

Abstract
Inflammation, the process aimed at restoring homeostasis after an insult, can be more damaging than the insult itself if uncontrolled, excessive, or prolonged. The inflammasome is an intracellular multimeric protein complex that regulates the maturation and release of proinflammatory cytokines of the IL-1 family in response to pathogens and endogenous danger signals. Growing evidence indicates that the inflammasome plays a key role in the pathogenesis of acute and chronic respiratory diseases. The inflammasome can be activated by the pathogens that account for the most prevalent infectious diseases of the respiratory tract, such as influenza A virus, Streptococcus pneumoniae, Pseudomonas aeruginosa, and Mycobacterium tuberculosis. The inflammasome also plays a role in the chronic inflammation of the airways of patients with asthma and chronic obstructive pulmonary disease, as well as in the initiation and progression of the inflammatory process in pulmonary fibrosis. The aim of this review is to summarize the most relevant points of inflammasome activation in lung diseases.
Keywords: innate immunity, interleukin-1β, respiratory diseases
inflammation is an adaptive response to noxious stimuli (41). The innate immunity comprises a system of germline-encoded receptors that inspect the intracellular and extracellular compartments for signs of infection and recognize highly conserved microbial motifs or “pathogen-associated molecular patterns” (PAMPs). These pattern-recognition receptors (PRRs) are expressed by cells at the front line of host defense against infection, such as macrophages, monocytes, dendritic cells, and epithelial cells. PRRs are not clonally distributed; therefore, all cells expressing PRRs immediately identify PAMP-expressing microbes as a potential threat (34). Membrane-bound Toll-like receptors (TLRs) and C-type lectins are the PRRs that probe the extracellular milieu and the endosomal compartments for PAMPs, while the cytosol is constantly scanned by intracellular nucleic acid sensors, such as interferon-inducible protein (AIM2) and retinoic acid-inducible gene-like helicases. Activation of these receptors causes proinflammatory cytokine production and type I interferon-dependent antiviral responses via the transcription factor NF-κB (53).
Nucleotide oligomerization domain (NOD)-like receptors (NLRs) are a particular type of intracellular PRR that recognize PAMPs and the host-derived signals named DAMPs (danger-associated molecular patterns). NLRs are composed of a conserved central domain, which mediates nucleotide binding and oligomerization [NACHT, NOD, or nucleotide-binding site (NBS) domain], a COOH-terminal leucine-rich domain (LRR), which senses NLR agonists and has an autoinhibitory effect in their absence (14), and an NH2-terminal region, which is required for protein-protein interaction. The human NLR gene family is composed of 22 members, which, depending on their NH2-terminal domains, are classified into 4 subfamilies: NLRA, NLRB, NLRC, and NLRP. NLRA contains an acidic transactivation domain, NLRB a baculovirus inhibitory repeat domain, NLRC a caspase recruitment domain (CARD), and NLRP a pyrin domain (PYD). Activation of certain NLRs (NLRP1, NLRP3, and NLRC4) leads to assembly of the inflammasome, a high-molecular-weight platform for the activation of caspase-1, which is required for proteolytic maturation and release of the proinflammatory cytokines IL-1β and IL-18 (15, 28, 42, 51).
Historical Perspective
The link between mutations in NLR genes and inflammatory diseases was established by Hoffmann and colleagues (24) in 2001, when they described mutations in NLRP3 in individuals affected by Muckle-Wells syndrome, a rare autoinflammatory disease characterized by recurrent episodes of fever and rash associated with ocular and articular manifestations (46). In 2002, Martinon et al. (38) described, for the first time, an inducible high-molecular-weight complex containing NLRP3, an adaptor protein [apoptosis-associated speck-like protein containing a CARD domain (ASC)], and proinflammatory caspases, which they called the inflammasome. Two years later, Agostini et al. (2) demonstrated that constitutive production of active IL-1β observed in Muckle-Wells syndrome was the molecular basis of the NLRP3 inflammasome-dependent disorders. In 2004, Mariathasan et al. (36) demonstrated the requirement of the adaptor ASC within the inflammasome, since macrophages from ATP-challenged ASC−/− mice were unable to produce mature IL-1β and IL-18. Moreover, ASC−/− mice also showed defective caspase-1-dependent cell death, establishing a molecular link between inflammation and cell death pathways.
The NLRP3 Inflammasome
The NLPR3 inflammasome is the best characterized (51) and participates in immune responses to infectious and noninfectious agents. It consists of the aforementioned NLRP3 receptor, the adaptor protein ASC, and caspase-1 (Fig. 1).
Fig. 1.
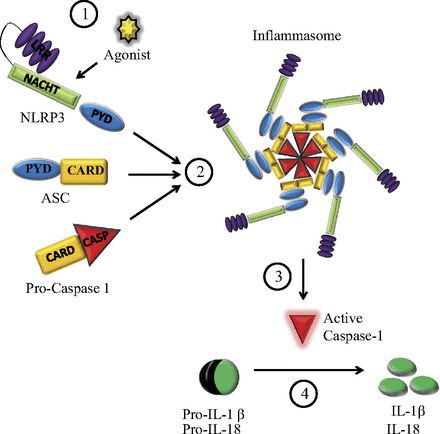
Nucleotide oligomerization domain (NOD)-like receptor containing a pyrin domain (NLRP3) inflammasome. Upon proper stimulation (1), NLRP3 recruits procaspase-1 through the adaptor protein ASC [apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)] to form the inflammasome (2). Within the inflammasome, procaspase-1 undergoes autocatalytic processing, resulting in active caspase-1 (3), which in turn cleaves the precursors pro-IL-1β and pro-IL-18 into the mature and active forms (4). PYD, pyrin domain; LRR, leucine-rich repeat.
ASC, also known as Pycard, is an adaptor protein that bridges NLRP3 and caspase-1. In resting monocytes and macrophages, ASC is sequestered in the nucleus; inflammatory stimuli induce ASC redistribution to the cytosol (9), where it interacts with NLRP3 through its PYD and with caspase-1 through its CARD.
Caspases are cysteine proteases involved in inflammatory or apoptotic pathways (57). Caspase-1, the most fully characterized of the proinflammatory caspases, is synthesized as an inactive zymogen. Upon appropriate stimulation, NLRP3 recruits ASC and procaspase-1 to form the inflammasome, where procaspase-1 is cleaved into a p10 and a p35 fragment. The latter is subsequently processed into the p20 subunit and its CARD, and two molecules of p20 heterodimerize with two molecules of p10 to form the mature and active enzyme (38, 50). Active caspase-1, in turn, cleaves the precursors of two potent proinflammatory cytokines, IL-1β and IL-18, in the cytoplasm; IL-1β and IL-18 are then released to the extracellular milieu by a yet undefined mechanism (35, 47).
NLRP3 inflammasome can be activated by multiple stimuli: whole pathogens (bacteria, viruses, and fungi), PAMPs, DAMPs (extracellular ATP and monosodium urate crystals), and environmental irritants (silica, asbestos, and UVB radiation). Two signals are thought to be required for activation: a “priming signal,” which induces the transcription of pro-IL-1β and pro-IL-18 and the expression of NLRP3 after TLR stimulation (29), and a “second signal,” which activates the inflammasome. However, the molecular mechanism that triggers inflammasome assembly remains unclear (Fig. 2). One of the proposed models of activation is purinergic P2X7 receptor-dependent pore formation by pannexin-1 hemichannel in response to extracellular ATP and K+ efflux, which allows extracellular PAMPs and/or DAMPs to access the cytosol and directly activate NLRP3 (10, 27). Another model is lysosomal rupture. After engulfment of particulate or crystalline agonists, such as silica and asbestos, the phagosome destabilizes and releases its content into the cytosol, which is sensed by NLRP3 and causes inflammasome activation (25). Finally, production of reactive oxygen species (ROS) appears to be a crucial event upstream of inflammasome assembly (66). ROS are sensed by thioredoxin and the thioredoxin-interacting protein (TXNIP) complex, causing dissociation of the complex and subsequent binding of TXNIP to NLRP3. This leads to recruitment of ASC and procaspase-1 by NLRP3 and assembly of the inflammasome (65). A recent study showed that ROS act as priming signals required for transcriptional upregulation of NLRP3, rather than its oligomerization (5).
Fig. 2.
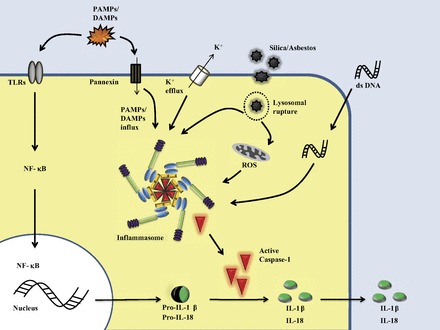
Activation of the inflammasome. Maturation and secretion of IL-1β requires 2 signals: the “priming signal” leads to synthesis of pro-IL-1β, pro-IL-18, and other components of the inflammasome, such as NLRP3, and the second signal results in assembly of the inflammasome, activation of caspase-1, and release of mature cytokines IL-1β and IL-18 into the extracellular milieu. Currently, the nature of the second signal is debated. The 3 proposed models of activation are shown: 1) extracellular ATP, which activates the purinergic P2X7 receptor and causes subsequent recruitment of pannexin-1 hemichannel to the plasma membrane and K+ efflux; 2) lysosomal rupture after engulfment of crystalline or particulate agonists; and 3) reactive oxygen species (ROS), which upregulate NLRP3 expression and activate the inflammasome. PAMP, pathogen-associated molecular pattern; DAMP, danger-associated molecular pattern; TLR, Toll-like receptor; dsDNA, double-stranded DNA.
Other Inflammasomes
Other inflammasomes, such as NLRP1, NLRC4, and AIM2, have also been characterized (Fig. 3, Table 1). The NLRP1 inflammasome, the first to be described, is activated by anthrax lethal toxin (7). Unlike most NLRs, NLRP1 activates caspase-1 directly, and ASC is not required for production of mature IL-1β (20). The NLRC4 or interleukin-converting enzyme protease-activating factor (IPAF) inflammasome is activated by cytosolic flagellin or by the basal body rod component of the type 3 secretion system found in Salmonella typhimurium, Shigella flexneri, Legionella pneumophila, and Pseudomonas aeruginosa (55). Differences from the NLRP3 and NLRP1 inflammasomes include the ability to activate caspase-1 without the adaptor ASC (56) and the constitutive expression of NLRC4 without a TLR-mediated priming signal (29). Finally AIM2, a PRR that senses cytosolic double-stranded DNA (dsDNA) (Fig. 2), is also capable of forming inflammasomes. Since ligand requirements for AIM2 include dsDNA from viruses, bacteria, or the host itself, it may also contribute to autoimmune responses (49).
Fig. 3.
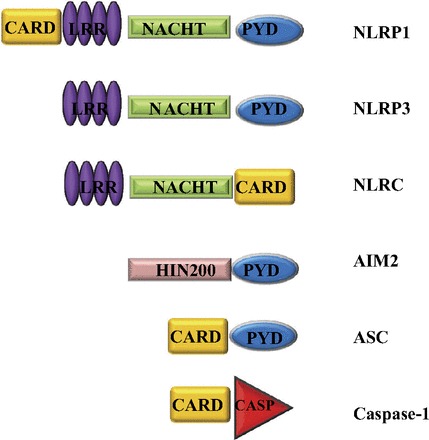
Human inflammasome-related proteins. NLRPs contain an NH2-terminal PYD, a conserved NACHT domain, and a COOH-terminal LRR domain. NLRP1 also contains a COOH-terminal CARD. NLRC4 contains an NH2-terminal CARD, which interacts directly with caspase-1. AIM2, a DNA-interacting human interferon-inducible 200 protein (HIN200) with an NH2-terminal PYD, can also form a caspase-1-activating inflammasome. With the exception of NLRP1, which can bind directly to procaspase-1 and does not require the adaptor protein ASC, inflammasomes are also composed of ASC and caspase-1.
Table 1.
Selected triggers for inflammasome activation
| Inflammasome | Activated by | Source | Reference |
|---|---|---|---|
| NLRP1 | Bacillus anthracis lethal toxin | Bacillus anthracis | Boyden et al. (7) |
| NLRP3 | ATP | Injured cells | Mariathasan et al. (37) |
| MSU | Injured cells | Martinon et al. (38a) | |
| ROS | Cellular stress | Zhou et al. (66) | |
| Whole pathogens | Candida albicans Saccharomyces cerevisiae | Schroder and Tschopp (53) | |
| Virus | Sendai virus | Kanneganti et al. (27) | |
| Adenovirus | |||
| Influenza virus | |||
| Bacterial toxins | Streptococcus pneumoniae | Mariathasan et al. (37) | |
| Listeria monocytogenes | |||
| Staphylococcus aureus | |||
| Environmental irritants | Silica | Hornung et al. (25) | |
| Asbestos | Dostert et al. (18) | ||
| NLRC4 | Flagellin | Salmonella typhimurium | Franchi et al. (20a) |
| Type II/IV secretion systems | Shigella flexneri | Suzuki et al. (56) | |
| Legionella pneumophila | Case et al. (9a) | ||
| Pseudomonas aeruginosa | Sutterwala et al. (55) | ||
| AIM2 | dsDNA | Fransicella tularensis | Rathinam et al. (49) |
| Cytomegalovirus | |||
| Listeria monocytogenes |
NLR, nucleotide oligomerization domain (NOD)-like receptor; NLRP, NLR subfamily containing a pyrin domain; NLRC, NLR subfamily containing a caspase recruitment domain; MSU, monosodium urate; ROS, reactive oxygen species; dsDNA, double-stranded DNA.
Regulation of Inflammasome Activity
Inflammasome activity requires precise regulation to avoid an excessive production of cytokines and its deleterious effects. Regulation takes place at transcriptional and posttranscriptional levels. For instance, NLRP3 is expressed at limited levels in macrophages and is highly inducible in response to proinflammatory stimuli such as LPS, cytokines, or ROS (5). Moreover, differential splicing of ASC can generate protein variants with an inhibitory function, instead of the classical adaptor molecule (8). Another level of regulation is the subcellular location of the inflammasome components; one example is the aforementioned redistribution of ASC from the nucleus to the cytoplasm in activated inflammatory cells (9). Additional regulation of the inflammasome activity can be achieved by secreted factors. In fact, type I interferons can suppress inflammasome activation and its subsequent production of IL-1β, which may contribute to the increased risk of secondary bacterial infections after influenza or other viral infections (23). Autophagy, the lysosomal-mediated process required to maintain cell homeostasis in response to stress, also plays a role in the regulation of inflammasome activity. Saitoh and colleagues (52) demonstrated that cells deficient in autophagy-specific proteins have an enhanced inflammasome activation in response to stimuli, which can be explained by the increase in cellular ROS levels caused by insufficient clearance of defective mitochondria (66). Another possible explanation for this phenomenon is the cytosolic translocation of mitochondrial DNA in dysfunctional mitochondria, which could not only engage the AIM2 inflammasome but also signal downstream an AIM2-independent pathway (45).
Role of the Inflammasome in Lung Diseases
Lung infections.
Community-acquired pneumonia (CAP) is the most common cause of severe sepsis and the leading cause of death from infection in the United States (60). Since the mortality rate from CAP has not changed significantly in the past four decades (44), a thorough comprehension of its pathogenesis is mandatory to find a suitable treatment and effective vaccines.
Many bacterial pathogens that can cause CAP have been shown to activate the NLRP3 inflammasome; the most common mechanism involves the secretion of pore-forming toxins. The virulence of Streptococcus pneumoniae, the leading cause of life-threatening infections such as CAP, meningitis, and sepsis, depends on the polysaccharide capsule and pore-forming toxins, such as cytolysin pneumolysin (PLY) (61). The disruption of the plasma membrane caused by PLY induces K+ efflux, one of the proposed mechanisms of activation of the NLRP3 inflammasome. Listeriolysin O, a pore-forming toxin produced by Listeria monocytogenes, and the α-hemolysin toxin produced by Staphylococcus aureus activate the NLRP3 inflammasome in human and mouse monocytic cells in a similar manner (13, 37).
Infection by certain viruses also results in inflammasome activation. dsDNA viruses can activate the AIM2 inflammasome, while DNA and RNA viruses can trigger assembly of the NLRP3 inflammasome. Influenza A virus (IAV), a major cause of lung infections and mortality, is known to activate the NLRP3 inflammasome (4, 58, 62), but the mechanism is unclear. Recent reports indicate that the IAV ion channel M2, which is involved in fusion during viral entry and in synthesis of new virions, can trigger inflammasome assembly and activation (26, 48). Infection of mice with IAV induces secretion of ATP into the bronchoalveolar lavage fluid (1), and ATP released from influenza-infected dying cells may also trigger the inflammasome-dependent response (1).
Active tuberculosis is primarily a disease of the lung, but it can progress to a generalized inflammatory disease. Mycobacterium tuberculosis (MTB) is a peculiar pathogen, because it resides within a phagosome-like compartment of host macrophages during infection, where it can suppress inflammasome activation (39). However, MTB has the ability to activate the NLRP3 inflammasome, depending on the expression of a functional protein secretion system (ESX-1). (17) The pathways that lead to inflammasome activation in this pathogen are not completely understood, but the export of ESAT-6 (early secreted antigenic target, 6 kDa, a family of small proteins secreted by MTB) via ESX-1 appears to be of critical importance (43).
Airway diseases.
Although the incidence and severity of asthma and chronic obstructive pulmonary disease (COPD) are increasing worldwide (32), there is no treatment to slow the progression of the latter, and a significant group of asthmatic patients remain resistant to available therapies. Hence, understanding the pathophysiology of these diseases to find effective therapies is of prime importance. Chronic inflammation of the airway is the common feature of both diseases, and a growing body of evidence suggests a role for the NLRP3 inflammasome in the pathogenesis of this inflammation.
Extracellular ATP is strongly and persistently upregulated in the airways of patients with COPD (31), and this is associated with a decline in lung function (12) and an increase in airway infiltration by inflammatory cells. As we previously mentioned, extracellular ATP activates the NLRP3 inflammasome by engaging the purinergic P2X7 receptor. This receptor is also upregulated in alveolar macrophages and blood neutrophils from patients with COPD (31), as well as in blood and airway neutrophils and alveolar macrophages in the mouse model of lung inflammation induced by cigarette smoke (CS) (33).
The role of the inflammasome components NLRP3 and ASC in CS models has not been fully investigated. However, there is evidence of inflammasome activation, since levels of caspase-1 are increased in lung tissue after CS challenge in mice. Caspase-1 levels are also higher in lung tissue from COPD patients and smokers than from nonsmoking donors (19). Selective inhibition of caspase-1 significantly decreased inflammation after CS challenge in animal models (11). These data support a role for the inflammasome in the airway inflammation in COPD and asthma, although further studies are required.
The role of the NLRP3 inflammasome in the development of allergic airway disease is controversial. As with COPD, ATP levels are also elevated in the airways of asthmatic patients and in animal models in response to allergen challenge (6). Moreover, IL-1β levels are higher in serum of asthmatic than nonasthmatic patients (59), and induced sputum contains higher levels of IL-1β in symptomatic than in nonsymptomatic asthmatic patients, suggesting a putative role of the NLRP3 inflammasome in chronic disease and acute exacerbations. However, recently, Allen et al. (3) failed to see a difference in clinical outcome of four allergic asthma models in NLRP3−/− mice compared with controls.
Pulmonary fibrosis.
The term “pulmonary fibrosis” includes a broad range of lung disorders characterized by progressive and irreversible destruction of normal lung architecture by excessive accumulation of collagen and other extracellular matrix (ECM) components in basement membranes and interstitial tissues. ECM expansion impairs effective gas exchange and leads to death due to extreme respiratory failure (30). However, the pathophysiological mechanisms underlying pulmonary fibrosis are not completely understood.
The importance of inflammation in initiation and progression of pulmonary fibrosis has been established (63). Silica, asbestos, and bleomycin, among other irritants, can injure lung epithelial cells and activate the NLRP3 inflammasome in macrophages, leading to IL-1β secretion (18, 21, 25). IL-1β secretion is also increased in macrophages treated with pravastatin, as demonstrated by Xu and colleagues (64), suggesting a role for statins in pulmonary fibrosis. IL-1β promotes production of TGF-β, the most potent and ubiquitous profibrotic cytokine (30), which triggers activation, proliferation, and transdifferentiation of epithelial cells and resident fibroblasts into collagen-producing myofibroblasts. IL-1β also promotes the secretion of neutrophil-attracting CXC chemokines, which favor the influx of neutrophils and exacerbate the damage to epithelial cells, and PDGF, which further induces fibrosis. Finally, TGF-β and IL-1β can increase the expression of plasminogen activator inhibitor 1, which inhibits ECM degradation, promotes recruitment of more inflammatory cells (67), and suppresses the release of antifibrogenic growth factors (30). The initial proinflammatory scenario of inflammasome activation can rapidly progress to a profibrotic one, leading to the chronic and devastating disease.
Acute respiratory distress syndrome.
Acute lung injury and its most severe form, acute respiratory distress syndrome (ARDS), are frequent complications of patients admitted to the intensive care unit and are associated with a mortality of ∼25% (54). ARDS can be triggered by different disorders, such as pneumonia, sepsis, ischemia, and trauma; however, there is neither an effective tool to predict patients' susceptibility to ARDS nor an effective therapy. Pathophysiologically, the disease is characterized by dysregulated inflammation, with excessive permeability of epithelial and endothelial barriers leading to lung edema formation and severe hypoxemia (40). Inflammasome-regulated cytokines appear to play a major role in the development of ARDS, as demonstrated by Dolinay and colleagues (16), since increases in the levels of circulating IL-18 in critically ill patients correlate with disease severity and mortality, even after adjustment for important confounders, such as Acute Physiology and Chronic Health Evaluation (APACHE) II score.
Conclusion
The lung is exposed to a variety of insults that can be detected by different PRRs, such as microbial molecules (LPS and flagellin), inhaled particles (asbestos and silica), and cell injury-associated endogenous molecules (ATP and potassium). The activation of different PRRs and the resultant signaling pathways play a critical role in host protection and in the pathology of lung diseases. New reports (21) implicate the inflammasome in the sensing of danger/stress signal, leading to increased levels of active IL-1β. Furthermore, the role of IL-1β as a critical inflammatory mediator of acute inflammation and tissue remodeling has been well established (22). Over the last 10 years, the discovery and characterization of the inflammasome have led to a comprehensive insight into the innate and adaptive immune responses to different lung insults. However, our knowledge of activation and regulation of the inflammasome is incomplete. Further understanding of the inflammasome complex would be advantageous in the development of new treatment modalities in acute lung injury and chronic lung disease.
GRANTS
This work was supported by a Merit Review grant from the Department of Veterans Affairs (K. M. Ridge).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.d.S. drafted the manuscript; M.A.K. edited and revised the manuscript; K.M.R. approved the final version of the manuscript.
REFERENCES
- 1. Aeffner F, Traylor ZP, Yu EN, Davis IC. Double-stranded RNA induces similar pulmonary dysfunction to respiratory syncytial virus in BALB/c mice. Am J Physiol Lung Cell Mol Physiol 301: L99–L109, 2011. [DOI] [PubMed] [Google Scholar]
- 2. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20: 319–325, 2004. [DOI] [PubMed] [Google Scholar]
- 3. Allen IC, Jania CM, Wilson JE, Tekeppe EM, Hua X, Brickey WJ, Kwan M, Koller BH, Tilley SL, Ting JP. Analysis of NLRP3 in the development of allergic airway disease in mice. J Immunol 188: 2884–2893, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30: 556–565, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187: 613–617, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Birrell MA, Eltom S. The role of the NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacol Ther 130: 364–370, 2011. [DOI] [PubMed] [Google Scholar]
- 7. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38: 240–244, 2006. [DOI] [PubMed] [Google Scholar]
- 8. Bryan NB, Dorfleutner A, Kramer SJ, Yun C, Rojanasakul Y, Stehlik C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflamm 7: 23, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182: 3173–3182, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a. Case CL, Shin S, Roy CR. Asc and Ipaf inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun 77: 1981–1991, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chaudhuri N, Paiva C, Donaldson K, Duffin R, Parker LC, Sabroe I. Diesel exhaust particles override natural injury-limiting pathways in the lung. Am J Physiol Lung Cell Mol Physiol 299: L263–L271, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Churg A, Zhou S, Wang X, Wang R, Wright JL. The role of interleukin-1β in murine cigarette smoke-induced emphysema and small airway remodeling. Am J Respir Cell Mol Biol 40: 482–490, 2009. [DOI] [PubMed] [Google Scholar]
- 12. Cicko S, Lucattelli M, Muller T, Lommatzsch M, De Cunto G, Cardini S, Sundas W, Grimm M, Zeiser R, Durk T, Zissel G, Boeynaems JM, Sorichter S, Ferrari D, Di Virgilio F, Virchow JC, Lungarella G, Idzko M. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol 185: 688–697, 2010. [DOI] [PubMed] [Google Scholar]
- 13. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA. Staphylococcus aureus α-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLos One 4: e7446, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dagenais M, Skeldon A, Saleh M. The inflammasome: in memory of Dr. Jurg Tschopp. Cell Death Differ 19: 5–12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: 519–550, 2009. [DOI] [PubMed] [Google Scholar]
- 16. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, Massaro AF, Rogers A, Gazourian L, Nakahira K, Haspel JA, Landazury R, Eppanapally S, Christie JD, Meyer NJ, Ware LB, Christiani DC, Ryter SW, Baron RM, Choi AM. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med 185: 1225–1234, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, Oberbeck-Muller D, Duque-Correa MA, Reece ST, Ruland J, Brosch R, Tschopp J, Gross O, Kaufmann SH. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol 42: 374–384, 2012. [DOI] [PubMed] [Google Scholar]
- 18. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674–677, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eltom S, Stevenson CS, Rastrick J, Dale N, Raemdonck K, Wong S, Catley MC, Belvisi MG, Birrell MA. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLos One 6: e24097, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25: 713–724, 2007. [DOI] [PubMed] [Google Scholar]
- 20a. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Nunez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in Salmonella-infected macrophages. Nat Immunol 7: 576–582, 2006. [DOI] [PubMed] [Google Scholar]
- 21. Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, Ryffel B, Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest 117: 3786–3799, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, Couillin I. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 179: 903–913, 2009. [DOI] [PubMed] [Google Scholar]
- 23. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34: 213–223, 2011. [DOI] [PubMed] [Google Scholar]
- 24. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29: 301–305, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9: 847–856, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11: 404–410, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, Nunez G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26: 433–443, 2007. [DOI] [PubMed] [Google Scholar]
- 28. Kepp O, Galluzzi L, Kroemer G. Mitochondrial control of the NLRP3 inflammasome. Nat Immunol 12: 199–200, 2011. [DOI] [PubMed] [Google Scholar]
- 29. Kupz A, Guarda G, Gebhardt T, Sander LE, Short KR, Diavatopoulos DA, Wijburg OL, Cao H, Waithman JC, Chen W, Fernandez-Ruiz D, Whitney PG, Heath WR, Curtiss R, 3rd, Tschopp J, Strugnell RA, Bedoui S. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8 T cells. Nat Immunol 13: 162–169, 2012. [DOI] [PubMed] [Google Scholar]
- 30. Liu RM. Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxid Redox Signal 10: 303–319, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lommatzsch M, Cicko S, Muller T, Lucattelli M, Bratke K, Stoll P, Grimm M, Durk T, Zissel G, Ferrari D, Di Virgilio F, Sorichter S, Lungarella G, Virchow JC, Idzko M. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 181: 928–934, 2010. [DOI] [PubMed] [Google Scholar]
- 32. Lopez AD, Murray CC. The global burden of disease, 1990–2020. Nat Med 4: 1241–1243, 1998. [DOI] [PubMed] [Google Scholar]
- 33. Lucattelli M, Cicko S, Muller T, Lommatzsch M, De Cunto G, Cardini S, Sundas W, Grimm M, Zeiser R, Durk T, Zissel G, Sorichter S, Ferrari D, Di Virgilio F, Virchow JC, Lungarella G, Idzko M. P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am J Respir Cell Mol Biol 44: 423–429, 2011. [DOI] [PubMed] [Google Scholar]
- 34. Manfredi AA, Rovere-Querini P. The mitochondrion—a Trojan horse that kicks off inflammation? N Engl J Med 362: 2132–2134, 2010. [DOI] [PubMed] [Google Scholar]
- 35. Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7: 31–40, 2007. [DOI] [PubMed] [Google Scholar]
- 36. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430: 213–218, 2004. [DOI] [PubMed] [Google Scholar]
- 37. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232, 2006. [DOI] [PubMed] [Google Scholar]
- 38. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10: 417–426, 2002. [DOI] [PubMed] [Google Scholar]
- 38a. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241, 2006. [DOI] [PubMed] [Google Scholar]
- 39. Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, Timmins GS, Sander P, Deretic V. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3: 224–232, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 122: 2731–2740, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Medzhitov R. Origin and physiological roles of inflammation. Nature 454: 428–435, 2008. [DOI] [PubMed] [Google Scholar]
- 42. Medzhitov R, Janeway C., Jr Innate immunity. N Engl J Med 343: 338–344, 2000. [DOI] [PubMed] [Google Scholar]
- 43. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, Anes E. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol 12: 1046–1063, 2010. [DOI] [PubMed] [Google Scholar]
- 44. Mizgerd JP, Skerrett SJ. Animal models of human pneumonia. Am J Physiol Lung Cell Mol Physiol 294: L387–L398, 2008. [DOI] [PubMed] [Google Scholar]
- 45. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neven B, Prieur AM, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol 4: 481–489, 2008. [DOI] [PubMed] [Google Scholar]
- 47. Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell 126: 659–662, 2006. [DOI] [PubMed] [Google Scholar]
- 48. Pang IK, Iwasaki A. Inflammasomes as mediators of immunity against influenza virus. Trends Immunol 32: 34–41, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11: 395–402, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol 13: 333–332, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rubartelli A, Gattorno M, Netea MG, Dinarello CA. Interplay between redox status and inflammasome activation. Trends Immunol 32: 559–566, 2011. [DOI] [PubMed] [Google Scholar]
- 52. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456: 264–268, 2008. [DOI] [PubMed] [Google Scholar]
- 53. Schroder K, Tschopp J. The inflammasomes. Cell 140: 821–832, 2010. [DOI] [PubMed] [Google Scholar]
- 54. Spragg RG, Bernard GR, Checkley W, Curtis JR, Gajic O, Guyatt G, Hall J, Israel E, Jain M, Needham DM, Randolph AG, Rubenfeld GD, Schoenfeld D, Thompson BT, Ware LB, Young D, Harabin AL. Beyond mortality: future clinical research in acute lung injury. Am J Respir Crit Care Med 181: 1121–1127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204: 3235–3245, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3: e111, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang PS, Mura M, Seth R, Liu M. Acute lung injury and cell death: how many ways can cells die? Am J Physiol Lung Cell Mol Physiol 294: L632–L641, 2008. [DOI] [PubMed] [Google Scholar]
- 58. Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30: 566–575, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thomas SS, Chhabra SK. A study on the serum levels of interleukin-1β in bronchial asthma. J Indian Med Assoc 101: 282, 284,, 286, 2003. [PubMed] [Google Scholar]
- 60. Waterer GW, Rello J, Wunderink RG. Management of community-acquired pneumonia in adults. Am J Respir Crit Care Med 183: 157–164, 2011. [DOI] [PubMed] [Google Scholar]
- 61. Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, Holmes A, Trendelenburg G, Heimesaat MM, Bereswill S, van der Linden M, Tschopp J, Mitchell TJ, Suttorp N, Opitz B. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol 187: 434–440, 2011. [DOI] [PubMed] [Google Scholar]
- 62. Wu W, Patel KB, Booth JL, Zhang W, Metcalf JP. Cigarette smoke extract suppresses the RIG-I-initiated innate immune response to influenza virus in the human lung. Am J Physiol Lung Cell Mol Physiol 300: L821–L830, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med 208: 1339–1350, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu JF, Washko GR, Nakahira K, Hatabu H, Patel AS, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, Ross JC, Estepar RS, Diaz AA, Li HP, Qu JM, Himes BE, Come CE, D'Aco K, Martinez FJ, Han MK, Lynch DA, Crapo JD, Morse D, Ryter SW, Silverman EK, Rosas IO, Choi AM, Hunninghake GM. Statins and pulmonary fibrosis: the potential role of NLRP3 inflammasome activation. Am J Respir Crit Care Med 185: 547–556, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11: 136–140, 2011. [DOI] [PubMed] [Google Scholar]
- 66. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011. [DOI] [PubMed] [Google Scholar]
- 67. Zmijewski JW, Bae HB, Deshane JS, Peterson CB, Chaplin DD, Abraham E. Inhibition of neutrophil apoptosis by PAI-1. Am J Physiol Lung Cell Mol Physiol 301: L247–L254, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]