs targeted by the cytoplasmic 5′-3′ exoribonuclease Xrn1 (Berretta et al., 2008, van Dijk et al., 2011), and the Nrd1-Unterminated Transcripts (NUTs) that accumulate upon nuclear depletion of the RNA-binding factor Nrd1 (Schulz et al., 2013). Besides the unstable species, Stable Unannotated Transcripts (SUTs) were identified as a class of exosome-insensitive lncRNAs that are detectable in WT cells, hence their definition as stable transcripts (Xu et al., 2009).
Beyond the definition of these classes of yeast lncRNAs, there is a considerable overlap between them. For instance, many NUTs overlap CUTs (Schulz et al., 2013), while SUTs largely overlap XUTs (van Dijk et al., 2011). There is also overlap between some CUTs and XUTs, indicating that a same transcript might be targeted by two distinct RNA surveillance machineries (Thompson and Parker, 2007, van Dijk et al., 2011).
What determines instability of yeast lncRNAs? In the case of NUTs and CUTs, early termination of transcription by the Nrd1-Nab3-Sen1 (NNS) complex promotes recruitment of the TRAMP4 complex (Tudek et al., 2014), formed by the Trf4 non-canonical poly(A)-polymerase, the RNA-binding protein Air1 or Air2, and the DExH-box RNA helicase Mtr4 (LaCava et al., 2005). Trf4-mediated poly-adenylation stimulates Rrp6/exosome recruitment and subsequent RNA degradation (LaCava et al., 2005), although Rrp6/exosome can also be recruited independently through a direct interaction with Trf4 (Tudek et al., 2014). On the other hand, the determinants of XUTs instability remain unclear. XUTs are synthesized by RNA polymerase II and poly-adenylated, as mRNAs (van Dijk et al., 2011), and they are degraded by the 5′-3′ exoribonuclease Xrn1, which carries the major exoribonuclease activity responsible for mRNA turnover in the cytoplasm. Several mRNA decay pathways lead to degradation by Xrn1 (Parker, 2012). The general mRNA decay pathways involve shortening of the poly(A) tail (Muhlrad and Parker, 1992), a process named deadenylation, that commonly leads to decapping, exposing the decapped transcript to 5′-3′ degradation by Xrn1 (Decker and Parker, 1993). Alternatively, deadenylation can also be followed by 3′-5′ degradation by the cytoplasmic exosome (Anderson and Parker, 1998). In addition, specialized decay pathways coupled to translation also promote Xrn1-mediated mRNA degradation. These are the Nonsense-Mediated Decay (NMD) and the No-Go Decay (NGD) involved in cytoplasmic quality control of mRNAs. NMD targets mRNAs with aberrant translation termination or recognized as such, including mRNAs with premature stop codon (Muhlrad and Parker, 1994) or long 3′-UTR (Muhlrad and Parker, 1999). NGD induces endonucleolytic cleavage of mRNAs with ribosome stalled in translation elongation, followed by degradation of the 5′ and 3′ fragments by the cytoplasmic exosome and Xrn1, respectively (Doma and Parker, 2006). Interestingly, recent data have revealed the role of NMD in control of cryptic lncRNAs (Berretta et al., 2008, Malabat et al., 2015, Smith et al., 2014), but the determinants of their sensitivity to NMD remained unknown.
Here, we report that 3′-ss extension determines XUTs sensitivity to NMD. We show that the stable/unstable lncRNAs antisense to ARG1 are distinct transcripts and that a 3′ extension confers NMD-sensitivity to the unstable isoform. The combination of extensive total RNA-sequencing (seq) and Cap-Analysis Gene Expression (CAGE)-seq with original bioinformatics pipelines leads to the assembly of the exhaustive XUT landscape in budding yeast. Globally, we show that a 3′-extension distinguishes XUTs from stable lncRNAs, insensitive to Xrn1. Genome-wide mapping of double-stranded (ds)RNA revealed that antisense (as)XUTs form dsRNA in vivo. Comparative analyses show that NMD preferentially targets extended asXUTs with 3′-ss end. NMD-sensitive XUTs are bound by ribosomes and are destabilized by RNA helicases Mtr4 and Dbp2. Finally, expression of anticomplementary transcripts in trans protects intergenic solo XUTs from NMD. We propose that asXUTs form dsRNA and can be translated then targeted by NMD only if dissociated by Mtr4 and Dbp2. In addition to its well-known substrates including aberrant mRNAs species, NMD targets XUT lncRNAs, i.e., those pervasive transcripts endowed with a regulatory potential, and thereby contributes in buffering genome expression.
Results
Unstable XUT1678 Is a 3′-Extended Isoform of the Stable SUT768 Targeted by the NMD
The observation from the original catalog of XUTs that a significant portion of them overlapped stable lncRNAs (SUTs) raised a fundamental question: do overlapping SUTs and XUTs correspond to the same transcripts that would be detectable in WT cells and would accumulate upon Xrn1 inactivation or are they distinct isoforms? To address this question, we characterized by northern blot the SUT768/XUT1678 pair, antisense to ARG1. On the basis of their annotation, XUT1678 is expected to display a 3′ extension (Figure 1A). A probe complementary to both transcripts revealed one RNA species in the WT and two in the xrn1Δ strain and the largest was only detected in the mutant (Figure 1B). On the other hand, a probe complementary to the predicted 3′ extension of the XUT detected only the largest isoform in xrn1Δ (Figure 1B). Thus, XUT1678 and SUT768 are two distinct transcripts and a 3′ extension discriminates the XUT from the stable, Xrn1-insensitive SUT.
Figure 1.

XUT1768 Is 3′-Extended Unstable Isoform of SUT768 Targeted by the NMD Pathway
(A) Representation of overlapping SUT768 (gray) and XUT1678 (red) antisense to ARG1 (blue). The probes I and II correspond to AMO2209 and AMO1595 (see Table S2), respectively, used in (B). Other images used probe II for detection of SUT768 and XUT1678.
(B) SUT768 and XUT1678 are distinct isoforms. The YAM1 (WT) and YAM6 (xrn1Δ) cells were grown to mid-log phase in yeast extract peptone dextrose medium (YPD) at 30°C. The total RNA was extracted and analyzed by northern blot.
(C) XUT1678 accumulation is independent from Nrd1. The YAM92 (WT), YAM97 (xrn1Δ), YAM143 (PGAL1-NRD1), and YAM985 (PGAL1-NRD1 xrn1Δ) cells were grown to mid-log phase in YPGal (Galactose) and then shifted for 4H30 in YPD (Glucose). The SUT768/XUT1678, RTL (Berretta et al., 2008), NEL025C, and scR1 detection was as above. The numbers represent XUT1678/scR1 ratios, set to 1 in xrn1Δ, in each medium (not determined: ND). See also Figure S1.
(D) XUT1678 and SUT768 are poly-adenylated. The poly(A)+ RNA was purified from YAM1 (WT) and YAM6 (xrn1Δ) total RNA. The total, purified poly(A)+, and unbound RNA were loaded in a 1:50:1 ratio.
(E) NMD targets XUT1678. The YAM1 (WT), YAM6 (xrn1Δ), YAM124 (rrp6Δ), YAM125 (trf4Δ), YAM127 (trf5Δ), YAM128 (trf4Δ rrp6Δ), YAM129 (trf5Δ rrp6Δ), YAM199 (ccr4Δ), YAM200 (dom34Δ), YAM202 (upf1Δ), YAM203 (upf2Δ), YAM204 (upf3Δ), YAM225 (lsm1Δ), and YAM226 (lsm7Δ) cells were grown as in (B). The numbers represent levels of XUT1678 and SUT768 normalized on scR1 and set to 1 in the WT.
(F) XUT1678 is sensitive to decapping. The WT (YAM1) and dcp2-7 (YAM2283) cells were grown in YPD to mid-log phase at 25°C and then shifted at 37°C for 15, 30, 60, and 120 min. In parallel, WT (YAM1) and xrn1Δ (YAM6) cells were grown as in (B).
See also Figure S1.
Nuclear depletion of Nrd1 leads to the accumulation of transcripts named NUTs, a large portion of which overlap annotated lncRNAs, including some SUTs and XUTs (Schulz et al., 2013). NUTs were proposed to be extended isoforms of these lncRNAs, targeted by the NNS-dependent termination pathway (Schulz et al., 2013). To determine whether this would also be the case for XUT1678, we depleted Nrd1 in XRN1 and xrn1Δ cells, using a PGAL1-NRD1 construct (PGAL1 is rapidly turned off upon transfer from galactose- to glucose-containing medium). We observed that in contrast to NEL025C, a well-known target of Nrd1, Nrd1 depletion in xrn1Δ cells had minor effect on XUT1678 accumulation (Figure 1C), indicating that the NNS complex is not crucial for termination of XUT1678. This is consistent with the observation that XUT1678 is not primarily targeted by the nuclear RNA decay (see below).
Rather, we postulated that termination of SUT768 and XUT1678 requires the canonical Pap1 poly(A)-polymerase. To test this, we combined a pap1-1 thermosensitive allele with xrn1Δ. At the non-permissive temperature, SUT678 level strongly dropped down and XUT1678 totally disappeared (Figure S1), indicating that synthesis of both transcripts requires Pap1. Consistent with this, they were recovered into similar levels in the poly(A)+ fraction of the xrn1Δ strain (Figure 1D). Note that in contrast to the PHO84 antisense lncRNA (Castelnuovo et al., 2013), XUT1678 did not accumulate when Pap1 was inactivated in rrp6Δ cells (Figure S1). Thus, synthesis of SUT768 and the 3′-extended XUT1678 depends on the canonical Pap1 poly(A)-polymerase, both transcripts being poly-adenylated, as mRNAs. We anticipated the discrimination between the stable and 3′-extended unstable transcript isoforms to occur post-transcriptionally.
To identify the pathway targeting XUT1678, we screened mutants of RNA decay factors, including the 3′-5′ exonuclease Rrp6 and the poly(A)-polymerases Trf4 and Trf5 (nuclear 3′-5′ RNA decay); the Ccr4 deadenylase; Dom34 (NGD pathway); the Upf1-3 factors (NMD pathway); and the Lsm1 and Lsm7 subunits of the heteroheptameric Lsm1-7 complex (required for efficient decapping). Northern blot analysis revealed a >11-fold accumulation of XUT1678 in the NMD mutants, while SUT768 was not affected (Figure 1E). XUT0741 antisense to ADH2 was also sensitive to NMD (Figure 1E). In contrast, inactivation of the other RNA decay factors had weak or no effect on XUT1678 and XUT0741 (Figure 1E), indicating that both XUTs are mainly targeted by NMD and Xrn1.
Finally, we tested the role of decapping in XUT1678 decay. Eukaryotic mRNAs carry a 7-methyl-guanosine (m7G) cap that protects them from 5′-3′ degradation by Xrn1. Consequently, decapping is a prerequisite for Xrn1-dependent mRNA degradation (Parker, 2012). In S. cerevisiae, the decapping enzyme complex is formed by Dcp1 and the catalytic subunit Dcp2 (Dunckley and Parker, 1999). We therefore tested XUT1678 sensitivity to decapping using a thermosensitive dcp2-7 mutant (Wilson et al., 2007), the choice of which was motivated by the very slow growth phenotype of dcp2Δ. As two other XUTs, XUT1678 accumulated rapidly upon decapping inactivation, while SUT768 was not affected (Figure 1F).
In summary, the stable SUT768 and unstable XUT1678 are distinct aslncRNA isoforms synthesized as poly(A)+ transcripts from the same locus and a 3′ extension confers the XUT sensitivity to NMD, decapping, and Xrn1. These data obtained from single-gene investigations prompted us to perform genome-wide analyses on a larger set of overlapping SUT/XUT pairs in order to get robust mechanistic insights into what determines XUT lncRNAs instability.
Exhaustive Landscape of XUT lncRNAs in S. cerevisiae
The original catalog of XUTs was established by single-end sequencing of RNA-seq libraries from WT and xrn1Δ cells of the reference strain S288C (van Dijk et al., 2011), but with a limited accuracy due to the sequencing protocols at that time (Wery et al., 2013). As this might dramatically impact the genome-wide comparative analysis of overlapping stable and unstable lncRNAs, we decided to assemble a refined catalog based on high-coverage RNA-seq to define more precisely the XUT landscape in yeast. Our rationale was to (1) analyze several laboratory strains of S. cerevisiae; (2) perform paired-end sequencing of strand-specific RNA-seq libraries with overall >1,500× coverage; (3) select the most robust XUTs among the segments reassembled from RNA-seq signals using complementary criteria; and (4) reannotate transcription start sites (TSS) of XUTs using a genome-wide map of capped transcripts TSS obtained by CAGE-seq (Figure S2).
Strand-specific RNA-seq libraries were prepared from biological duplicates of haploid WT and xrn1Δ cells of S288C, of the closely related W303 strain and of the more distant SK1 strain (Liti et al., 2009). For SK1, a common model for meiosis studies, we also analyzed diploid WT and xrn1Δ/xrn1Δ cells. We identified 1,813 XUTs stabilized in the xrn1Δ mutant of at least one of the strains analyzed (Figure S2). Among them, 1,198 were common to the four strains. In parallel, we defined specific signatures for one strain (highly strain specific), for two strains, and for the three haploid strains (Figure S2).
In parallel, we aimed to use CAGE-seq to precisely map TSS of XUTs. However, CAGE relies on a specific treatment of m7G capped RNAs (Takahashi et al., 2012), and most RNAs, including XUTs, that accumulate in the absence of Xrn1 are decapped. This is shown by the sensitivity of XUTs to Terminator 5′-Phosphate-Dependent Exonuclease (Terminator), a processive 5′-3′ exonuclease that digests RNA with 5′-monophosphate ends, but not with m7G cap (Figure 2A). On the other hand, RNA-seq analysis of dcp2-7 cells shifted for 2 hr at 37°C revealed significant stabilization of 1,046 XUTs upon decapping inactivation (Figure 2B), which is spectacular in comparison to the 101 Dcp2-sensitive lncRNAs previously shown to accumulate in a dcp2Δ context (Geisler et al., 2012), half of which are XUTs (Figure S2). Furthermore, XUTs in the dcp2-7 context were not affected by Terminator treatment (Figure 2C), indicating that they accumulate as capped transcripts in decapping-deficient cells. We therefore constructed CAGE-seq libraries using total RNA from WT and Dcp2-inactivated cells (Figure S2). Then we set up a peak-calling algorithm to define the CAGE peaks in the WT and dcp2-7 strains, which identified TSS for 78% of mRNA and 50% of SUTs expressed in the dcp2-7 mutant (Figure 2D). Notably, 625 XUTs had ≥1 peak at the proximity of their predicted 5′ end. Consequently, TSS of these XUTs were reannotated according to the position of these CAGE peaks (Figure S2). In addition, 15 pairs of tandem XUTs, for which peak(s) were detected for the first XUT only, were fused into a single, large XUT. Note that comparison of the CAGE signals in a decapping-deficient context with those obtained by an alternative technique in upf1Δ cells (Malabat et al., 2015) showed similar distributions around the annotated TSS of mRNAs, SUTs, and XUTs (Figure S2), despite differences of few nucleotides that might be the consequence of the techniques and/or strains used.
Figure 2.

Comprehensive XUT Landscape in S. cerevisiae
(A) XUTs in xrn1Δ cells lack m7G cap. The box-plot of RNA-seq signal ratio for XUTs in WT (YAM1) and xrn1Δ (YAM6) is shown. For each strain, libraries were prepared using the same total RNA submitted to rRNA depletion (Ribo-) or Terminator digestion (Ter). The density (tag/nt) was computed for the 1,681 XUTs of S288C.
(B) Sensitivity of XUTs to decapping. The scatter plot of tag density for ORFs (gray) and XUTs of S288C (red, n = 1,681) in WT (YAM1) and dcp2-7 (YAM2283) cells shifted for 2 hr at 37°C is shown. The libraries were prepared from rRNA-depleted RNA. The results are presented as log2 of density (dashed line: x = y line).
(C) XUTs accumulate as capped RNA in decapping-deficient cells. The same as (A), for WT (YAM1) and dcp2-7 (YAM2283) cells shifted for 2 hr at 37°C.
(D) Proportion of mRNAs and SUTs for which TSS were identified by peak-calling in WT (YAM1) and dcp2-7 (YAM2283) cells, shifted 2 hr at 37°C. Only the mRNAs (n = 5,460) and SUTs (n = 672) with RNA-seq signal ≥1 RPKM in dcp2-7 were considered. To evaluate the peak-calling noise, the same analysis was performed on 6,641 sequences (200 nt long) randomly selected from the set of mRNA and SUTs described above.
(E) Overlap between the 1,781 XUTs annotated here and other lncRNAs, including XUTs from the original catalog (∗XUT). The overlap had to cover ≥50% of each transcript to be counted.
(F) Snapshot of XUT0685 (S288C-specific) and XUT0686 (common to all strains). The RNA-seq signals are visualized as a heatmap for WT and xrn1Δ cells from the four laboratory strains and for WT (YAM1) and dcp2-7 (YAM2283) cells shifted for 2 hr at 37°C. The signals for the + and − strands, respectively, are shown (upper and lower). The color turns from yellow to dark blue as the RNA-seq signal increases. The CAGE-seq signal in dcp2-7 is visualized in a separate panel in blue (+ strand) and pink (− strand). The small red arrows indicate the CAGE peaks detected by the peak-calling algorithm in the dcp2-7 condition. The ORFs, SUT, NUTs, and XUTs are represented as blue, gray, brown, and red arrows, respectively. The snapshots were produced using VING (Descrimes et al., 2015).
See also Figure S2.
In summary, we assembled a refined catalog of 1,798 XUT lncRNAs significantly stabilized upon Xrn1 inactivation in at least one S. cerevisiae laboratory strain: 1,153 are antisense to open reading frame (ORFs); 825 overlap XUTs from the original catalog, while 798 had not been annotated previously; and 424 overlap SUTs (Figure 2E). Figure 2F shows snapshots of the RNA-seq and CAGE-seq signals for two XUTs.
XUTs Are 3′-Extended Isoforms of SUTs
To further investigate the basis of XUTs instability, we focused on the 292 pairs of SUT/XUT showing ≥75% of overlap (using the present annotation) to determine whether they correspond to the same transcripts or to distinct isoforms, as the SUT768/XUT1678 (Figure 1B). We anticipated that in the case of distinct isoforms, overlapping SUTs and XUTs might display distinct 5′ and/or 3′ extremities. To test this, we measured the distance between the annotated 5′ coordinates of these overlapping SUTs and XUTs and observed a distribution centered on 0 (Figure 3A), with a median distance of 2 nt (Figure S3), suggesting that overlapping SUTs and XUTs start at the same positions. This was further supported by the observation that the CAGE peaks identified in the WT and in the dcp2-7 mutant around the annotated 5′ end of the SUTs are similarly distributed in the two conditions (Figure 3B), i.e., no additional CAGE-seq peaks appear when XUTs are stabilized. As an illustrative example, Figure 3C shows a snapshot of the RNA-seq and CAGE-seq signals for the overlapping SUT768/XUT1678 pair with the CAGE peaks detected in the WT and dcp2-7 conditions.
Figure 3.

Overlapping XUTs and SUTs Share the Same TSS, but XUTs Are 3′-Extended
(A) Distribution of the distance between the annotated 5′ (black) and 3′ (red) coordinates of overlapping SUTs and XUTs (≥75% of overlap and n = 292).
(B) Global distribution of CAGE peaks detected for the WT (black) and the dcp2-7 mutant (gray) in the ±100 nt around the annotated 5′ coordinates of SUTs overlapped by XUTs (same set as above).
(C) Snapshot of overlapping SUT768 and XUT1678. The RNA-seq signals are visualized as in Figure 2F for WT (YAM1) and xrn1Δ (YAM6) at 30°C and for WT (YAM1) and dcp2-7 (YAM2283) shifted for 2 hr at 37°C. The CAGE-seq signals are shown in separate panels for the WT (YAM1) and dcp2-7 (YAM2283). The annotation and CAGE peaks are represented as above. For SUT768 and XUT1678, the two peaks at positions 220559 and 220536 (− strand) were detected in both conditions.
See also Figure S3.
In contrast, a similar comparison of the annotated 3′ coordinates of the 292 pairs of overlapping SUT/XUT revealed a shift of the distribution (Figure 3A), with a median distance of 29 nt (Figure S3), indicating that XUTs are longer in 3′ than their overlapping SUTs.
This global analysis confirms the results obtained at the single-gene level with the SUT768/XUT1678 pair and indicates that overlapping SUTs and XUTs are distinct isoforms sharing the same TSS, but with a 3′ extension specific to the XUT family.
XUTs Are Targeted by the Translation-Dependent NMD Pathway
To generalize the observation that XUTs are sensitive to NMD (Figure 1E), we performed total RNA-seq in WT and upf1Δ cells of the S288C strain. Globally, 73% of S288C XUTs (1,229/1,681) significantly accumulated in the absence of Upf1 (upf1Δ/WT ratio ≥2, p -value ≤0.05; Figures 4A and S4), which is more than the previously estimated number of NMD-sensitive lncRNAs in yeast (Smith et al., 2014) (Figure S4). In addition, 875 of these XUTs (including XUT1678) were also sensitive to Dcp2 (Figure S4). Reanalysis of RNA-seq data from upf1Δ, xrn1Δ, and upf1Δ xrn1Δ mutants (Malabat et al., 2015) using the present XUTs annotation confirmed that the majority of them are stabilized in the two single and in the double mutants (Figure S4), indicating that XUTs are primarily targeted by NMD.
Figure 4.

XUT lncRNAs Are NMD Targets
(A) Scatter plot of tag density for ORFs (gray) and XUTs of the S288C strain (red, n = 1,681) in WT and upf1Δ cells. The results are presented as in Figure 2B.
(B) Density plot of polysome-seq/RNA-seq signals in upf1Δ cells for mRNAs (blue, n = 5,167), XUTs (red, n = 426), SUTs (gray, n = 414), and sn(o)RNAs (green, n = 75). The libraries and raw data were previously described (Smith et al., 2014).
(C and D) Metagene representation of ribosome footprints (solid lines) and fragmented RNA signal (dashed lines) for 245 Upf1-sensitive (C) and 30 Upf1-sensitive (D) XUTs in WT (gray) and upf1Δ (black) cells. The TSS and TTS correspond to TS and termination sites, respectively. The x axis is in virtual nt. The libraries and raw data were previously described (Smith et al., 2014).
(E) Configuration of NMD-sensitive and NMD-insensitive XUTs. The XUTs of S288C (n = 1,681) were classified into solo (type 1; no overlap with mRNA expressed in upf1Δ, i.e., RPKM ≥1) and asXUTs (overlap ≥1 nt). The asXUTs were further classified into full antisense (type 2), free 5′ end (type 3), free 3′ end (type 4), and free 5′ and 3′ ends (type 5). For each of these types as for the whole set of S288C XUTs, we determined the proportion of long and short XUTS (< and ≥381 nt, respectively) that are sensitive and insensitive to NMD.
See also Figure S4.
The observation that most XUTs are sensitive to NMD suggests that they are scanned by ribosomes when they reach the cytoplasm. In this regard, recent ribosome profiling data support the idea that yeast lncRNAs bear small (s)ORFs that are actively translated (Smith et al., 2014). To test whether it is the case for XUTs, we reanalyzed published polysome-seq and ribosome profiling data (Smith et al., 2014). Importantly, as mRNAs, but in contrast to sn(o)RNAs, XUTs and SUTs showed significant association to polysomes (Figure 4B). Furthermore, significant ribosome binding was detected for 275 XUTs, among which 245 are NMD-sensitive. For these NMD-sensitive XUTs, ribosome footprints were exclusively detected within the TSS-proximal region, followed by a long ribosome-free 3′ UTR (Figure 4C). On the other hand, NMD-insensitive XUTs showed ribosomes binding on >50% of their sequence (Figure 4D). As an example, ribosome footprints were detected up to position 370/1,420 of the NMD-sensitive XUT1678, in upf1Δ. Manual inspection of the corresponding sequence identified three putative sORFs at positions 4–36, 73–81, and 145–363, the latter coding a 72 amino acids long peptide, followed by a >1 kb long 3′ UTR (Figure S4).
Together, these data strongly indicate that XUTs are actively translated and that those bearing long 3′ UTR are specifically targeted by the NMD, which is consistent with the well-known role of NMD in targeting aberrant mRNAs with long 3′ UTR (Muhlrad and Parker, 1999).
NMD Preferentially Targets Long XUTs with Single-Stranded 3′ End
Assuming that lncRNA size correlates with 3′ UTR length, we speculated that it should contribute to discriminate between XUTs that are targeted or not by NMD. Indeed, Upf1-insensitive XUTs tend to be smaller than Upf1-sensitive XUTs (data not shown), with 381 and 731 nt of median size, respectively. However, size is not sufficient to explain NMD-sensitivity of XUTs, neither is the presence of a putative specific motif since extensive computational searches failed to identify any robust motif in NMD-sensitive XUTs (data not shown).
In a recent study of mRNA isoforms half-lives, formation of dsRNA at the mRNAs 3′ end has been proposed to increase stability (Geisberg et al., 2014). Conversely, we expected the inability of a transcript to form dsRNA at its 3′ end to induce instability. To explore whether dsRNA plays a role in the determination of XUTs sensitivity to NMD, we analyzed the configuration of Upf1-sensitive and -insensitive XUTs (Figure 4E). Importantly, XUTs insensitive to NMD were significantly underrepresented among asXUTs with a free 3′ end (types 4 and 5, p = 1.16e−03 and 2.06e−02, respectively; chi-square test of independence), but not full asXUTs or with a free 5′ end (types 2 and 3, p = 0.13 and 0.79, respectively). This indicates that XUT with a 3′-ss end are preferentially targeted by NMD. The higher proportion of NMD-sensitive XUTs in type 5 (89%) compared to type 4 (82%) suggests that having both extremities free might be cumulative with regards to NMD sensitivity. Intriguingly, 163/512 solo XUTs (32%) were insensitive to NMD, which is more than expected (type 1, p = 3.00 × 10−03). Indeed, most of them are expected to be NMD-sensitive since their 3′ end is not engaged in dsRNA. However, 97 of these 163 (60%) solo NMD-insensitive XUTs are short (<381 nt) and might escape NMD due to their small size.
We conclude that NMD preferentially targets long XUTs, with 3′-ss end. Accordingly, XUTs with a 3′ end engaged in a dsRNA structure would be expected to be NMD-insensitive. However, the observation that a large portion of such XUTs are sensitive to NMD (Figure 4C) raises the possibility that although antisense, these XUTs do not form dsRNA and therefore escape NMD, prompting us to address whether asXUTs form dsRNA in vivo.
asXUTs Form dsRNA In Vivo
S. cerevisiae has lost RNAi (Drinnenberg et al., 2009). However, expression of DCR1 and AGO1 from S. castellii in S. cerevisiae reconstitutes a functional RNAi pathway (Drinnenberg et al., 2009) that targets dsRNA structures and produces small RNAs (Drinnenberg et al., 2011).
To test whether asXUTs form dsRNA in vivo, we sequenced small RNAs from WT and xrn1Δ strains of S. cerevisiae, expressing or not RNAi factors (Figure 5A). Upon RNAi reconstitution, both XRN1 and xrn1Δ strains accumulated 19–23 nt small RNAs (Figure S5), as expected (Drinnenberg et al., 2011, Sinturel et al., 2015). Notably, 64% and 72% of the uniquely mapped reads from the RNAi+ and xrn1Δ RNAi+ strains, respectively, originated from regions of the genome with sense/antisense transcription, which represent only 17.5% of the genome (Figure 5B). Furthermore, as retrotransposons and transcripts from other repeated regions, XUTs were largely targeted by RNAi, with up to 22% of the small reads mapping on XUTs in the xrn1Δ RNAi+ context (Figure S5). The idea that asXUT form dsRNA even in XRN1 cells was supported by the observation that 47% of asXUTs, but only 5% of solo XUTs, showed significant enrichment for small RNA (Figure 5C). In the absence of Xrn1, the proportion of XUTs showing significant enrichment for small RNAs reached 80% for the antisense and only 32% for the solo (Figure 5C). Snapshots for the TAT1/XUT0051 and FAR1/XUT0521 pairs showed that small RNAs production is restricted to the region of overlap between mRNAs and asXUTs. Indeed, no read mapped to the 3′ end of XUT0051, which is not overlapped by TAT1 mRNA (Figure 5D). On the other hand, the full antisense XUT0521 was completely covered by the small RNA signal (Figure S5). In both cases, the small RNAs were abundant enough in the xrn1Δ RNAi+ context to be detected by northern blot as a discrete 23 nt band (Figure S5).
Figure 5.

asXUTs Form dsRNA In Vivo
(A) RNAi reconstitution in S. cerevisiae. The WT (YAM1730), RNAi+ (YAM1725), xrn1Δ (YAM2271), and xrn1Δ RNAi+ (YAM1982) cells were grown to mid-log phase in YPD at 30°C. The libraries were constructed using purified small RNAs.
(B) Proportion of 19–23 nt reads uniquely mapped into genome regions showing sense/antisense, sense only, or no transcription (coverage of each nt of the genome by a transcript was determined in a strand-specific manner using the official annotation combined to the segmentation used here to assemble the refined XUTs catalog). (arrows: transcription units and black: strand to which reads mapped) (grey: antiparallel strand).
(C) Proportion of solo and asXUTs of the W303 strain showing significant 19–23 nt small RNA production for both the XUT and the anticomplementary strand (RNAi+/RNAi- ratio ≥2).
(D) Snapshot of small RNAs along the TAT1/XUT0051 locus. The densities of 19–23 nt uniquely mapped reads for the + and − strands are shown (upper and lower), respectively. The ORFs, CUT, and XUT are represented by blue, green, and red arrows, respectively. The thin blue lines correspond to UTRs. The 3′ region of XUT0051 not overlapped by TAT1 mRNA is highlighted. The blue stars indicate the position of probes used for northern blot (Figure S5). The snapshot was produced using VING (Descrimes et al., 2015).
See also Figure S5.
In conclusion, a large proportion of asXUTs form dsRNA in vivo.
Formation of dsRNA Protects XUTs from NMD
Our data indicate that most XUTs are targeted to NMD following translation of sORFs in their 5′ portion, and also that asXUTs form dsRNA, raising a mechanistic question: how could ribosomes translate a sORF on a XUT engaged in a dsRNA? In addition, XUTs with free 3′ end are preferentially targeted by NMD. For example, XUT0051 with a 3′-ss end was more sensitive to NMD than the full antisense XUT0521 (Figure S6; see also Figures 5 and S5 for snapshots of small RNAs).
We postulated that RNA helicases might help to unwind the dsRNA structures and release asXUTs as single-stranded molecules, thereby allowing them to be translated and targeted by NMD. There were two of the yeast RNA helicases that appeared to be strong candidates: Mtr4, the ATP-dependent 3′-5′ helicase activity of which could unwind dsRNA with a 3′ extension (Bernstein et al., 2008), and the dsRNA-specific helicase Dbp2, that was reported to physically interact with Upf1 (Bond et al., 2001). Importantly, inactivation of Mtr4 and Dbp2 resulted into stabilization of NMD-sensitive XUTs (Figures 6A and 6B). This stabilization was specific to Dbp2 and Mtr4, since null mutants of the Ski2, Dhh1, and Dbp1 RNA helicases had no effect on the tested XUTs (data not shown). The direct involvement of Mtr4 in unwinding mRNA/asXUTs duplexes is further supported by the analysis of CRAC data (Tuck and Tollervey, 2013) showing that Mtr4 binds to the 5′ region of mRNAs that form dsRNAs with asXUTs, while its binding to the rest of the mRNA population was very low (Figure S6).
Figure 6.

Formation of dsRNA Protects XUTs from NMD
(A) XUTs sensitivity to Mtr4. The WT (YAM1) and xrn1Δ (YAM6) cells were grown as above. The WT (YAM115) and tetOFF::MTR4 (BSY1756) cells were grown as in Figure 1B, then doxycycline (dox, 10 μg/ml) was added. The cells were harvested 0, 6, and 9 hr after dox addition (see growth curves in Figure S6).
(B) XUTs sensitivity to Dbp2. The WT (YAM1) and dbp2Δ (YAM2627) cells were grown as in Figure 1B.
(C) Schematic representation expression in trans of the transcript anticomplementary (black) to a solo XUT (red).
(D and E) WT (YAM1), xrn1Δ (YAM6), and upf1Δ (YAM202) cells transformed with the pMD2 empty vector (D) or expressing the anticomplementary transcript of solo XUT1150 in trans (E) were grown in CSM-U to mid-log phase at 30°C. The XUT1150 level was determined by strand-specific RT-quantitative (q)PCR from total RNA and normalized on scR1. The data are presented as mean values ± SD, calculated from three independent biological replicates.
See also Figure S6.
Finally, to provide additional evidence that dsRNA formation contributes to protect XUTs from NMD, we selected NMD-sensitive solo XUTs with predicted 5′ sORFs bound by ribosomes (Smith et al., 2014). We then artificially expressed in trans their antisense from a plasmid in WT, xrn1Δ, and upf1Δ cells (Figure 6C). Importantly, upon expression of their antisense, these solo XUTs were stabilized and lost their sensitivity to NMD (Figures 6D, 6E, and S6).
From these data, we conclude that dsRNA formation protects asXUTs from NMD and that the Mtr4 and Dbp2 RNA helicases contribute to asXUTs destabilization, presumably through dsRNA unwinding.
Discussion
In this work, we assembled the exhaustive XUT landscape in S. cerevisiae. The production of a refined catalog of XUTs was motivated by the considerable technical development for library preparation and the increased performances of sequencers, which combined with original bioinformatics pipelines, improve transcripts detection. Hence, 798 of the 1,781 XUTs described here had not been previously annotated. We note that XUTs represent the second largest class of transcripts in the yeast transcriptome, after mRNAs. Most XUTs were common to all laboratory yeast strains. However, we identified a subset of strain-specific transcripts, reminiscent of lncRNAs with strong cell-type specificity in human cells (Djebali et al., 2012). Building on our finding that XUTs are capped and accumulate upon decapping inactivation, we used CAGE-seq to accurately reannotate the TSS of 35% of XUTs. The CAGE data also showed that overlapping XUTs and SUTs share the same TSS. In contrast, XUTs overlapping SUTs have 3′ extensions that promote their instability, as previously reported for two individual cases (Marquardt et al., 2011).
Using genome-wide approaches and single-gene characterization, we provided insights into XUTs metabolism and revealed a major pathway responsible for their degradation. XUTs are synthesized by RNA polymerase II (van Dijk et al., 2011), capped, and poly-adenylated by the canonical poly(A)-polymerase Pap1, as mRNAs. A large proportion of XUTs are then specifically targeted by the NMD, leading to decapping by Dcp2 and ultimate degradation of the decapped XUTs by Xrn1. However, the observation that some XUTs were not sensitive to NMD suggests that other pathways might contribute to direct them to Xrn1-mediated degradation.
NMD is tightly associated to translation and the finding that >70% of XUTs are sensitive to this pathway supports the idea that they are translated (at least for a pioneer round of translation). Indeed, we showed that XUTs associate to polysomes and carry putative sORFs that are bound by ribosomes. In NMD-sensitive XUTs, ribosomes are restricted to a short region close to XUTs 5′ end, followed by a long ribosome-free 3′ UTR, a signal known to activate NMD (Muhlrad and Parker, 1999).
Another major finding of this work is that asXUTs form dsRNA in vivo. Using a reconstituted RNAi system (Drinnenberg et al., 2011, Sinturel et al., 2015), we found that 80% of asXUTs engaged in dsRNA. Interestingly, a fraction of solo XUTs yet shows small RNAs production in the RNAi+ context, indicating that, for those exceptions, the solo XUTs might be overlapped by uncharacterized antisense cryptic transcripts or, alternatively, adopt secondary structures proficient for Dicer processing.
Mechanistically, we provide evidence that dsRNA protects asXUTs from NMD and that the presence of a 3′-ss extension induces NMD-sensitivity. In keeping with that NMD depends on translation, but that dsRNA would impede ribosome binding to sORFs of asXUTs, we found that the Mtr4 and Dbp2 RNA helicases contribute to degrade NMD-sensitive XUTs. The fact that neither Mtr4 nor Dbp2 loss completely recapitulates the effect of upf1Δ, in terms of XUTs level, might be due to redundancy and/or sickness of the helicases mutants. Alternatively, in a heterogeneous population, a NMD-sensitive XUT might be single-stranded in some cells and form dsRNA in others; RNA helicases would then act only in the second case to provide access to NMD, while Upf1 would target both populations. Direct binding of Mtr4 to mRNA/asXUT is supported by CRAC data, and the insensitivity of XUTs to Trf4 and Trf5 suggests that Mtr4 act on XUTs independently of the TRAMP4/5 complex. Since Mtr4 is nuclear, these observations suggest that mRNA/asXUT duplexes exist in the nucleus where they are targeted by Mtr4. Dbp2 localizes in the nucleus and the cytoplasm depending on the conditions (Beck et al., 2014). Dbp2 has also been proposed to repress cryptic transcription (Cloutier et al., 2012). According to this idea, in our model gene, we would expect both SUT768 and XUT1678 levels to increase in the dbp2 mutant. However, the fact that only the XUT isoform was affected is not consistent with the hypothesis of transcriptional derepression. Also not consistent with a global derepression of XUTs, analysis of recent RNA-seq data obtained in dbp2Δ cells revealed up and downregulation for 386 and 686 antisense transcripts, respectively (Beck et al., 2014), both classes including XUTs (225 and 114). Future high-resolution RNA-seq and native elongating transcript (NET)-seq experiments will be required to quantitatively assess the sensitivity of XUTs to Dbp2 at the RNA and transcriptional level.
Together, our data lead to a model where Mtr4 (in the nucleus) and/or Dbp2 (in the nucleus and/or the cytoplasm) would preferentially unwind mRNA/XUT duplexes for which the XUT 3′ end remains single-stranded, such as XUT1678 (Figure 7). This 3′ extension could act as a platform for the helicases and/or serve as a primer to initiate the unwinding of the duplex. Helicases activity would result in the release of the single-stranded asXUT that ribosomes would rapidly bind for translation, in the cytoplasm. During this round of translation, detection of a long 3′ UTR would activate NMD, leading to decapping and Xrn1-mediated degradation of the XUT. In contrary, Mtr4/Dbp2 binding and/or activity would not be favored on antisense lncRNAs fully engaged in dsRNA (such as SUT768), the persistence of the dsRNA thus preventing translation and protecting against NMD.
Figure 7.

Model
See main text for details.
At the first glance, the fact that a fraction of full asXUTs was found to be NMD-sensitive would be inconsistent with this model. Nonetheless, it is possible that an asXUT fails to form dsRNA with its sense mRNA because both transcripts are not co-expressed in the same cells, as proposed for the PHO84 antisense lncRNA (Castelnuovo et al., 2013). However, our finding that 80% of asXUTs show significant production of small RNAs in RNAi+ xrn1Δ cells indicates that they co-exist and interact with their sense partner, at least in some cells. Further characterization using single-cell approaches will be required to understand these exceptions.
We and others revealed the role of NMD in the clearance of pervasive lncRNAs transcripts in yeast. Our work provides another layer of complexity by highlighting that NMD targets exclusively XUTs, i.e., those lncRNAs endowed with a regulatory potential and contributes in that way in buffering genome expression. In mammalian cells, NMD regulated physiological gene expression, but it is also controlled during cell differentiation or in response to cellular stress (Mühlemann and Jensen, 2012). This opens the exciting perspective that physiological environmental conditions could affect intracellular levels of lncRNAs with regulatory potential. The extent of pervasive transcription in human cells and the conservation of NMD raise the question of a conserved role of this pathway in controlling regulatory lncRNAs. Of particular interest would be the extent and impact of dsRNA in protecting lncRNAs from NMD in mammalian cells and the role of mechanisms that are absent in yeast, but contribute to NMD activation in these systems, such as the exon-junction complex-associated NMD activation (Le Hir et al., 2001). Future work on mammalian lncRNAs will be required to further reveal the complexity of pervasive transcripts regulation and physiological functions.
Experimental Procedures
Yeast strains are listed in Table S1. Total RNA was extracted from exponentially growing cells using standard hot phenol procedure. RNA detection by northern blot was performed using 32P-labeled oligonucleotides (Table S2). Strand-specific RT were performed from three independent biological replicates.
Strand-specific total RNA-seq libraries were prepared from rRNA-depleted or Terminator-treated RNA from biological duplicates of each strain. All bioinformatics analyses used uniquely mapped reads. Tags densities were normalized on snoRNAs levels. Segmentation parameters were selected among 3,880 combinations to give the best compromise between SUT and ORF detection (see Figure S7). CAGE-seq was performed using total RNA extracted from biological duplicates of WT and decapping-deficient cells. Peak-calling used filtered 25–27 nt uniquely mapped reads. Small RNA libraries were prepared from purified 10–40 nt small RNAs and bioinformatics analysis used 19–23 mapped reads.
Supplemental Experimental Procedures provide detailed descriptions of experiments and bioinformatics analyses.
Author Contributions
M.W. and A.M. designed the project. M.W. performed all the experiments and N.V. and A.S.D. provided technical assistance. M.D. performed bioinformatics analysis, with D.G.’s support. M.W. and A.M. analyzed the data. M.W. and A.M. wrote the manuscript.
Acknowledgments
We thank D. Bartel, D. Libri, A. Nicolas, E. Tran, and A. van Hoof for yeast strains. We are also grateful to A. Lebreton and B. Séraphin for sharing with us their unpublished tetOFF::MTR4 (BSY1756) strain. We thank T. Rio Frio, S. Baulande, and P. Legoix-Né (NGS platform) and A. Lermine (bioinformatics platform) from Institut Curie and the eBio bioinformatics platform (Orsay) for support; E. Heard, H. Le Hir, M. Springer, L. Bénard, D. Hermand, and members of our labs for discussions and critical reading of the manuscript. High-throughput sequencing was performed by the NGS platform of Institut Curie, supported by grants ANR-10-EQPX-03 and ANR10-INBS-09-08 from the Agence Nationale de la Recherche (investissements d’avenir) and by the Canceropôle Ile-de-France. This work has also benefited from the facilities and expertise of the high-throughput sequencing platforms of IMAGIF (http://www.i2bc.paris-saclay.fr). A.M.’s lab is supported by the ANR “REGULncRNA”, ERC “EpincRNA” starting, and ERC “DARK” consolidator grants.
Published: January 21, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2015.12.020.
Contributor Information
Daniel Gautheret, Email: daniel.gautheret@u-psud.fr.
Antonin Morillon, Email: antonin.morillon@curie.fr.
Accession Numbers
Sequence data can be accessed at the NCBI Gene Expression Omnibus using accession numbers GEO: GSE64090 and GSE69384. Genome browsers for visualization of processed total/small RNA-seq and CAGE-seq data are accessible at http://vm-gb.curie.fr/mw2.
Supplemental Information
References
- Anderson J.S., Parker R.P. The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 1998;17:1497–1506. doi: 10.1093/emboj/17.5.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista P.J., Chang H.Y. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck Z.T., Cloutier S.C., Schipma M.J., Petell C.J., Ma W.K., Tran E.J. Regulation of glucose-dependent gene expression by the RNA helicase Dbp2 in Saccharomyces cerevisiae. Genetics. 2014;198:1001–1014. doi: 10.1534/genetics.114.170019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein J., Patterson D.N., Wilson G.M., Toth E.A. Characterization of the essential activities of Saccharomyces cerevisiae Mtr4p, a 3′->5′ helicase partner of the nuclear exosome. J. Biol. Chem. 2008;283:4930–4942. doi: 10.1074/jbc.M706677200. [DOI] [PubMed] [Google Scholar]
- Berretta J., Morillon A. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009;10:973–982. doi: 10.1038/embor.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta J., Pinskaya M., Morillon A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev. 2008;22:615–626. doi: 10.1101/gad.458008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond A.T., Mangus D.A., He F., Jacobson A. Absence of Dbp2p alters both nonsense-mediated mRNA decay and rRNA processing. Mol. Cell. Biol. 2001;21:7366–7379. doi: 10.1128/MCB.21.21.7366-7379.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelnuovo M., Rahman S., Guffanti E., Infantino V., Stutz F., Zenklusen D. Bimodal expression of PHO84 is modulated by early termination of antisense transcription. Nat. Struct. Mol. Biol. 2013;20:851–858. doi: 10.1038/nsmb.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier S.C., Ma W.K., Nguyen L.T., Tran E.J. The DEAD-box RNA helicase Dbp2 connects RNA quality control with repression of aberrant transcription. J. Biol. Chem. 2012;287:26155–26166. doi: 10.1074/jbc.M112.383075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker C.J., Parker R. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev. 1993;7:1632–1643. doi: 10.1101/gad.7.8.1632. [DOI] [PubMed] [Google Scholar]
- Descrimes M., Ben Zouari Y., Wery M., Legendre R., Gautheret D., Morillon A. VING: a software for visualization of deep sequencing signals. BMC Res. Notes. 2015;8:419. doi: 10.1186/s13104-015-1404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma M.K., Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg I.A., Weinberg D.E., Xie K.T., Mower J.P., Wolfe K.H., Fink G.R., Bartel D.P. RNAi in budding yeast. Science. 2009;326:544–550. doi: 10.1126/science.1176945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg I.A., Fink G.R., Bartel D.P. Compatibility with killer explains the rise of RNAi-deficient fungi. Science. 2011;333:1592. doi: 10.1126/science.1209575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley T., Parker R. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J. 1999;18:5411–5422. doi: 10.1093/emboj/18.19.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisberg J.V., Moqtaderi Z., Fan X., Ozsolak F., Struhl K. Global analysis of mRNA isoform half-lives reveals stabilizing and destabilizing elements in yeast. Cell. 2014;156:812–824. doi: 10.1016/j.cell.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S., Lojek L., Khalil A.M., Baker K.E., Coller J. Decapping of long noncoding RNAs regulates inducible genes. Mol. Cell. 2012;45:279–291. doi: 10.1016/j.molcel.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCava J., Houseley J., Saveanu C., Petfalski E., Thompson E., Jacquier A., Tollervey D. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell. 2005;121:713–724. doi: 10.1016/j.cell.2005.04.029. [DOI] [PubMed] [Google Scholar]
- Le Hir H., Gatfield D., Izaurralde E., Moore M.J. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001;20:4987–4997. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G., Carter D.M., Moses A.M., Warringer J., Parts L., James S.A., Davey R.P., Roberts I.N., Burt A., Koufopanou V. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabat C., Feuerbach F., Ma L., Saveanu C., Jacquier A. Quality control of transcription start site selection by nonsense-mediated-mRNA decay. eLife. 2015;4:e06722. doi: 10.7554/eLife.06722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S., Hazelbaker D.Z., Buratowski S. Distinct RNA degradation pathways and 3′ extensions of yeast non-coding RNA species. Transcription. 2011;2:145–154. doi: 10.4161/trns.2.3.16298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Mühlemann O., Jensen T.H. mRNP quality control goes regulatory. Trends Genet. 2012;28:70–77. doi: 10.1016/j.tig.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Parker R. Mutations affecting stability and deadenylation of the yeast MFA2 transcript. Genes Dev. 1992;6:2100–2111. doi: 10.1101/gad.6.11.2100. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Parker R. Premature translational termination triggers mRNA decapping. Nature. 1994;370:578–581. doi: 10.1038/370578a0. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Parker R. Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA. 1999;5:1299–1307. doi: 10.1017/s1355838299990829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil H., Malabat C., d’Aubenton-Carafa Y., Xu Z., Steinmetz L.M., Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–1042. doi: 10.1038/nature07747. [DOI] [PubMed] [Google Scholar]
- Parker R. RNA degradation in Saccharomyces cerevisae. Genetics. 2012;191:671–702. doi: 10.1534/genetics.111.137265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn J.L., Chang H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn J., Guttman M. RNA Function. RNA and dynamic nuclear organization. Science. 2014;345:1240–1241. doi: 10.1126/science.1252966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D., Schwalb B., Kiesel A., Baejen C., Torkler P., Gagneur J., Soeding J., Cramer P. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell. 2013;155:1075–1087. doi: 10.1016/j.cell.2013.10.024. [DOI] [PubMed] [Google Scholar]
- Sinturel F., Navickas A., Wery M., Descrimes M., Morillon A., Torchet C., Benard L. Cytoplasmic control of sense-antisense mRNA pairs. Cell Rep. 2015;12:1853–1864. doi: 10.1016/j.celrep.2015.08.016. [DOI] [PubMed] [Google Scholar]
- Smith J.E., Alvarez-Dominguez J.R., Kline N., Huynh N.J., Geisler S., Hu W., Coller J., Baker K.E. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7:1858–1866. doi: 10.1016/j.celrep.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft R.J., Pheasant M., Mattick J.S. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays. 2007;29:288–299. doi: 10.1002/bies.20544. [DOI] [PubMed] [Google Scholar]
- Taft R.J., Pang K.C., Mercer T.R., Dinger M., Mattick J.S. Non-coding RNAs: regulators of disease. J. Pathol. 2010;220:126–139. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Lassmann T., Murata M., Carninci P. 5′ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat. Protoc. 2012;7:542–561. doi: 10.1038/nprot.2012.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D.M., Parker R. Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007;27:92–101. doi: 10.1128/MCB.01023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisseur M., Kwapisz M., Morillon A. Pervasive transcription - Lessons from yeast. Biochimie. 2011;93:1889–1896. doi: 10.1016/j.biochi.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Tuck A.C., Tollervey D. A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell. 2013;154:996–1009. doi: 10.1016/j.cell.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudek A., Porrua O., Kabzinski T., Lidschreiber M., Kubicek K., Fortova A., Lacroute F., Vanacova S., Cramer P., Stefl R., Libri D. Molecular basis for coordinating transcription termination with noncoding RNA degradation. Mol. Cell. 2014;55:467–481. doi: 10.1016/j.molcel.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudek A., Candelli T., Libri D. Non-coding transcription by RNA polymerase II in yeast: Hasard or nécessité? Biochimie. 2015;117:28–36. doi: 10.1016/j.biochi.2015.04.020. [DOI] [PubMed] [Google Scholar]
- van Dijk E.L., Chen C.L., d’Aubenton-Carafa Y., Gourvennec S., Kwapisz M., Roche V., Bertrand C., Silvain M., Legoix-Né P., Loeillet S. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011;475:114–117. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- Wery M., Kwapisz M., Morillon A. Noncoding RNAs in gene regulation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011;3:728–738. doi: 10.1002/wsbm.148. [DOI] [PubMed] [Google Scholar]
- Wery M., Descrimes M., Thermes C., Gautheret D., Morillon A. Zinc-mediated RNA fragmentation allows robust transcript reassembly upon whole transcriptome RNA-Seq. Methods. 2013;63:25–31. doi: 10.1016/j.ymeth.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Wilson M.A., Meaux S., van Hoof A. A genomic screen in yeast reveals novel aspects of nonstop mRNA metabolism. Genetics. 2007;177:773–784. doi: 10.1534/genetics.107.073205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyers F., Rougemaille M., Badis G., Rousselle J.C., Dufour M.E., Boulay J., Régnault B., Devaux F., Namane A., Séraphin B. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.