

Abstract
Listeriosis is considered as an important public health issue. Sortase A (srtA) is an enzyme with catalytic role in L. monocytogenes that breaks the junction between threonine and glycine in the LPXTG motif (a key motif in internalin A (InlA) that plays an important role in listeriosis). Inactivation of srtA was shown to inhibit anchoring of the invasion protein InIA. This is in addition to inhibiting peptidoglycan-associated LPXTG proteins. Therefore, it is of interest to inhibit strA using potential molecules. Here, we describe the design of an inhibitor with high binding affinity to srtA with the ability to prevent the attachment of srtA to the LPXTG proteins such as InIJ. A homology model of Listeria monocytogenes Sortase A was developed using MODELLER (version 9.12). We screened StrA to 100,000 drug-like ligands from the Zinc database using Molecular docking and virtual screening tool PyRX). Pharmacokinetic analysis using the FAFDrugs3 web server along with ADME and toxicity analysis based on Lipinski rule of five were adopted for the screening exercise followed by oral toxicity check using PROTOX (a server) for every 10 successive hits. The results from PROTOX server indicated that Lig #1 (with LD50 of 2000mg/kg) and Lig #7 (with LD50 of 2000mg/kg) have toxicity class 4 and Lig #3 (with LD50 of 14430mg/kg) has toxicity class 6. Subsequent modifications of these structures followed by FAFDrugs3 analysis for high bioavailability value selected Lig #7 according to Lipinski rules of five. Thus, Lig #7 with IUPAC name ((R)-4-{(S)-1-[(S)-2-Amino-4-methylvaleryl]-2-pyrrolidinyl}-1-[(S)-1-(ethylamino) carbonyl-propylamino] -2-propyl-1, 4- butanedione) is suggested as a potential candidate for srtA inhibition for further consideration.
Keywords: Listeria monocytogenes, sortase, drug design, ligands, docking, virtual screening and toxicity
Background
Listeriosis is a food-borne bacterial disease caused by Listeria monocytogenes [1]. L. monocytogenes is responsible for severe infections in those who suffered from impaired immune systems as well as in pregnant women leading to fetus abortion [2, 3]. Some studies have suggested an increasing rate of drug resistance with substantial growth not only in environmental isolates [4] but also in clinical strains in comparison to the precedence [5]. Current treatment is based on amino-penicillin (ampicillin or amoxicillin) and gentamicin [6] what affect translation pathways in the host cells. Except amino-peniciliins, L. Monocytogenes is also sensitive to Cephalosporins, Fosfomycins and Fluoroquinolons. Cephalosporins can mimic the D-Ala-D-Ala sequence at the end of peptidoglycan precursors. Penicillin binding proteins (PBPs) that facilitated the synthesis of peptidoglycan of cell wall is inhibited by mistaken antibiotic with peptidoglycan. Third and fourth generations of Fluoroquinolons are more suitable for the grampositive bacteria like Listeria because they can affect topoisomerase IV, Fosfomycin and inhibit UDP-N-acetylglucosamine– 3–enol-pyrovyl-transferase (MurA). It catalyses key reactions in the peptido-glycan biosynthesis [7–9].
Sortase A (srtA) is an enzyme with catalytic role in L. monocytogenes that breaks the junction between threonine and glycin in the LPXTG motif having a vital role in listeriosis. LPXTG motif is near the carboxyl terminal followed by a hydrophobic region and a positively charged tail. The cell wall anchoring of LPXTG proteins such as InIJ using a transpeptidation reaction catalyzed by srtA. SrtA connects the peptide bond of threonine to the amino group of the peptidoglycan for cell wall attachment using the internalin protein [10–12].
A study conducted by Bierne et al. (2002) [13], revealed that inactivation of srtA not only can inhibit anchoring of the invasion protein InIA but also can inhibit other peptidoglycanassociated LPXTG proteins [13]. Garandeau et al. (2002) indicated that srtA mutant is affected by the expression of internalin at the bacterial surface. Moreover, this is less invasive and finally, it is attenuated for virulence in the mouse model [10]. Another study suggested that mutation in srtA can both result in partial loss of the proteins bound to the cell wall peptido-glycan and support cell wall association in some member of LPXTG surface proteins family [14]. Interestingly, inactivation of srtA showed less colonization than that of InlA mutant in a mouse model [10, 13]. Sortase substrates have deniable functions in adherent, internalins, blood clotting, and immune evasion factors and also in transporting nutrition substances alongside to cell wall that without this enzyme the virulence of the bacterium is more likely to be disappeared [15, 16& 17]. The crucial role of srtA in bacterial growth and its viability and because it is located on the cell wall membrane, it is a promising target for anti-listeriosis drugs. The bacterium is pathogenic when InIJ binds to the cell wall surface. Inhibiting the srtA by an inhibitor results in the inability of protein transferring. Thus, L. monocytogenes will be inactivated due to loss of connection in the network [18, 19]. Therefore, it is of interest to design molecules for the inhibition strA in controlling listeriosis.
Methodology
Protein target and ligand structures:
The crystal structure of Listeria monocytogenes sortase A is not available at the protein data bank (PDB). Hence, a homology model of sortase A was constructed using MODELLER (version 9.12). Small molecule structures for 100,000 drug-like ligands from the Zinc database (http://zinc.docking.org/) [20] were used in the study. The ligands were selected from drug like category with pH 7.0.
Molecular docking, virtual screening and drug design:
Molecular docking simulation using PyRX [21] consisting of AutoDock [22] and AutoDockvina [23] with a Lamrkian genetic algorithm as scoring function was completed. AutoDock is a molecular docking tool. Hyperchem™ was used to perform modifications in the successive hit structures.
Pharmacokinetic analysis:
Successive hits retrieved from virtual screening were then analyzed for absorption, distribution, metabolism and excretion using the FAFDrugs3 web server [24] for ADME analysis with Lipinski rule of five [25]. Moreover, oral toxicity was checked using the PROTOX server for every 10 successive hits [26].
Result & Discussion
A homology model of sortase A was constructed using Bacillus anthracis sortase (pdb|2OQW|A) as template for virtual screening. Moreover, the predicted model was then energy minimized using MODELLER (version 9.12). It is essential to design a high binding affinity ligand to prevent interaction between srtA and LPXTG proteins. Binding affinity of the top seven hits has binding values from -4.1 to -7. The hit #1 score is -6.9 and scores of -4.9, -6.1, -5.3, -5.7, -4.1 and -7.0 was achieved for hit #2-#7, respectively.
We analyzed the selected top 3 hits based on their binding affinity using FAFDrugs3 for Lipinski rule of 5. Results from this analysis are given in Table 1 (see supplementary material). Lig #3 has acceptable bioavailability and lower rotatable bonds (<= 10 rotatable bonds are suitable for ligand binding to proteins) among the selected ligands. However, it showed a low binding value of -6.1 with an error tag “low risk hemiketal” in FAFDrug3 analysis. Deletion of two OH groups showed acceptable FAFDrug3 with -5.9 binding score. Hence, other hits were considered for further analysis.
For Lig #1, a binding affinity of -6.9 and errors of “low risk and covalent: carbamatethicarbamate / high-risk crown2_2 / lowrisk n acylated azoles/low risk thiol/and rotatable bonds” were shown. S atoms were deleted to remove Thiol error. The new ligand reached a binding affinity of -7.82. Replacing N atom with C atom eliminated the crown 2_2 error in the structure. Finally a binding affinity of -7.8 was reached. We got Thio-carbamate error by repeating step 2 for another N atom and a binding affinity of -7.4 was reached for the new ligand. The new ligand was rejected in FAFDrugs because of high rotatable bonds. We created a double bond in iso-butane substituent and converted it to isobutene to correct this error. Thus, the designed ligand was found accepted by FAFDrugs3 (Figure 1).
Figure 1.
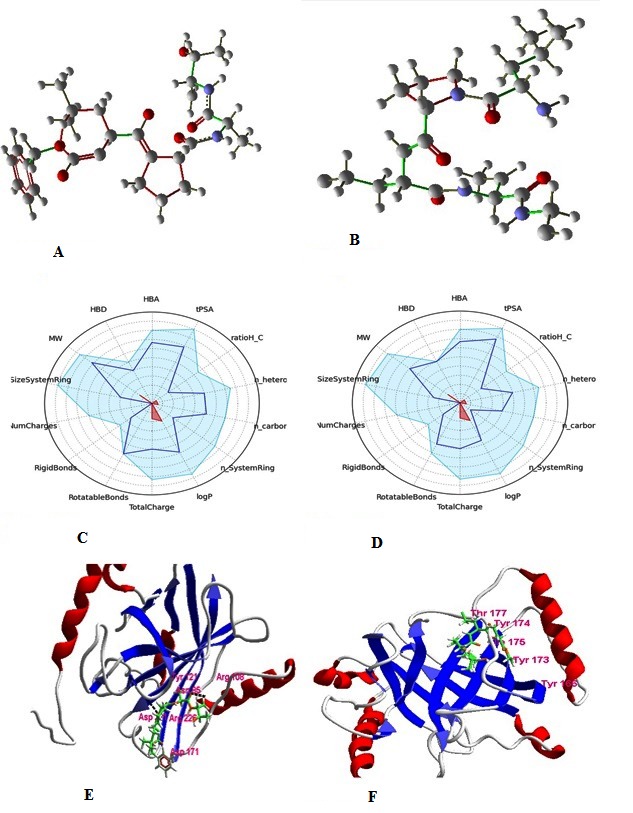
The rationally designed ligands based on ligand 1 (successive hit 1) and ligand 7 (successive hit 7) could theoretically reach acceptable pharmacological properties: (A): The structure of the rationally designed ligand based on the structure of hit 1; (B): The structure of rationally designed ligand based on the structure of hit 7; (C): The pharmacological filters indicates that rationally designed ligand 1 has acceptable drug like properties; (D): The pharmacological filters indicates that the rationally designed ligand 7 has acceptable drug like properties; (E): ligand 1 interacts with Asp 171, Arg 226, Asn 85, Arg 108 and Tyr 121: (F): ligand 7 interacts with Tyr 166, Tyr 173, Tyr 174 and Thr 175.
Ligand #7 with the binding affinity equal to -7.8 is observed. The errors of “High risk and covalent aldehyde/Crown 2_2/Hemiketal/n acylated azoles/number of rotatable bonds/number of HBD/PSA” were noted in this case. For the first step, one of the “O” atoms was deleted. As a result of this action, aldehyde error was eliminated and the binding affinity of -6.4 was achieved for the designed ligand. In addition, by deleting two OH atoms hemiketal and HBD and PSA errors with the binding affinity of -7.2 were eliminated. Also, by replacing N atom with C, we eliminated crown 2_2 error and the binding affinity of -7.1 was reached. As the final step, an aldehyde functional group was removed: two hydroxyl groups and one methyl group from the base structure of lig# 7. Eventually, errors related to HBA, HBD and tPSA were eliminated. Then five single bonds changed to double bond in order to decrease rotatable bonds' errors. One of this five double bonds converted amine to nitrile, which decreased HBD (Figure 1). The changed ligands passed FAFDrug3 successfully as drug-like filter and we selected this ligand as the candidate for inhibition of Sortase protein from Listeria (Figure 1). Further, the increasing of bioavailability was considered in Lig #7 and was found accepted based on Lipinski rule of five. Therefore, lig #7 was selected for further analysis of pharmacological properties. The toxicity of designed ligands was checked using PROTOX server. Lig #1 with LD50 of 2000mg/kg and Lig #7 with LD50 of 2000mg/kg have toxicity class of 4 and Lig3 with LD50 of 14430mg/kg has toxicity class 6 (1 is for the most and 6 is for the least toxicity level) is reported.
Conclusion
New molecular strategies based on drug design can offer promising approach to treat listeriosis. Ligand #7 derived from virtual screening analysis that was previously known drug-like as a potent inhibitor for the target protein sortase is reported in this article. We performed several modifications on its structure to make it a ppropriate for better binding. The new ligand with the IUPAC name of “((R)-4-{(S)-1-[(S)-2- Amino-4- methylvaleryl] -2-pyrrolidinyl} -1-[(S)-1 -(ethylamino) carbonylpropyl- amino]-2-propyl-1, 4-butanedione)” is shown as a potent inhibitor of sortase protein in this report. It should be noted that this requires in-vitro and in-vivo confirmation for further consideration.
Supplementary material
Footnotes
Citation:Rashidieh et al. Bioinformation 11(11): 501-505 (2015)
References
- 1.Ailes E. Clin Infect Dis. 2012;54:5. [Google Scholar]
- 2.Vázquez-Boland JA. Clin Microbiol Rev. 2001;14:3. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swaminathan B, et al. Microbes Infect. 2007;9:10. doi: 10.1016/j.micinf.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 4.Walsh D. J Appl Microbiol. 2001;90:4. doi: 10.1046/j.1365-2672.2001.01273.x. [DOI] [PubMed] [Google Scholar]
- 5.Safdar A, Armstrong D. Clin Infect Dis. 2003;37:359. doi: 10.1086/376631. [DOI] [PubMed] [Google Scholar]
- 6.Temple ME, Nahata MC. Ann Pharmacother. 2000;34:656. doi: 10.1345/aph.19315. [DOI] [PubMed] [Google Scholar]
- 7.Cotter PD. Antimicrob Agents Chemother. 2002;46:2784. doi: 10.1128/AAC.46.9.2784-2790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mather R, et al. Am J Ophthalmol. 2002;133:463. doi: 10.1016/s0002-9394(02)01334-x. [DOI] [PubMed] [Google Scholar]
- 9.Walker RC. Mayo Clin Proc. 1999;74:1030. doi: 10.4065/74.10.1030. [DOI] [PubMed] [Google Scholar]
- 10.Garandeau C, et al. Infect Immun. 2002;70:1382. doi: 10.1128/IAI.70.3.1382-1390.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cossart P, Jonquières R. Proc Natl Acad Sci. 2000;97:5013. doi: 10.1073/pnas.97.10.5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaspar AH. J Bacteriol. 2005;187:4646. doi: 10.1128/JB.187.13.4646-4655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bierne H. Mol Microbiol. 2002;43:869. doi: 10.1046/j.1365-2958.2002.02798.x. [DOI] [PubMed] [Google Scholar]
- 14.Mariscotti JF. Int Microbiol. 2012;15:43. doi: 10.2436/20.1501.01.157. [DOI] [PubMed] [Google Scholar]
- 15.Dussurget O. Annu Rev Microbiol. 2004;58:587. doi: 10.1146/annurev.micro.57.030502.090934. [DOI] [PubMed] [Google Scholar]
- 16.Sabet C. Infect Immun. 2005;73:6912. doi: 10.1128/IAI.73.10.6912-6922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebrun M. Mol Microbiol. 1996;21:579. doi: 10.1111/j.1365-2958.1996.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 18.Selvaraj C. J Recept Signal Transduct Res. 2014;34:221. doi: 10.3109/10799893.2013.876044. [DOI] [PubMed] [Google Scholar]
- 19.Braun L, et al. Mol Microbiol. 1998;27:1077. doi: 10.1046/j.1365-2958.1998.00750.x. [DOI] [PubMed] [Google Scholar]
- 20.Irwin JJ, Shoichet BK. J ChemInf Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dallakyan S, Olson AJ. Methods Mol Biol. 2015;1263:243. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- 22.Morris GM, et al. Curr Protoc Bioinformatics. 2008;8:8. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 23.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagorce D, et al. BMC Bioinformatics. 2008;9:396. doi: 10.1186/1471-2105-9-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipinski CA. Drug Discov Today Technol. 2004;1:337. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Drwal MN, et al. Nucleic Acids Res. 2014;42:W53. doi: 10.1093/nar/gku401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.