

Abstract
Autophagy plays a pivotal role in cellular homeostasis and adaptation to adverse environments, although the regulation of this process remains incompletely understood. We have recently observed that caveolin-1 (Cav-1), a major constituent of lipid rafts on plasma membrane, can regulate autophagy in cigarette smoking-induced injury of lung epithelium, although the underlying molecular mechanisms remain incompletely understood. In the present study we found that Cav-1 interacted with and regulated the expression of ATG12-ATG5, an ubiquitin-like conjugation system crucial for autophagosome formation, in lung epithelial Beas-2B cells. Deletion of Cav-1 increased basal and starvation-induced levels of ATG12-ATG5 and autophagy. Biochemical analyses revealed that Cav-1 interacted with ATG5, ATG12, and their active complex ATG12-ATG5. Overexpression of ATG5 or ATG12 increased their interactions with Cav-1, the formation of ATG12-ATG5 conjugate, and the subsequent basal levels of autophagy but resulted in decreased interactions between Cav-1 and another molecule. Knockdown of ATG12 enhanced the ATG5-Cav-1 interaction. Mutation of the Cav-1 binding motif on ATG12 disrupted their interaction and further augmented autophagy. Cav-1 also regulated the expression of ATG16L, another autophagy protein associating with the ATG12-ATG5 conjugate during autophagosome formation. Altogether these studies clearly demonstrate that Cav-1 competitively interacts with the ATG12-ATG5 system to suppress the formation and function of the latter in lung epithelial cells, thereby providing new insights into the molecular mechanisms by which Cav-1 regulates autophagy and suggesting the important function of Cav-1 in certain lung diseases via regulation of autophagy homeostasis.
Keywords: caveolin-1, ATG12-ATG5, ATG16L, autophagy, lung diseases
autophagy is a dynamic process responsible for the turnover of cellular organelles and long-lived proteins. During this process, cytosolic proteins and organelles (e.g., mitochondria and endoplasmic reticulum) are engulfed into double-membrane-bound vesicles, autophagosomes. The outer membrane of the autophagosome subsequently fuses with lysosomes to form autolysosomes in which the engulfed components are degraded by lysosomal hydrolases, regenerating metabolic precursors that are recycled for macromolecular synthesis and ATP generation (20, 42). Autophagy is induced above basal levels in response to diverse stimuli including nutrient starvation, genotoxic agents, cytokines, and oxidative stress. This process provides an essential function in the maintenance of cellular homeostasis and adaptation to adverse environments (16, 29, 35).
More than 30 autophagy-related genes and gene products critical in the regulation of autophagy, designated “ATG,” have been identified heretofore in yeast and higher mammals (13). Autophagosome formation requires two ubiquitin-like conjugation systems, the ATG12 and the microtubule-associated protein 1 light chain 3 B (LC3B, yeast ATG8) systems, which are tightly associated with expansion of autophagosomal membrane. The ATG12 system is located upstream of the LC3B system in the context of ATG protein organization. ATG12 is finally conjugated to ATG5, forming the irreversible ATG12-ATG5 complex, which strongly enhances the formation of LC3B-phosphatidylethanolamine conjugation (12). In mammals, the conversion of LC3B from LC3B-I (free form) to LC3B-II (phosphatidylethanolamine-conjugated form) represents a key step in autophagosome formation (15).
Caveolae are 50- to 100-nm omega-shaped invaginations of the cell surface plasma membrane that are enriched in glycosphingolipids and cholesterol (6, 17). Caveolae occur in a variety of cell types including epithelial cells, endothelial cells, fibroblasts, smooth muscle cells, and adipocytes. Caveolin-1 (Cav-1), a 21- to 24-kDa protein, is a major resident scaffolding protein constituent of caveolae that participates in vesicular trafficking and signal transduction events (6, 7, 17). This molecule has been known to interact with various pathophysiologically important molecules such as inflammatory regulator Toll-like receptor (40), apoptotic factors Fas and Bid (43), and antioxidant molecule heme oxygenase-1 (14). We have recently observed that Cav-1 can regulate autophagy in contexts of cigarette smoking-induced chronic obstructive pulmonary disease (COPD) and hypoxia-induced pulmonary hypertension (5, 19, 36); however, the detailed molecular mechanisms by which Cav-1 regulates autophagy remain incompletely understood.
In the present study, we found that Cav-1 could interact with ATG5 and ATG12, and their active complex ATG12-ATG5, thereby suppressing the formation and activity of ATG12-ATG5 conjugate and subsequent autophagy in lung epithelial cells. These data demonstrated an alternative mechanism for Cav-1 to suppress autophagy in addition to previous studies (5, 19, 36) and reemphasized the important role of Cav-1 in regulation of autophagy homeostasis in certain lung diseases.
MATERIALS AND METHODS
Cell culture.
Beas-2B lung epithelial cells were purchased from American Type Culture Collection (CRL-9609), and mouse lung fibroblasts were isolated from wild-type C57BL6 or Cav-1 knockout mice (Jackson Laboratory, no. 007083) as described previously (32, 41). All cells were maintained in DMEM containing 10% fetal bovine serum and antibiotics. For starvation, full culture medium was replaced by Hanks' balanced salt solution (HBSS, Invitrogen, no. 24020-117) for indicated times. For autophagic flux assay, bafilomycin A1 (BafA1, Sigma, B1793) was used to inhibit the autophagosome-lysosome fusion.
Transfection of siRNA and cDNA.
Human Cav-1-siRNA was from Dharmacon (L-003467-00), and ATG12-siRNA was obtained from Qiagen (SI02655289). The siRNA was transfected into cells by using the transfection reagent Lipofectamine 2000 (Invitrogen, no. 11668019). Human wild-type ATG12 cDNA was obtained from Origene (SC117215), and the monomeric red fluorescent protein (RFP)-Cav-1 was from Addgene (www.addgene.org). The mutant derivative of ATG12 was performed with the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, no. 200555), following the supplier's protocol. All cDNAs were transfected into Beas-2B cells by using Lipofectamine 2000 (Invitrogen, no. 11668019).
Immunoprecipitation and immunoblotting analysis.
Immunoprecipitation (IP) and immunoblotting (IB) were performed essentially as previously described (32, 41). Antibodies against LC3B (L7543) and β-actin (A5441) were from Sigma. The Cav-1 antibody was obtained from BD Transduction Laboratories (no. 610406). ATG5 antibodies were from Santa Cruz Biotechnology (sc-33210) or Cell Signaling (no. 2630). ATG12 antibodies were purchased from Abgent (AP1816a) or Cell Signaling (no. 2010). ATG16L antibody was also a product of Cell Signaling (no. 8089).
Real-time PCR.
Total RNA was extracted from cells by using TRIzol (Invitrogen) according to the manufacturer's instructions and reverse transcribed into cDNA with random hexamers and Superscript II reverse transcriptase (Invitrogen). The relative levels of ATG5 and ATG12 mRNA transcripts to control GAPDH were determined by quantitative real-time PCR using the SYBR Green system (Takara) on a spectrofluorometric thermal cycler (ABI 7500 system).
Sucrose-gradient subcellular fractionation.
Sucrose gradient-derived fractions were separated as described previously (41). In brief, cells were lysed in ice-cold MBS buffer (25 mM Mes, pH 6.5, 150 mM NaCl, 1% Triton X-100, 1 mM Na3VO4, and protease inhibitors). Lysates were adjusted to 4 ml of 40% sucrose by mixing with 2 ml of 80% sucrose and overlaid with 4 ml of 35% sucrose and 4 ml of 5% sucrose in MBS buffer. Samples were ultracentrifuged at 39,000 rpm for 18 h [SW41 rotor (Beckman Instruments)] and fractionated into 12 subfractions.
Confocal imaging study.
After treatment, cells were fixed with 4% paraformaldehyde and analyzed with the immunofluorescence staining protocol as described previously (32). Samples were viewed with an Olympus Fluoview 300 confocal laser scanning head with an Olympus IX70 inverted microscope.
Image quantification and statistics.
Immunoblots were quantified by use of the ImageJ software downloaded from the NIH website. All data of at least three independent experiments are expressed as means ± SD and analyzed by Student's t-test with SPSS 13.0 software. Values of P < 0.05 were considered to be statistically significant.
RESULTS
Cav-1 regulates the expression of ATG12-ATG5 in addition to LC3B.
Because we have recently observed that Cav-1 can modulate cigarette smoking- or hypoxia-induced autophagy (5, 19, 36), the initial purposes of the present study are to examine whether Cav-1 can also regulate autophagy in classical models such as starvation and to investigate whether it can modulate expression of other autophagic molecules. In agreement with our previous findings in other autophagy models, siRNA-dependent knockdown of Cav-1 also significantly increased starvation-induced autophagy in lung epithelial Beas-2B cells (Fig. 1A). More interestingly, the knockdown of Cav-1 not only upregulated the levels of LC3B-II but also increased the starvation-induced expression of ATG12-ATG5 (Fig. 1A), an important upstream complex responsible for LC3B lipidation. Furthermore, Cav-1-siRNA exerted no appreciable effects on the basal expression of p-S-6 (Fig. 1A), a hallmark reflecting the activity of mammalian target of rapamycin (mTOR) which is the crucial upstream signaling of autophagy. In case of starvation, the level of p-S-6 in Cav-1-siRNA-treated cells was even a little higher than that in control cells, which was not consistent with the enhanced levels of autophagy in Cav-1 deficiency cells. These data suggested that Cav-1 regulation of autophagy was not likely through modulation of mTOR activity, but most likely through the autophagy machinery itself.
Fig. 1.
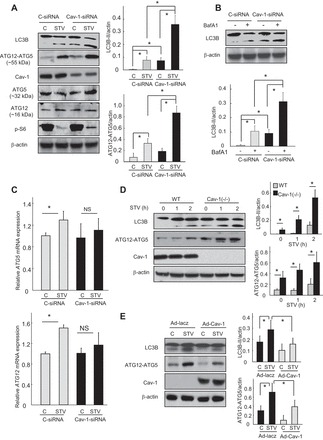
Caveolin-1 (Cav-1) regulates the expression of ATG12-ATG5 and LC3B. A–C: Beas-2B cells were transfected with control- or Cav-1-siRNA for 48 h, and followed by starvation with Hanks' balanced salt solution (HBSS) for 2 h (A and C), or bafilomycin A1 (BafA1; 100 nM) for 2 h (B). Cells were then harvested for immunoblotting analysis of indicated proteins (A and B) or RT-PCR analysis of expression of ATG5 or ATG12 mRNA transcripts (C). D: lung fibroblasts from wild-type (WT) and Cav-1(−/−) mice were starved with HBSS for indicated time. E: Cav-1(−/−) fibroblasts were infected with adenovirus Lacz or Cav-1 for 48 h, followed by starvation with HBSS for 2 h. Cells were then harvested for immunoblotting analysis of indicated proteins. Blots were quantified from 3 independent results. C, control; STV, starvation. *Significant difference (P < 0.05); NS, not significant.
The expression of LC3B-II in Cav-1-siRNA-treated cells was further increased by bafilomycin A1 (BafA1, Fig. 1B), a well-characterized lysosome inhibitor, suggesting that the increased levels of LC3B-II by Cav-1 deficiency were indeed induced, rather than due to an impaired autophagosome-lysosome fusion. The mRNA transcripts of both ATG5 and ATG12 were notably induced by starvation in control cells but were not induced in Cav-1-siRNA-treated cells (Fig. 1C), which strongly indicated that the increased level of ATG12-ATG5 in Cav-1 deficiency cells was not likely due to an increased protein synthesis. Similarly, starvation-induced autophagy was also enhanced in Cav-1 deficiency lung fibroblasts, again with increased levels of both LC3B-II and ATG12-ATG5 (Fig. 1D). Furthermore, the increased autophagy in Cav-1 deficiency fibroblasts appeared to be reversible, as “knock-in” back of Cav-1 using adenovirus in these cells decreased the expression of both LC3B-II and ATG12-ATG5 (Fig. 1E).
Cav-1 physically interacts with the ATG12-ATG5 system.
We next sought to explore the mechanisms how Cav-1 regulates the levels of ATG12-ATG5. Since Cav-1 is readily to physically interact with various proteins and regulate their function and expression, we then examined the possible interactions between Cav-1 and ATG5 or ATG12. IP studies revealed that both ATG5 and ATG12 appeared to interact with Cav-1 under basal conditions, and these basic interactions were disrupted by starvation (Fig. 2A). Reversed IP assays further confirmed that both free forms of ATG12 and ATG5 effectively reacted with Cav-1, and these reactions were decreased during starvation (Fig. 2A). Intriguingly, the IP assays also demonstrated that the interaction between Cav-1 and the ATG12-ATG5 conjugate was declined during starvation, despite that the cellular level of ATG12-ATG5 was slightly increased (Fig. 2A).
Fig. 2.
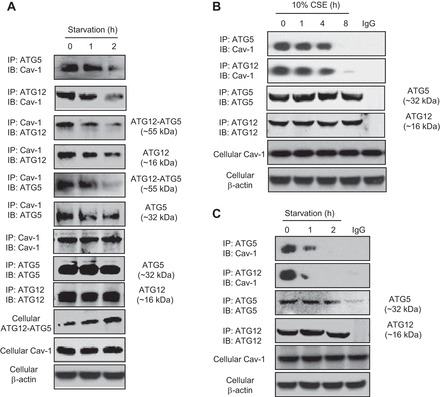
Cav-1 physically interacts with the ATG12-ATG5 system. A and B: Beas-2B cells were starved with HBSS (A) or treated with cigarette smoke extract (CSE; B) for indicated times. C: wild-type lung fibroblasts were starved with HBSS for indicated times. Samples were immunoprecipitated (IP) and subjected to immunoblotting (IB) analysis as indicated in the figures. Cellular Cav-1 and β-actin derived from same cell lysates of IP experiments served as loading controls.
We also examined the reactions between Cav-1 and the ATG12-ATG5 system in other autophagy model and in other cell type, and similar kinetics has been observed in cigarette smoke extract (CSE) treated Beas-2B cells (Fig. 2B) and in starvation treated wild-type lung fibroblasts (Fig. 2C), suggesting that ubiquitous interactions exist between these molecules.
ATG12-ATG5 interacts with Cav-1 in cytosol.
Our recent studies have demonstrated that partial LC3B interacts with Cav-1 and Fas to form a triple death complex in the plasma membrane lipid rafts (5, 19), we therefore attempted to examine whether the ATG12-ATG5 system also partially localizes to lipid rafts. Subcellular fractionation experiments showed that a very small amount of ATG12-ATG5 localized to low-density Cav-1 containing fractions 3–5 under basal conditions in Beas-2B cells and lung fibroblasts, whereas most of the free ATG5 and ATG12-ATG5 conjugate were in fractions 9–12 (Fig. 3A). Interestingly, although we have confirmed that the free form of ATG5 can also react with Cav-1, it appeared not to localize to float fractions 3–5, since in both Beas-2B cells and fibroblasts inappreciable free ATG5 existed in these fractions (Fig. 3A). However, confocal imaging analysis revealed that the reactions between Cav-1 and ATG12-ATG5 system in Beas-2B cells exhibited mostly all over the cytosol and even in nucleus but not much on plasma membrane (Fig. 3B). In fact, we have recently observed that, in Beas-2B cells, Cav-1 distributed majorly in cytosol as well as in nucleus (22).
Fig. 3.
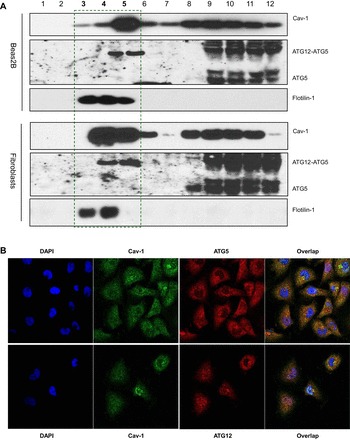
ATG12-ATG5 interacts with Cav-1 in cytosol. A: Beas-2B cells or wild-type fibroblasts were subfractioned and subjected to Western blot analysis for Cav-1, flotillin-1, and ATG5. B: confocal imaging analysis of ATG5-Cav-1 and ATG12-Cav-1 interactions in Beas-2B cells. Cells were fixed and stained with indicated antibodies and were subjected to confocal analysis.
Competitive reactions between ATG5, ATG12, and Cav-1.
To further clarify the complicated interactions between ATG5, ATG12, and Cav-1, we tried to modulate their reactions using plasmid cDNA or siRNA. As expected, overexpression of ATG5 resulted in an increased ATG12-ATG5 conjugation and therefore enhanced the basal autophagy levels (Fig. 4A), which is consistent with other recent reports (34, 39). The BafA1 flux assay further demonstrated that ATG5 overexpression increased the autophagosome formation rather than decreased the autophagosome maturation (Fig. 4B). Interestingly, overexpression of ATG5 increased the interaction between ATG5 and Cav-1, whereas it decreased that between ATG12 and Cav-1 (Fig. 4C). Genetic knockdown of ATG12 by siRNA markedly decreased the cellular ATG12-ATG5 conjugate as well as its interaction with Cav-1, but further increased the interaction between Cav-1 and the free form of ATG5 (Fig. 4D). These data altogether strongly suggested a competitive mechanism of the interactions among ATG5, ATG12, and Cav-1.
Fig. 4.
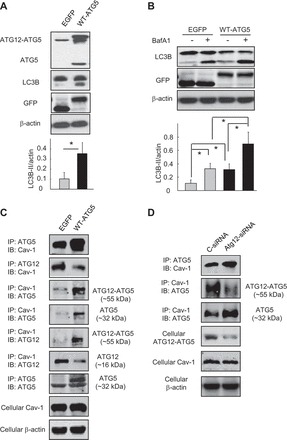
Competitive reactions between ATG5, ATG12, and Cav-1. A–C: Beas-2B cells were transfected with wild-type ATG5 or its control vector EGFP for 48 h (A and C), followed by BafA1 (100 nM) treatment for 2 h (B). Samples were then subjected to Western blot analysis (A and B) or IP assays (C) as indicated in the figures. Blots were quantified from 3 independent results. *Significant difference (P < 0.05). D: Beas-2B cells were transfected with ATG12-siRNA or its control for 48 h and subjected to IP assays as indicated. Cellular Cav-1 and β-actin derived from same cell lysates of IP experiments served as input controls.
Mutation of the Cav-1-binding motif on ATG12 disrupts the interaction between these molecules.
Proteins that bind Cav-1 typically contain canonical Cav-1-binding motifs (CBMs), ΦXΦXXXXΦ or ΦXXXXΦXXΦ, where Φ refers to an aromatic amino acid (W, F, or Y) and X is any nonaromatic amino acid, although proteins without such motifs are also capable of binding to Cav-1 (5, 40). A detailed analysis of the amino acid sequence of ATG5 and ATG12 revealed that ATG5 has no such typical CBM, whereas ATG12 contains a sequence 101FIYVNQSFAP110, an exact consensus of CBM. Overexpression of wild-type ATG12 also increased the levels of autophagy, as revealed by the enhanced ATG12-ATG5 conjugation, the increased LC3B-II expression (Fig. 5A), and the autophagic flux results (Fig. 5B). To examine whether the CBM sequence on ATG12 mediates the ATG12-Cav-1 interaction, we generated amino acid substitution mutants of ATG12 at position Y103. Interestingly, overexpression of wild-type ATG12 increased its interaction with Cav-1, which was effectively attenuated by Y103A mutation, an aromatic-to-nonaromatic amino acid substitution (Fig. 5C). Moreover, this Y103A mutation further increased the levels of starvation-induced autophagy (LC3B-II) relative to the wild-type ATG12 control (Fig. 5D).
Fig. 5.
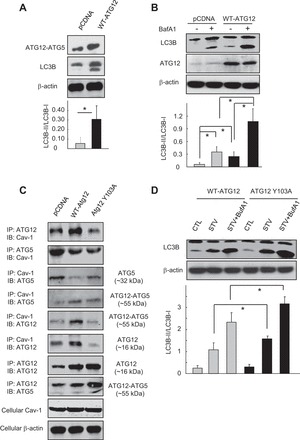
Mutation of the Cav-1 binding motif in ATG12 disrupts their interaction and modulates autophagy. A and B: Beas-2B cells were transfected with wild-type ATG12 or their vector control pCDNA for 48 h (A and B), and followed by BafA1 (100 nM) treatment for 2 h (B). Samples were then subjected to Western blot analysis. Blots were quantified from 3 independent results. C: control plasmid, wild-type ATG12 (WT-ATG12), or mutated ATG12Y103A were transfected into Beas-2B cells for 48 h. Samples were subjected to IP assays as indicated. Cellular Cav-1 and β-actin derived from same cell lysates of IP experiments served as input controls. D: Beas-2B cells were transfected with WT-ATG12 or ATG12Y103A for 48 h. Cell were then starved by HBSS for 2 h, with or without BafA1, and analyzed by Western blot for LC3B. Blots were quantified from 3 independent results. *Significant difference (P < 0.05).
Cav-1 regulates the expression of ATG16L.
It has been well established that the ATG12-ATG5 conjugate forms a complex with ATG16L in mammals (13, 20, 42), and this triplex plays an essential role in autophagosome formation. We were therefore interested in examining whether Cav-1 could also regulate the expression of ATG16L in Beas-2B cells. Notably, siRNA-dependent knockdown or cDNA-based overexpression of Cav-1 significantly increased or decreased the expression of ATG16L, respectively (Fig. 6A), which was in complete agreement with the function of Cav-1 in regulation of autophagy. Moreover, RT-PCR analysis revealed that the mRNA expression of ATG16L were not affected by Cav-1 (Fig. 6B), suggesting a posttranscriptional regulation mechanism.
Fig. 6.
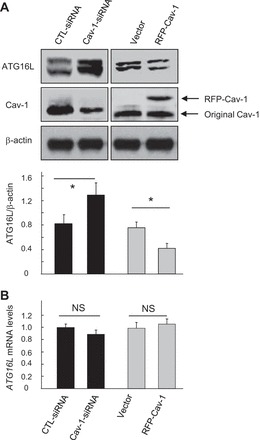
Cav-1 modulates expression of ATG16L. Beas-2B cells were transfected with indicated siRNAs or cDNAs for 48 h and harvested for Western blot analysis of ATG16L or Cav-1 (A) or RT-PCR analysis of ATG16L (B). Blots were quantified from 3 independent results. *Significant difference (P < 0.05).
DISCUSSION
Cav-1 has been known to regulate a variety of signaling pathways, primarily through its physical interaction with target proteins. The typical Cav-1-protein interaction occurs through the scaffolding domain in Cav-1 and the CBM in target proteins, such as endothelial nitric oxide synthase (11), Toll-like receptor 4 (40), and activin receptor-like kinase 1 (37). We have recently observed that the autophagy protein LC3B and transcription factor nuclear erythroid 2 p45-related factor-2 (Nrf2), respectively, contain amino acid sequences very similar to CBM, and these similar structures mediate the interactions of Cav-1-LC3B (4) and Cav-1-Nrf2 (22). In the present study, we have also found that a typical CBM is presented in ATG12, which also mediates its interaction with Cav-1, as mutation of the CBM in ATG12 effectively disrupted their association. We have carefully looked at several autophagy-related proteins such as Beclin-1, ATG4B, ATG7, ATG16L, and ATG5 and found no typical CBM in these proteins. Other autophagy proteins, however, have not been checked.
Certain proteins without such typical CBM can also interact with Cav-1 (6, 8, 17). For example, we have found that the apoptotic factor Fas has no CBM but binds to Cav-1 through the palmitoylation domains, and in such a case the mutation of the Cav-1 scaffolding domain has little effect on the Cav-1-Fas interaction (5). In the present study, we have also observed that ATG5 can effectively interact with Cav-1, although it does not contain a typical or similar CBM. The free form of ATG5 appeared to be not localized in the lipid raft fractions (Fig. 3A), but it can be effectively pulled down by Cav-1 (Fig. 2A), and knockdown of ATG12 further increased the ATG5-Cav-1 interaction (Fig. 4D), strongly suggesting that an interaction indeed occurs between ATG5 and Cav-1 that is not medicated by ATG12.
Therefore, ATG5, ATG12, and Cav-1 form a variety of complex forms, ATG12-ATG5, ATG5-Cav-1, ATG12-Cav-1, and a triplex ATG12-ATG5-Cav-1, and Cav-1 effectively modulates the expression and function of ATG12-ATG5. The interactions between ATG5, ATG12, and Cav-1 appear to be competitive with each other. Cav-1 deficiency resulted in an increased ATG12-ATG5 interaction (Fig. 1, A and C), and, similarly, knockdown of ATG12 led to an increased ATG5-Cav-1 interaction (Fig. 4D). On the other hand, overexpression of ATG5 increased the levels of ATG5-Cav-1 and ATG12-ATG5 but decreased the interaction of ATG12-Cav-1 (Fig. 4, A and C). All these data strongly demonstrate a competitive mechanism of the interactions among these three proteins.
It might be noteworthy that after overexpression or knockdown of the cellular basal levels of ATG12 or ATG5 (Figs. 4C, 4D, and 5C), the interaction between Cav-1 and another molecule was reversely regulated despite the correspondingly changed ATG12-ATG5 conjugate (e.g., in case of ATG5 overexpression, co-IP of Cav-1 and ATG12 was decreased in spite of the increased ATG12-ATG5 conjugate, Fig. 4C). Furthermore, the interactions between Cav-1 and ATG12 or ATG5 declined during starvation, also regardless of the increased cellular level of ATG12-ATG5 (Fig. 2A). These data altogether suggest that Cav-1 may interact with free from of ATG12 or ATG5 stronger than with their ATG12-ATG5 conjugate.
Unlike the irreversible conjugation of ATG12-ATG5, the interactions between Cav-1 and ATG5, ATG12, or ATG12-ATG5 are disassociated during the autophagy process when stimulated by starvation or CSE (Fig. 2, A–C), and this kinetic is very similar to that of Cav-1-LC3B interaction (Fig. 2D in Ref. 5). Interestingly, overexpression of either ATG5 or ATG12 not only enhanced the levels of ATG12-ATG5 but also increased their interactions with Cav-1 (Figs. 4C and 5C). Similarly, overexpression of LC3B also increased the levels of Cav-1-LC3B at basal conditions (Fig. 3B in Ref. 5). These findings suggest that Cav-1 functions as an important regulator that spontaneously interacts with the autophagic proteins ATG5, ATG12, ATG12-ATG5 conjugate, and LC3B, thereby trying to maintain the autophagy homeostasis (Fig. 7). In addition to our studies (5, 19, 36), others have also observed that significantly increased levels of autophagy in a variety of Cav-1 deficiency cells and tissues, including epithelial, endothelial, stromal, and cancer cells, and adipocytes (3, 18, 25, 26), which emphasizes a ubiquitous function of Cav-1 in suppression of autophagy.
Fig. 7.
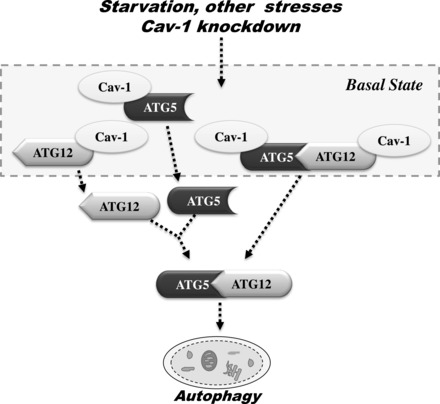
Schemata showing the proposed role of the Cav-1 in the regulation of ATG12-ATG5 system during the autophagic process. Under basal conditions, Cav-1 forms complexes with ATG5, ATG12, and the ATG12-ATG5 conjugate. Stimulation of lung epithelial cells with starvation or other stresses, or knockdown of Cav-1, results in the rapid dissociation of these complexes, and subsequently the ATG12-ATG5 conjugate triggers the autophagic process.
It should be noted that in our present study we demonstrated that overexpression of ATG5 or ATG12 eventually enhanced autophagy in lung epithelial Beas-2B cells, whereas in a previous study Fujita et al. (9) observed that in PC12 cells overexpression of Atg5 did not affect autophagy and overexpression of Atg12 or Atg16L even inhibited LC3 lipidation and autophagy. However, recently it has been reported that in MCF-7 cells overexpression of ATG5 could rescue the autophagic activity (39), and overexpression of ATG5 in mice could activate autophagy and extend lifespan in vivo (34). Thus the effects of overexpression of these ATG proteins in regulation of autophagy may vary between different cells and study models.
It might be of interests to note that in the fractionation experiment a small part of ATG12-ATG5 existed in the float fractions 3–5 (Fig. 3A), whereas the confocal analysis revealed little interactions occurred on the plasma membrane. One plausible explanation is that during fractionation some of the autophagosomes, especially the small-sized ones, were not disrupted, so that these undestroyed autophagosomes might exist in the float fractions. The sizes of autophagosomes are usually 0.5–1.5 μm (28), whereas in some conditions they could be as small as 100–200 nm in diameters (1), similar to the sizes of caveolae. The highly lipidated LC3B-II in autophagosomes may lead these vesicles have the same physicochemical characters as caveolae.
Our studies demonstrated that the expression of ATG16L was modulated by Cav-1 in Beas-2B cells as well (Fig. 6), which was consistent with the levels of autophagy. Thus the function of Cav-1 in autophagy homeostasis might also be due in part to its regulation of ATG16L, although the detailed molecule mechanisms of this process need further investigation.
Cav-1 has been shown to play important roles in a number of human diseases; however, whether the role of Cav-1 in these diseases is due to its regulation of autophagy requires further investigation, probably depending on how importantly autophagy plays a role in the pathogenesis of the diseases. We have demonstrated that autophagy mediates cigarette smoking-induced lung epithelial cell apoptosis and contributes to the development of COPD (4, 5, 36). In agreement with this, Cav-1 knockout mice displayed increased levels of autophagy and were more susceptible to cigarette smoking-induced COPD (5). In a systemic sclerosis model, Castello-Cros and colleagues (3) have also observed enhanced autophagy in Cav-1 null mice; they suggest that inhibition of autophagy and aerobic glycolysis may represent a new promising therapeutic strategy for halting fibrosis in systemic sclerosis patients. In these disease models, the functions of Cav-1 in disease pathogenesis appear to be mediated, at least in part, by autophagy. In lipopolysaccharide (LPS)-induced lung inflammation and sepsis models, deletion of Cav-1 resulted in decreased production of inflammatory mediators and animal mortality (10, 27), but whether this function of Cav-1 is due to autophagy remains unclear, although recently autophagy has been shown to suppress LPS-induced inflammation and LC3B-deficient mice were more susceptible to LPS-induced mortality (31).
In addition to our recent studies showing that autophagy mediates cigarette smoking-induced lung epithelial damage (2, 4, 5, 36), other works have also demonstrated that autophagy positively regulates lung injury induced by influenza H5N1 (33, 38), nanomaterials (21, 23), and ventilation (24). Thus inhibition of autophagy might represent a valuable therapeutic target in certain lung diseases (30), and preservation of Cav-1 might acquire therapeutic benefits in these diseases.
In conclusion, our present study represents initial efforts to explore the molecular mechanisms by which Cav-1 suppresses autophagy through physical interactions with the ATG12-ATG5 system and regulation of ATG16L in lung epithelial cells. Thus Cav-1 functions as a spontaneous suppressor of autophagy by association with autophagic proteins ATG5, ATG12, ATG12-ATG5 conjugate, and LC3B, as well as through modulation of ATG16L expression. Cav-1 plays important role in certain human diseases, possibly through regulation of autophagy.
GRANTS
This work was supported in part by the Distinguished Young Investigator Award by the National Natural Science Foundation of China (H.-H. Shen, 30825019), the General Project of National Natural Science Foundation of China (Z.-H. Chen, 31170859), and the Distinguished Young Investigator Award by the Natural Science Foundation of Zhejiang Province (Z.-H. Chen, R2110323).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Z.-H.C. and H.-H.S. conception and design of research; Z.-H.C., J.-F.C., J.-S.Z., H.L., L.-Q.C., K.M., and W.L. performed experiments; Z.-H.C., J.-F.C., A.M.C., and H.-H.S. analyzed data; Z.-H.C., J.-F.C., A.M.C., and H.-H.S. interpreted results of experiments; Z.-H.C. prepared figures; Z.-H.C. drafted manuscript; Z.-H.C., A.M.C., and H.-H.S. edited and revised manuscript; Z.-H.C., J.-F.C., J.-S.Z., H.L., L.-Q.C., K.M., W.L., A.M.C., and H.-H.S. approved final version of manuscript.
REFERENCES
- 1.Abeliovich H, Dunn WA, Jr, Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 151: 1025–1034, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An CH, Wang XM, Lam HC, Ifedigbo E, Washko GR, Ryter SW, Choi AM. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol 303: L748–L757, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castello-Cros R, Whitaker-Menezes D, Molchansky A, Purkins G, Soslowsky LJ, Beason DP, Sotgia F, Iozzo RV, Lisanti MP. Scleroderma-like properties of skin from caveolin-1 deficient mice: Implications for new treatment strategies in patients with fibrosis and systemic sclerosis. Cell Cycle 10: 2140–2150, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, Choi AM. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 3: e3316, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA 107: 18880–18885, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res 86: 219–225, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev 84: 1341–1379, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem 272: 6525–6533, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19: 2092–100, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, Malik AB. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol 177: 4853–4860, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S, Gachhui R, Crooks C, Wu C, Lisanti MP, Stuehr DJ. Interaction between caveolin-1 and the reductase domain of endothelial nitric-oxide synthase. Consequences for catalysis. J Biol Chem 273: 22267–22271, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The ATG12-ATG5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282: 37298–37302, 2007. [DOI] [PubMed] [Google Scholar]
- 13.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43: 67–93, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin Y, Kim HP, Chi M, Ifedigbo E, Ryter SW, Choi AM. Deletion of caveolin-1 protects against oxidative lung injury via up-regulation of heme oxygenase-1. Am J Respir Cell Mol Biol 39: 171–179, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klionsky DJ, Abdallam FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 40: 280–293, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol 185: 381–385, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Lay S, Briand N, Blouin CM, Chateau D, Prado C, Lasnier F, Le Liepvre X, Hajduch E, Dugail I. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy 6: 754–763, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, Feghali-Bostwick C, Ryter SW, Kim HP, Rabinovitch M, Choi AM. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 183: 649–658, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6: 463–477, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Liu H, Sun Y, Wang H, Guo F, Rao S, Deng J, Zhang Y, Miao Y, Guo C, Meng J, Chen X, Li L, Li D, Xu H, Wang H, Li B, Jiang C. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol 1: 37–45, 2009. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Liu H, Zhou J, Cao JF, Zhou XB, Choi AM, Chen ZH, Shen HH. Caveolin-1 inhibits expression of antioxidant enzymes through direct interaction with nuclear erythroid 2 p45-related factor-2 (Nrf2). J Biol Chem 287: 20922–20930, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu HL, Zhang YL, Yang N, Zhang YX, Liu XQ, Li CG, Zhao Y, Wang YG, Zhang GG, Yang P, Guo F, Sun Y, Jiang CY. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death Dis 2: e159, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López-Alonso I, Aguirre A, González-López A, Fernández ÁF, Amado-Rodríguez L, Astudillo A, Batalla-Solís E, Albaiceta GM. Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-κB pathway. Am J Physiol Lung Cell Mol Physiol 304: L844–L852, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Caro J, Lisanti MP, Sotgia F. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 9: 3515–3533, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, Lisanti MP, Sotgia F. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle 10: 1784–1793, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirza MK, Yuan J, Gao XP, Garrean S, Brovkovych V, Malik AB, Tiruppathi C, Zhao YY. Caveolin-1 deficiency dampens Toll-like receptor 4 signaling through eNOS activation. Am J Pathol 176: 2344–2351, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct 27: 421–429, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 24(8): 105– 137, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakahira K, Choi AM. Autophagy: a potential therapeutic target in lung diseases. Am J Physiol Lung Cell Mol Physiol 305: L93–L107, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12, 222–230, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Wang X, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 203: 2377–2389, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan H, Zhang Y, Luo Z, Li P, Liu L, Wang C, Wang H, Li H, Ma Y. Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung inflammation through NF-κB and p38 MAPK signaling pathways. Am J Physiol Lung Cell Mol Physiol 306: L183–L195, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4: 2300, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabinowitz JD, White E. Autophagy and metabolism. Science 330: 1344–1348, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryter SW, Lam HC, Chen ZH, Choi AM. Deadly triplex: smoke, autophagy and apoptosis. Autophagy 7: 436–437, 2011. [DOI] [PubMed] [Google Scholar]
- 37.Santibanez JF, Blanco FJ, Garrido-Martin EM, Sanz-Rodriguez F, del Pozo MA, Bernabeu C. Caveolin-1 interacts and cooperates with the transforming growth factor-beta type I receptor ALK1 in endothelial caveolae. Cardiovasc Res 77: 791–799, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, Guo F, Liu H, Nan W, Zhao Y, Yan Y, Tang J, Zhao C, Yang P, Liu K, Wang S, Lu H, Li X, Tan L, Gao R, Song J, Gao X, Tian X, Qin Y, Xu KF, Li D, Jin N, Jiang C. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal 5: ra16, 2012. [DOI] [PubMed] [Google Scholar]
- 39.Tekirdag KA, Korkmaz G, Ozturk DG, Agami R, Gozuacik D. MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy 9: 374–385, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol 182: 3809–3818, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N, Choi AM. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 203: 2895–2906, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9: 1102–1109, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Zhang M, Lee SJ, An C, Xu JF, Joshi B, Nabi IR, Choi AM, Jin Y. Caveolin-1 mediates Fas-BID signaling in hyperoxia-induced apoptosis. Free Radic Biol Med 50: 1252–1262, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]