

Abstract
Globoid cell leukodystrophy (GLD, Krabbe disease) is due to autosomal recessive mutations in the lysosomal enzyme galactosylceramidase (GALC). Many GLD patients develop infantile-onset of progressive neurologic deterioration and death by 2 years of age, whereas others have a later-onset, milder disease. Cord blood transplant slows disease progression much more effectively when performed presymptomatically, highlighting the importance of early diagnosis. Current diagnosis is based on reduced GALC activity, DNA sequence, and clinical examination. However, presymptomatic diagnosis is hampered by imperfect genotype-GALC activity-phenotype correlations. In addition, three polymorphisms in the GALC gene are variably associated with disease mutations and have unknown effects on GALC activity and disease outcome. Here, we study mutations that cause infantile or later-onset GLD, and show that GALC activity is significantly lower in infantile versus later-onset mutants when measured in the lysosomal fraction, but not in whole-cell lysates. In parallel, infantile-onset mutant GALCs showed reduced trafficking to lysosomes and processing than later-onset mutant GALCs. Finally, the cis-polymorphisms also affected trafficking to the lysosome and processing of GALC. These differences potentially explain why the activity of different mutations appears similar in whole-cell extracts from lymphocytes, and suggest that measure of GALC activity in lysosomes may better predict the onset and severity of disease for a given GLD genotype.
SIGNIFICANCE STATEMENT Globoid cell leukodystrophy (GLD, Krabbe disease) is diagnosed by measuring galactosylceramidase (GALC) activity and DNA analysis. However, genotype and phenotype often do not correlate due to considerable clinical variability, even for the same mutation, for unknown reasons. We find that altered trafficking to the lysosome and processing of GALC correlates with GLD severity and is modulated by cis-polymorphisms. Current diagnosis of GLD is based on GALC activity of total cell lysates from blood, which does not discriminate whether the activity comes from the lysosome or other subcellular organelles. Measurement of GALC activity in lysosomes may predict which infants are at high risk for the infantile phenotype while distinguishing other children who will develop later-onset phenotypes without onset of symptoms for years.
Keywords: galactosylceramidase, globoid cell leukodystrophy, Krabbe disease, lysosomal processing, lysosomal storage disorders, protein trafficking
Introduction
Globoid cell leukodystrophy (GLD, Krabbe disease) is a demyelinating, neurodegenerative disease that affects both the central and peripheral nervous systems, and is caused by mutations in the galactosylceramidase (GALC) gene. GALC (EC 3.2.1.46) is a lysosomal enzyme involved in the catabolism of galactosylceramide, a major lipid in myelin, kidney, and epithelial cells of the small intestine and colon (Chen YQ et al., 1993). More than 85% of patients with GLD have the infantile form of the disease and present with symptoms between the ages of 3 and 12 months, which include extreme irritability, spasticity, developmental delay, and ultimately death within a few years. The remaining 10%–15% of patients have a later-onset form of the disease, in which symptoms present between 13 months of age and the seventh decade of life. Later-onset patients have a milder disease, which may include weakness, loss of vision, and intellectual regression (Wenger, 2011a).
Current therapy aims to reduce demyelination by hematopoietic cell transplantation (HCT), which generates cells that are thought to transfer GALC to myelinating cells. However, judging the efficacy of cord blood transplantation is complicated by variable genotype–phenotype relationships in these patients. Furthermore, for effective treatment in children with the infantile-onset phenotype, transplantation must be performed before the onset of symptoms (Duffner et al., 2012), but at this age HCT is associated with a 20% mortality rate. Therefore, accurate diagnosis and prognosis are extremely important for the care of GLD patients.
Unfortunately, neither measurements of GALC enzymatic activity nor documentation of GALC mutations reliably predicts the severity of phenotype in GLD patients. For example, in newborn screening, an infant with a low GALC activity and two documented mutant alleles may be diagnosed with the infantile form of GLD, when indeed, the child may have the later-onset form of the disease. This child would accordingly undergo HCT and increased risk of mortality, despite the fact that disease would have appeared significantly later.
The goal of this study is to understand why mutations with a range of predicted severity do not always correlate with differences in GALC activity, and why the same mutation in GALC can result in a broad spectrum of clinical variability. Three factors have been suggested as potential contributors to clinical variability: cis-polymorphisms (Raghavan et al., 2005), environmental influences including diet (Pannuzzo et al., 2010), and modifier genes such as saposinA/Psap (Yagi et al., 2004) or others (Lattanzi et al., 2010). The GALC precursor protein is generated in the endoplasmic reticulum (ER) and is transported to the lysosome, where it is processed into 50 and 30 kDa subunits (Nagano et al., 1998). Herein, we find that the trafficking and processing of GALC are impaired more by infantile-onset than later-onset mutations. In addition, we show that measuring GALC activity in lysosome fractions better distinguishes between infantile- and later-onset mutations. Furthermore, we find evidence that polymorphisms also contribute to the trafficking and processing of GALC, thereby affecting the functional GALC activity. Thus, the combination of cis-polymorphisms with known disease mutations may alter the clinical phenotype of GLD. Overall, we propose that the development of more accurate strategies for the diagnosis of GLD should include the measure of GALC activity in lysosomal fractions.
Materials and Methods
Plasmid construction and site-directed mutagenesis.
A full-length cDNA clone of the human GALC gene was obtained from Open Biosystems (#MHS1010-7295902) that harbors the c.1685C polymorphism. The coding sequence of GALC was then amplified by PCR from the full-length cDNA clone and ligated between the BamHI and XhoI sites of the pCMVTag4A vector (Stratagene) to generate GALC-FLAG or GALC only (with the GALC stop codon immediately upstream of the FLAG fusion). The primers used were hGalc-cDNA-BamHI-s (5′-GGGGG GGATCC GCCACC atg actgcggccg cgggttcggc-3′) and hGalc-cDNA-XhoI-as (5′-GGGGG CTCGAG gcgtgtggcttccacaagaaag-3′) for the FLAG fusion, and hGalc-cDNA-STOP-XhoI-as (5′-GGGGG CTCGAG TTA gcgtgtggcttccacaagaaag-3′) for GALC-only expression. The coding sequence was also ligated between the XhoI and BamHI sites of pEGFP-N1 (Clontech) for the generation of hGalc-eGFP. The primers used were hGalc-cDNA-XhoI-s (5′-GGGGG CTCGAG GCCACC atg actgcggccg cgggttcggc-3′) and hGalc-cDNA-BamHI-as (5′-GGGGGG GGATCC CG gcgtgtggcttccacaagaaag-3′). GALC-HA was generated by modifying the hGalc-FLAG clone with the primers HA-fusion-SalI-s (5′-TCGACT ACCCAT ACGAC GTCCC AGACT ACGCT TAGG-3′) and HA-fusion-SalI-as (5′-TCGAC CTAAG CGTAG TCTGG GACGT CGTAT GGGTA G-3′). The QuikChange mutagenesis kit (Stratagene) was used for site-directed mutagenesis, and the primers used were hGalc-change of glycine to aspartate at the amino acid residue number 286 (G286D)-s (5′-gacatgggtgcagactgctggggtcgc-3′), hGalc-G286D-as (5′-gcgaccccagcagtctgcacccatgtc-3′), hGalc-change of threonine to methionine at the amino acid residue number 529 (T529M)-s (5′-cctggcgagcatcacttcatgctacgccaag-3′), hGalc-T529M-as (5′-cttggcgtagcatgaagtgatgctcgccagg-3′), hGalc-change of tyrosine to serine at the amino acid residue number 567 (Y567S)-s (5′-tgactacaaagtgtgatgttagcatagagacccctgacacag-3′), hGalc-Y567S-as (5′-ctgtgtcaggggtctctatgctaacatcacactttgtagtca-3′), hGALC-change of glycine to serine at the amino acid residue number 57 (G57S)-s (5′-ccgcgccgatgctgtcgaactcccg-3′), hGALC-G57S-as (5′-cgggagttcgacagcatcggcgcgg-3′), hGALC-change of aspartate to asparagine at the amino acid residue number 544 (D544N)-s (5′-gattgtgttggatgcattggcagcccatgtaatgg-3′), hGALC-D544N-as (5′-ccattacatgggctgccaatgcatccaacacaatc-3′), hGALC-change of leucine to serine at the amino acid residue number 634 (L634S)-s (5′-aggtgaaatgacccttaatagttgacgtgagtgtataccattttttt-3′), and hGALC-L634S-as (5′-aaaaaaatggtatacactcacgtcaactattaagggtcatttcacct-3′) for the mutations. For the polymorphisms, hGalc-C550T-s (5′-ggattgtgggcgccaagtgttaccatgatttggac-3′), hGalc-C550T-as (5′-gtccaaatcatggtaacacttggcgcccacaatcc-3′), hGalc-G742A-s (5′-cgaactcttcaaggtggttaatgttataggggctcatt-3′), hGalc-G742A-as (5′-aatgagcccctataacattaaccaccttgaagagttcg-3′), hGalc-C1685T-s (5′-tacaactggaccaatctgactataaagtgtgatgtatacatagag-3′), and hGalc-C1685T-as (5′-ctctatgtatacatcacactttatagtcagattggtccagttgta-3′) were used. All the constructs were confirmed by DNA sequencing.
Animal cell culture and transfection.
All animal experimentation was performed in strict accord with Institutional Animal Care and Use Committee approved protocols (Roswell Park Cancer Institute and University at Buffalo). HEK-293T cells were maintained in DMEM supplemented with 10% FBS. Primary oligodendrocyte progenitor cell cultures were obtained based on the immunopanning method (Dugas et al., 2006; Shin et al., 2012). Oli-neu cells were cultured as previously described (Jung et al., 1995). Primary rat Schwann cells were prepared from sciatic nerve and passaged not more than 3 times as described previously (Feltri et al., 1992). Lipofectamine 2000 (Invitrogen) was used for all transfections. For the normalization of transfection, the pGL3-promoter luciferase plasmid (Promega) was cotransfected with the hGALC clones. Luciferase activities were measured as described previously (Shin et al., 2009).
Dermal fibroblast culture.
Dermal fibroblasts from GLD patients and normal age-matched controls were purchased from Telethon Biobank. The code numbers are FFF0181994 (K359AfsX3+Y567S), FFF0751981 (30kbΔ+T529M), FFF0352007 (30kbΔ+G286D), FFF0161996 (homozygous E136EfsX35), FFF0332007 (E130K+N295T), FFF0482013 (R69X+S94P), FFF0201984 (N295T+G609GfsX6), FFF0091993 (homozygous 30kbΔ), FFF1071983 (normal male; wild-type (WT)-1), and FFF0551986 (normal female; WT-2). They were grown in Medium 106 (Invitrogen) containing Low Serum Growth Supplement (Invitrogen) and maintained at 37°C in a humidified CO2 incubator.
Western blot analyses.
Total cell lysates or subcellular fractions in RIPA buffer containing protease inhibitors (Roche) were separated by 12% SDS-PAGE, transferred to PVDF membrane (Millipore), and blocked with 5% skim milk or BSA in TBS-Tween 20. The primary antibodies that were used were β-tubulin (Sigma-Aldrich), GALC (Lee et al., 2010), FLAG (Sigma-Aldrich), LAMP2 (Abcam), and GRP78 (Novus Biologicals). Protein band densities were quantified using ImageJ (National Institutes of Health) and normalized by cotransfected luciferase activities.
Subcellular fractionation.
Lysosomal and ER fractions were purified using the Lysosome enrichment kit (Thermo Scientific) and the Endoplasmic Reticulum enrichment kit (Novus Biologicals), respectively, according to the manufacturer's instructions.
Immunocytochemistry.
Forty-eight hours after transfection, cells were fixed with 4% PFA or 100% ice-cold methanol, permeabilized with Triton X-100, blocked with 10% FCS and 1% BSA, and incubated overnight with primary GALC (Lee et al., 2010), LAMP2 (BD Biosciences and Santa Cruz Biotechnology), KDEL (Enzo Life Sciences), 58k (Sigma-Aldrich), or EEA1 (Cell Signaling Technology) antibodies. After washing with PBS, samples were incubated with secondary antibodies. After an additional washing with PBS, coverslips were mounted with Vectashield (Vector Laboratories) mounting medium, and DAPI. All images were captured in 63× magnification using an Apotome.2 (Zeiss) and 100× magnification using a confocal TCS SP5 II microscope (Leica). z-axis series spanning 2–2.5 μm were acquired as 1024 × 1024 pixel images by sequentially scanning (between frames), using a step size of 0.13 μm. The diameter of lysosomes is in a range of 0.1–1.2 μm (Kuehnel, 2003). Nonspecific background staining was determined by omitting the primary antibody.
GALC enzyme assay.
GALC activity was determined by the method described previously (Martino et al., 2009).
Chemicals.
DMSO, α-lobeline, 4-phenylbutyrate (4-PBA), betaine, glycerol, sucrose, curcumin, proline, sarcosine, taurine, trehalose, and ectoine were all purchased from Sigma-Aldrich. Tauroursodeoxycholate was from EMD Millipore, and trimethylamine N-oxide dehydrate was from Cole-Parmer.
Results
Trafficking and processing of GALC are impaired by infantile- but not later-onset mutations
To explore possible causes of variable genotype-GALC activity-phenotype correlations in GLD, we generated WT and common missense mutants of GALC cDNA (Fig. 1A), which were fused to epitope tags at the carboxyl terminus. Tags of small size (e.g., V5 and 6xHis) on the carboxyl terminus of GALC were previously shown to have no influence on GALC function (Lee et al., 2010). We thus cloned the human GALC coding region into the mammalian expression vector under the CMV promoter and fused it to various carboxyl terminal tags, including FLAG, HA, or eGFP. Western blot studies of transfected HEK-293T cells hybridized with polyclonal anti-GALC antibody (Lee et al., 2010) confirmed that the expression and processing from FLAG fused GALC (GALC-FLAG) and HA fused GALC (GALC-HA) were comparable with that of GALC with no tag (GALC), whereas eGFP fused GALC (GALC-GFP) resulted in aberrant GALC expression (Fig. 1B). Furthermore, the enzymatic activities of GALC-FLAG and GALC-HA were similar to the nontagged GALC (range 15–16 nmol/h/mg in the whole cell), whereas GALC-GFP had lower enzymatic activity (Fig. 1B). We therefore used the GALC-FLAG construct for our experiments. Oligodendrocyte GALC enzymatic activities from wild-type (WT) and Galc-deficient twitcher mice, an authentic mouse model of GLD, were used for comparison. Interestingly, twitcher oligodendrocytes had higher GALC activity than nontransfected HEK-293T cells (Fig. 1B). Because nonsense-mediated mRNA decay suppresses Galc expression in the twitcher mice (Lee et al., 2006), the difference of basal activity level between both cell types may indicate the presence of another enzyme that can apparently increase GALC activity in oligodendrocytes, but not in HEK-293T cells.
Figure 1.

GALC trafficking and processing in transfected cells is impaired in infantile-onset mutants but not in a later-onset mutant. A, A scheme is shown of GALC structure and common missense mutations found in GLD of European ancestry (Wenger, 2011a). The two most common infantile-onset missense mutations of GLD are T529M (exon 14) and Y567S (exon 15). The most frequent later-onset mutation is G286D (exon 8). B, Forty-eight hours after transfection of HEK-293T cells, total cell lysates were analyzed with a polyclonal anti-GALC antibody (Lee et al., 2010) that detects the 80 kDa precursor, and both the 50 and 30 kDa processed forms of GALC protein. There were nonspecific bands ∼90 and 40 kDa (asterisks). β-Tubulin was used for loading control. FLAG and HA fused to the C terminus of GALC were comparable with the untagged protein, in contrast fusion with eGFP markedly decreased GALC expression. GALC-FLAG and GALC-HA had similar enzyme activities to the nontagged GALC, whereas GALC-GFP had lower enzymatic activity. Oligodendrocyte GALC enzymatic activities at 4 DIV from WT and Galc-deficient twitcher mice, an authentic mouse model of GLD, were used for comparison. C, Compared with wild-type, the later-onset mutation (G286D) is significantly processed, whereas the infantile forms (T529M and Y567S) are much less processed. D, Lysosomal fractions, which were purified from the lysate of GALC-transfected HEK-293T cells and analyzed by Western blotting with anti-GALC, β-tubulin, and LAMP2 antibodies, contain mostly the processed forms of GALC, but not the precursor form. In the lysosomal fractions, there are almost no processed forms of GALC for the infantile forms (T529M and Y567S) in contrast to the later-onset form (G286D). E, F, In total cell lysates, the GALC activity of the later-onset mutant (G286D) is indistinguishable from that of the infantile forms (T529M and Y567S), whereas in lysosomal fractions, there is residual GALC activity for the later-onset mutant, but almost no activity for the infantile-forms, revealing a correlation between lysosomal GALC activity and severity of disease. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). n.s., Not significant. G, Immunofluorescence for WT or the three mutant GALCs in HEK-293T cells with anti-GALC (red) and anti-KDEL, anti-58k, anti-EEA1, or anti-LAMP2, antibodies (all green) showed that WT and later-onset mutant GALC colocalized with all four subcellular markers, whereas the infantile-onset GALC mutants were not (or minimally) detectable in the lysosome, and mainly found in the ER and Golgi. The same analyses in transfected oli-neu cells at 4 DIV (H) and primary rat Schwann cells (I) with anti-GALC (red) and anti-LAMP2 (green) confirmed that the WT GALC and later-onset mutant are present in the lysosome, whereas the infantile-forms are not. Arrows indicate overlapping lysosomal puncta and GALC (yellow). Orthogonal views (XY, XZ, and YZ) of a z-stack are shown. White lines indicate section positions. Nuclei were labeled with DAPI. DIV, Days of in vitro differentiation. Scale bars: G–I, 5 μm.
To determine how three common missense mutations (G286D, 2%; T529M and Y567S, each 8%, in individuals of European ancestry; Fig. 1A) in patients with GLD (Wenger, 2011a) influence the expression, processing, and trafficking of GALC, we performed site-directed mutagenesis on the human GALC gene with the FLAG tag at its carboxyl terminus. The mutations are numbered according to Human Genome Variation Society (HGVS) nomenclature recommendations, in which numbering begins from the first methionine of the putative 42 residue signal sequence. These mutations have elsewhere been described as G270D, T513M, and Y551S, respectively.
Western blot analyses on cell lysates using a polyclonal anti-GALC antibody (Lee et al., 2010) showed that WT-GALC was normally processed from its 80 kDa precursor into 50 and 30 kDa fragments (Fig. 1C). In contrast, mainly the 80 kDa precursor protein was detected in the infantile mutations T529M and Y567S, suggesting that processing of GALC is impaired in infantile mutations. Interestingly, the later-onset mutation G286D was processed normally into the 50 and 30 kDa fragments; however, the amount of the processed forms was less than those of WT (Fig. 1C). Lysosomal fractions purified from cells transfected with WT GALC (Fig. 1D) showed that the processed GALC proteins (50 and 30 kDa) were indeed detected in the lysosomal fraction, whereas the precursor form (80 kDa) was barely detectable, indicating that the lysosome contains mostly processed GALC. Similarly, the later-onset G286D GALC mutant also showed processed GALC proteins in lysosomal fractions, whereas the infantile-onset mutations showed barely detectable processed GALC proteins (Fig. 1D). It has been postulated that later-onset patients might have minimal, yet clinically significant, residual GALC activity in the nervous system, which could theoretically explain their milder disease (Harzer et al., 2002; Wenger, 2011b). However, when total cell lysate GALC activity is measured in leukocytes from GLD, there is no apparent difference between the enzymatic activity of patients with the later-onset G286D mutation and patients with mutations causing the infantile-onset form (Duffner et al., 2012). We confirmed this finding by measuring GALC activity in total lysates from cells transfected with our site-directed mutated constructs (Fig. 1E). In contrast, by measuring the activity of GALC in lysosomal fractions, we found that the later-onset mutation has residual enzymatic activity in the lysosome, whereas the infantile-forms do not (Fig. 1F). These data suggest that trafficking to the lysosomes may be limited in a mutation-specific manner, and suggest, for the first time, a potential correlation between GALC activity in the lysosomes with severity of the phenotype.
To confirm that the trafficking of severe GALC mutants was impaired, we performed double-immunofluorescent labeling of GALC and subcellular markers. WT GALC and G286D GALC each showed some colocalization with ER (KDEL), Golgi (58k), and lysosomal markers (LAMP2 and EEA1) (Fig. 1G), which suggest that they are trafficked through the conventional ER/Golgi path to reach the lysosome. However, the infantile-onset mutants T529M and Y567S were present only in the ER and Golgi and were rarely found in the lysosome, suggesting that trafficking of these GALC mutants to the lysosome was disrupted (Fig. 1G). The same trafficking analyses in transfected oli-neu mouse oligodendrocytes (Jung et al., 1995) and primary rat Schwann cells (Feltri et al., 1992) stained for LAMP2 confirmed that the later-onset mutant is better translocated into the lysosome than the infantile forms (Fig. 1H,I). These findings suggest that the severity of infantile GLD mutations may be explained by impaired trafficking and processing of GALC in the lysosome, whereas the milder phenotype of later-onset mutations may be due to a partially retained ability to traffic and process GALC in lysosomes.
cis-polymorphisms that are commonly found with mutations affect the activity and trafficking of GALC
Although each parent of an affected child with GLD usually carries one normal and one mutated GALC allele, GALC enzyme activity can range widely in carriers. The reason for this variability is unknown, but it is possible that cis-polymorphisms that encode additional amino acid changes modulate GALC activity (Fig. 2A). To remain consistent with HGVS nomenclature, the polymorphisms previously noted as c.502T, c.694A, and c.1637C are here referred to as c.550T, c.742A, and c.1685C, respectively. These common polymorphisms have each been shown to reduce WT GALC activity by ∼10–50% (De Gasperi et al., 1996; Luzi et al., 1996; Furuya et al., 1997). The c.1685C polymorphism has also been shown to reduce later-onset G286D GALC activity (Hossain et al., 2014). These normal variants in the GALC coding region do not result in clinical disease in the homozygous state and are not thought to cause disease as compound heterozygotes with one copy of a disease-causing mutation (Wenger, 2011a). Interestingly, the G286D later-onset mutation is always found with either the c.550T or c.1685C polymorphism, the T529M infantile mutation with the c.550T, c.742A, or c.1685C polymorphism, and the Y567S with the c.1685C polymorphism (Fig. 2A). Because of these additional amino acid changes, it is possible that these normal variants may also affect GALC trafficking to the lysosome, which is indispensable for GALC processing and activation, thereby influencing the phenotype.
Figure 2.

Benign cis-polymorphisms affect total enzymatic activity and trafficking of GALC. A, Diagram of the genomic structure of GALC and common benign polymorphisms (blue) found in GLD patients. A new numbering classification, starting from the upstream translational start site, was used to define the residue numbers. The G286D later-onset mutation is always found with the c.550T or c.1685C polymorphism. The T529M infantile-onset mutation is always found with the c.550T, c.742A, or c.1685C polymorphism. The Y567S infantile-onset mutation is always found with the c.1685C polymorphism. These three common polymorphisms were introduced by site-directed mutagenesis. B, GALC activities were measured in total lysates prepared from cells transfected with wild-type or mutant GALC in combination with indicated cis-polymorphisms. Both c.550T and c.742A reduced WT GALC activity, whereas only c.550T or c.1685C reduced G286D or Y567S activity, respectively. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). C, HEK-293T cells transfected with WT or mutant GALCs in combination with indicated cis-polymorphisms were costained for GALC (red) and LAMP2 (green). Nuclei were labeled with DAPI. Orthogonal views (XY, XZ, and YZ) of a z-stack. White lines indicate section positions. D, Quantification of colocalized GALC and LAMP2 signals was obtained by counting the number of yellow puncta. C, D, All polymorphisms reduced lysosomal targeting of WT GALC. The c.550T and c.1685C polymorphisms reduced lysosomal targeting of G286D and Y567S, respectively. More than three independent experiments were performed with qualitatively similar results. Quantification was performed on 8–16 cells. Error bars indicate SD. **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). Scale bar: C, 5 μm.
To determine the effect of specific polymorphisms on GALC activity, human GALC constructs were engineered to contain all combinations of mutations and polymorphisms previously mentioned. All of the constructs previously tested in Figure 1 harbored the c.1685C polymorphism contained in the human cDNA clone. The new clones were then transfected into HEK-293T cells and used for measurement of GALC activity in total cell lysates. Both c.550T and c.742A polymorphisms were found to reduce WT GALC activity, whereas c.1685C did not affect WT GALC significantly (Fig. 2B). Interestingly, the effects of polymorphisms were disease mutation-specific. The c.550T polymorphism reduced GALC activity of the later-onset G286D. The infantile T529M mutation had a very low GALC activity, even without the addition of these polymorphisms, and was not decreased further when combined with additional polymorphisms. In contrast, the infantile-onset Y567S mutation was significantly reduced by the c.1685C polymorphism, indicating that the combination of certain cis-polymorphisms with certain disease mutations can significantly impair GALC activity. Parallel immunocytochemical analyses (Fig. 2C) revealed that the polymorphisms also affected lysosomal targeting of the GALC proteins. These changes were quantified by counting yellow puncta that represent GALC colocalized with the lysosomal marker LAMP2 (Fig. 2D). This quantification mirrored the results of the GALC activity experiment (Fig. 2B): all three polymorphisms reduced the lysosomal trafficking of WT GALC; c.550T reduced the lysosomal trafficking of G286D GALC; and c.1685C dramatically reduced the lysosomal trafficking of Y567S GALC (Fig. 2D). Together, it is clear that in combination with specific disease-causing mutations in GALC, cis-polymorphisms can indeed affect the trafficking and total enzymatic activity of GALC.
cis-polymorphisms change the total level and processing of GALC
To ask whether the polymorphisms affected the processing of GALC, we next performed Western blot analyses on total cell lysates of transfected HEK-293T cells. We found that the polymorphisms had various effects on the levels of precursor and cleaved forms of WT or mutant GALC (Fig. 3A). Total GALC levels in the cell were calculated by quantifying and summing the 80 kDa precursor and the 50 and 30 kDa cleaved forms of GALC (Fig. 3B). We found that both the c.550T polymorphism and the c.742A polymorphism reduced total WT GALC levels, whereas the c.1685C polymorphism did not. The c.1685C and c.550T polymorphisms also reduced the total level of the later-onset G286D GALC. In contrast, c.1685C only reduced total T529M GALC marginally, whereas this polymorphism dramatically reduced total Y567S GALC. Notably, no mutation, with or without combined polymorphisms, completely abolished GALC levels.
Figure 3.

cis-polymorphisms affect GALC processing. A, Lysates from HEK-293T expressing GALC mutations in combination with various cis-polymorphisms were analyzed by Western blot using anti-GALC antibody. Both the GALC mutations and some of the polymorphisms affect the level and processing of GALC. B, Quantification of levels in four independent experiments, normalized by cotransfection of luciferase plasmid, showed that both the c.550T and c.742A polymorphisms reduced total GALC (defined as the sum of the GALC levels for the 80, 50, and 30 kDa forms) in the WT context, but c.1685C did not have any effect. In the case of later-onset G286D mutant, both c.1685C and c.550T reduced total GALC levels. The c.1685C polymorphism reduced total GALCs of both T529M and Y567S. Interestingly, none of the mutations with (or without) polymorphisms completely abolished GALC expression. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). C, The ratio of 50 versus 80 kDa levels and (D) 30 versus 80 kDa levels showed that both c.550T and c.742A reduce the processing of WT. Both c.1685C and c.550T reduce processing of the later-onset G286D GALC. None of the polymorphisms had any effect on the infantile-onset T529M GALC, whereas c.1685C reduced the infantile-onset Y567S GALC processing, indicating a critical pathogenic role of this polymorphism in patients harboring the Y567S mutation. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
We defined the level of processed forms as the ratio of cleaved 50 kDa or 30 kDa GALC fragments relative to the 80 kDa precursor form. For example, the level of WT 50 kDa or 30 kDa relative to WT 80 kDa precursor showed that the c.1685C polymorphism only marginally reduced the processing of WT GALC, whereas the c.550T and c.742A polymorphisms reduced the processing of WT GALC significantly. Both the c.1685C and c.550T polymorphisms reduced the processing of the later-onset G286D GALC mutant. No polymorphisms had an effect on the processing of the T529M GALC infantile-onset mutant. Interestingly, the c.1685C polymorphism dramatically reduced the processing of the Y567S GALC mutant. Indeed, Y567S GALC has normal processing without the c.1685C polymorphism; albeit this combination has not been documented in human.
In addition to the processing, the cis-polymorphisms also affect the level of the 80 kDa precursor form of GALC, suggesting that the polymorphisms may change synthesis or stability of GALC as well (Fig. 3A). We confirmed the relative amounts of the 80 kDa precursor protein with monoclonal anti-FLAG antibody (Fig. 4A), and noted the similar pattern using either GALC or FLAG antibodies (compare Figs. 3A, 4A). We next quantified the 80 kDa GALC precursor shown in both Figures 3A and 4A (Fig. 4B). The c.550T polymorphism increased the level of 80 kDa WT GALC, whereas none of the polymorphisms affected the level of 80 kDa G286D GALC. Both c.1685C and c.742A polymorphisms reduced the level of 80 kDa T529M GALC, whereas the c.1685C polymorphism did not have much of an effect on the level of 80 kDa Y567S GALC. Together, these data suggest that cis-polymorphisms, in combination with specific mutations in GALC, can reduce the total level, trafficking, and processing of GALC. In this way, the main effect of the mutations may be to cause incorrect targeting and consequently a critical deficit in enzyme processing and activity.
Figure 4.
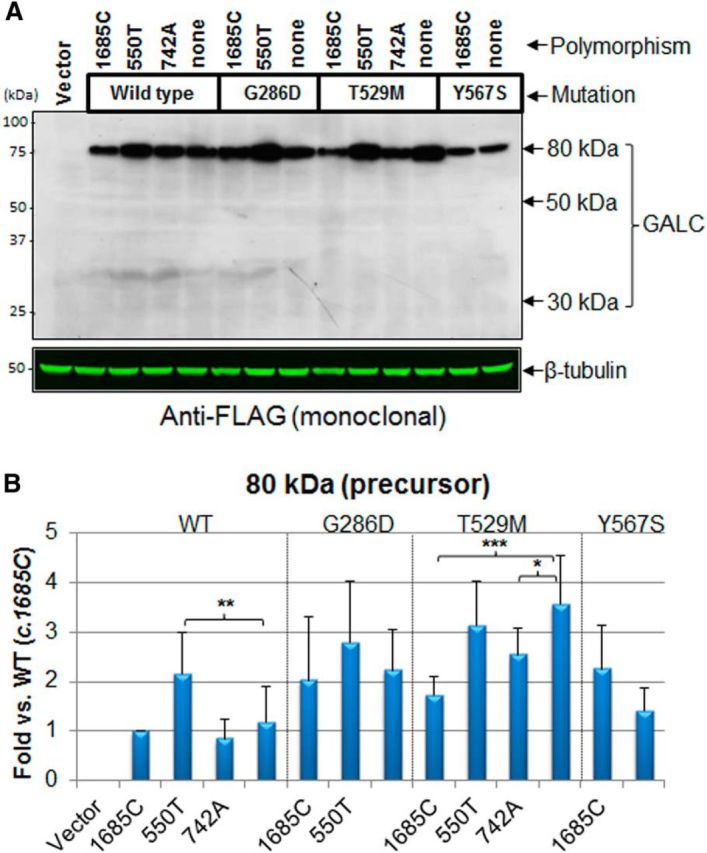
cis-polymorphisms affect the level of precursor 80 kDa GALC. A, Lysates of cells transfected with the GALC mutants and cis-polymorphisms were analyzed by Western blot with anti-FLAG antibody, which detected only the 80 kDa precursor form. The FLAG antibody did not detect the 30 kDa fragment, suggesting that the FLAG tag at the C terminus might slightly modify the 30 kDa structure and mask the epitope. B, Quantification of the 80 kDa GALC densities showed that the c.550T polymorphism increased the unprocessed WT GALC, whereas none of the polymorphisms significantly affected the unprocessed G286D GALC. Both c.1685C and c.742A polymorphisms reduced the unprocessed level of T529M GALC. The polymorphism c.1685C did not have much effect on the unprocessed Y567S GALC. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Additionally, we noticed that Western analysis using a FLAG antibody detected only the 80 kDa precursor form of GALC, even though the 30 kDa carboxyl terminal fragment should also contain FLAG (Fig. 4A). Similarly, a carboxyl HA tag was only detected on the 80 kDa form and not on the 30 kDa fragment by Western blot with HA antibody (data not shown). Instead, both 30 kDa epitope-tagged fragments were easily detected by the GALC antibody (Fig. 1B). This was previously observed also with the carboxyl terminal V5 tags, for unknown reasons (Lee et al., 2010). It is possible that short-sized tags (FLAG, V5, and HA) at the carboxyl terminus slightly modify the 30 kDa structure after GALC processing and mask the epitope tag, without affecting enzyme activity.
The amount of GALC precursor form in the ER has a substantial effect on total cell GALC activity in GALC mutants
Because the precursor form of GALC was almost absent in the lysosomal fractions (Fig. 1D), it may instead be localized in another organelle, where it may potentially have enzymatic activity. The cleavage site for GALC processing is in the loop of the β-sandwich domain, which is far from the active site (Deane et al., 2011), indicating that the cleavage event may have no relevance for enzyme activity. Indeed, it is known that secreted GALC in the 80 kDa precursor form retains GALC activity (Lee et al., 2010). Therefore, we hypothesized that GALC located outside of the lysosome could confound measures of GALC activity, derived from total cell lysates, as is currently done for diagnostic purposes.
To directly determine whether the precursor GALC in nonlysosomal organelles has enzymatic activity, ER or lysosomal fractions were purified from total cell lysates and analyzed by Western blot (Fig. 5A,C) and evaluated for GALC activity (Fig. 5B,D). The WT GALC activity generated from the ER was low, but not zero, and accounted for only 11%–24% of total WT cellular activity. Mutant GALC activities from the ER were similarly low but accounted for a more substantial 30%–68% (G286D), 32%–89% (T529M), and 35%–75% (Y567S) of total mutant cellular activity (Fig. 5E,F). These data indicate that GALC activity from nonlysosomal origins, for example the ER, could significantly contribute to GALC activity when measured in whole-cell lysates.
Figure 5.

GALC in the ER has substantial enzyme activity. A, Purified ER fractions from total cell lysates of transfected HEK-293T cells showed that the 80 kDa GALC precursor form is present in the ER. GRP78 was used as an ER loading control. B, GALC activity assay performed with ER fractions reveals that the precursor protein possesses some enzymatic activity. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). C, Purified lysosomal fractions contain mainly the processed GALCs. D, The same lysosomal fractions were also used for determination of GALC activities. Error bars indicate SD of duplicates. E, The GALC activities from ER (red bar) and lysosome (blue bar) were combined to show the relative contributions to actual cellular activity. F, Percentage of the activities from each subcellular organelle shows that the GALC in ER contributes significantly to the total activities in the mutants (G286D, T529M, and Y567S), but not in WT.
Additional later-onset GALC mutations also retain partial processing and trafficking to lysosomes
To provide further evidence for the difference in lysosomal targeting and GALC activity in lysosomes between later- and infantile-onset mutations, we generated two more later-onset (G57S and L634S) and one more infantile (D544N) mutant GALC constructs. The mutations are numbered according to HGVS, which have elsewhere been described as G41S, D528N, and L618S, respectively. G57S is responsible for the majority of later-onset GLD in the Catania region of Italy (Lissens et al., 2007), D544N is found in the infantile-onset Muslim Arab population of Israel (Rafi et al., 1996), and L634S is a prominent later-onset mutation in Japan (Xu et al., 2006; Hossain et al., 2014). All of these mutations are sometimes found in homozygosity among GLD patients, and in the homozygous state they cause later- or infantile- onset GLD respectively, suggesting they are clearly associated with the age of onset of GLD.
Measurements of GALC activity in total lysates from HEK-293T cells transfected with the mutated constructs showed that both later-onset G57S and L634S GALCs have higher enzymatic activities than that of the infantile D544N (Fig. 6A, right bar of each GALC), and this propensity is maintained more evidently in the lysosomal fraction (Fig. 6B, right bar of each GALC). Because these mutations are always found with the c.1685C polymorphism (Rafi et al., 1996; Satoh et al., 1997; Lissens et al., 2007), we also made additional mutants, each containing c.1685C as well (Fig. 6A,B, left bar of each GALC). The c.1685C polymorphism reduced the activities of G57S and D544N, but not that of L634S in total cell lysates (Fig. 6A, left bar of each GALC). However, in the lysosomal fractions, the c.1685C significantly reduced enzymatic activities of all of G57S, D544N, and L634S (Fig. 6B, left bar of each GALC). Interestingly, infantile D544N GALC has some residual activity without the c.1685C polymorphism in both total lysates and lysosomal fraction, but the activity was not detectable with the polymorphism (Fig. 6A,B). This suggests that the c.1685C polymorphism may augment the severity of the infantile-onset phenotype of D544N GALC mutation.
Figure 6.

Additional later-onset GALC mutations also retain partial trafficking and processing of GALC in lysosomes. Two more later-onset (G57S and L634S) and one more infantile (D544N) GALC mutations were generated with or without the c.1685C polymorphism. Measurements of GALC activities in both total lysates (A) and lysosomal fractions (B) from HEK-293T transfected with the mutant constructs show that both later-onset mutants G57S and L634S have significantly higher residual GALC activities compared with the infantile D544N form, revealing a strong correlation between residual GALC activity and severity of disease. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). n.s., Not significant. C, In total cell lysates, both later- and infantile-onset mutations are processed less than the WT. However, in lysosomal fractions, the later-onset forms (G57S and L634S) are more processed than the infantile form (D544N). D, Immunofluorescence for the three mutant GALCs in HEK-293T cells with anti-GALC (red) and anti-LAMP2 antibodies (green) shows that later-onset mutant GALCs colocalized (yellow) more with the lysosomal marker, whereas the infantile-onset GALC mutant was minimally detectable in the lysosome. Orthogonal sections (XY, XZ, and YZ) of a z-stack are shown. White lines indicate the position of sections. Nuclei were labeled with DAPI. Scale bar, 5 μm.
Western blot analyses on total cell lysates using a polyclonal anti-GALC antibody demonstrated that mainly the 80 kDa precursor protein was detected in both infantile- and later-onset mutations (Fig. 6C). However, the lysosomal fractions confirmed that the later-onset G57S or L634S GALC mutant was processed more than the infantile-onset D544N. The c.1685C polymorphism reduced the processing of all mutant GALC forms (Fig. 6C), in line with the lysosomal mutant GALC activities (Fig. 6B). Parallel immunocytochemical analyses revealed that both later-onset G57S and L634S GALCs colocalized with LAMP2 (Fig. 6D), whereas the infantile-onset mutant D544N was rarely found in the lysosome as already shown in the previous study (Lee et al., 2010). These findings confirmed that the milder phenotype of later-onset mutations may be due to a partially retained ability to traffic and process GALC in lysosomes.
Dermal fibroblasts from later-onset patients have minimal, yet significantly higher, GALC activities than those of infantile-onset patients
To validate that the differences that we observed in lysosomal GALC activity between infantile- and later-onset forms were relevant for GLD, we used cells from GLD patients. Eight different GLD dermal fibroblasts and two age-matched normal cells (Fig. 7A) were cultured and analyzed by lysosomal fractionation and GALC enzyme activity (Fig. 7B). Unfortunately, homozygous cells having the G286D, T529M, and Y567S mutations were not available, so we obtained the three mutants in compound heterozygosity with a large deletion mutant, such as the 30 kb GALC deletion (Δ) or K359AfsX3 (Tappino et al., 2010). We assumed that GALC produced from the large deletions in the GALC locus would be low due to nonsense-mediated mRNA decay associated with the premature stop codons, or to protein instability. Therefore, the GALC activity from the fibroblasts can be considered to derive mainly from the missense mutations. We also analyzed one more later-onset (N295T+G609GfsX6) and four more infantile-onset (homozygous E136EfsX35, E130K+N295T, R69X+S94P, and homozygous 30-kbΔ) GLD fibroblasts (Fig. 7A).
Figure 7.

Lysosomal fractions of dermal fibroblasts from later-onset patients have significantly higher GALC activities than those from infantile-onset patients. A, The table shows clinical and molecular information of GLD dermal fibroblasts used in this study, which were from published (Tappino et al., 2010) and unpublished samples (Mirella Filocamo, Telethon Biobank, Italy). K359AfsX3 (lysine 359 substituted by alanine followed by a frame-shift translational termination) is located close to the frequent 30 kb deletion GLD mutation and therefore will generate a truncation similar to the 30 kb deletion. B, GALC activities in total cell lysates or lysosomal fractions of the dermal fibroblasts from GLD patients and age-matched normal controls (<4 years old). GALC activities from total cell lysates of the later-onset mutants are many times indistinguishable from those of the infantile forms. However, GALC activities from the lysosomal fractions showed that the later-onset mutants have more enzyme activity in the lysosome and are more reliably distinguished from those of the infantile forms. WT-1 is male and WT-2 is female. Error bars indicate SD of triplicates. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). n.s., Not significant.
When GALC activities were measured in whole-cell extracts prepared from the GLD fibroblasts, there was no distinction between patients with later-onset mutations (G286D+30-kbΔ or N295T+G609GfsX6) and patients with the infantile-onset forms (T529M+30-kbΔ, Y567S+K359AfsX3, homozygous E136EfsX35, E130K+N295T, R69X+S94P, and homozygous 30-kbΔ) (Fig. 7B), as has been previously documented in blood samples (Fig. 7A; % GALC activity) (Tappino et al., 2010; Mirella Filocamo, personal communication). However, when GALC activities were measured in lysosomal fractions from the GLD fibroblasts, both later-onset mutations had low (0.31–0.32 nmol/h/mg) but significantly higher GALC activity than infantile forms (range, 0.04–0.23 nmol/h/mg), confirming that there is a correlation between GALC activity in the lysosome and the severity of the phenotype. As expected, homozygous 30 kbΔ fibroblast did not produce any GALC activity in total cell lysates and barely detectable activity in lysosomal fractions (Fig. 7B). In addition, there was a significant difference in the GALC activity from total cell lysates between the two normal controls (0.79 vs 3.05 nmol/h/mg). We do not have full DNA sequence of the GALC alleles from these controls, but there could be cis-polymorphisms that alter WT GALC activities. Also, the effect of cis-polymorphisms on the mutants was not testable with only eight mutant fibroblast lines. However, a future study with more patient and control lines can address this question.
4-PBA increases the level and activity of GALC mutants
Pharmacological small chaperones are promising therapeutics for misfolded protein diseases, which based on altered protein trafficking, could also include GLD. To promote trafficking of GALC mutants to the lysosome, we tested 14 chemicals that are known to behave as chaperones: DMSO, α-lobeline, 4-PBA, betaine, glycerol, sucrose, curcumin, proline, sarcosine, taurine, trehalose, ectoine, tauroursodeoxycholate, and trimethylamine N-oxide dehydrate, in transfected HEK-293T cells that express WT, G286D, T529M, or Y567S GALCs. Two different concentrations of each chaperone were used to treat cells for 24 hours, followed by Western analyses with a GALC antibody. Interestingly, we find that 4-PBA enhances levels of the precursor GALC (Fig. 8A). Treatment with 4-PBA increased only the precursor or sometimes increased both the precursor and processed forms of GALCs, in part dependent on associated cis-polymorphisms (Fig. 8A). 4-PBA also increased the amount of the precursor form of the infantile-onset T529 GALC mutants, but never its processed forms (Fig. 8A). As shown before, the c.1685T polymorphism increased remarkably the processing of the Y567S mutants, and 4-PBA did not appear to modify the Y567S processed forms, but to increase the overall amount of GALC.
Figure 8.

4-PBA enhances GALC levels and activity. A, GALC levels were measured from total lysates of transfected cells expressing WT or mutant GALC in combination with various cis-polymorphisms after treatment with 2 mm 4-PBA for 24 h. The chaperone increased the 80 kDa GALC precursor in WT and in all mutants, but not relative levels of processed GALC fragments. B, GALC activities were measured from transfected cells expressing the WT and G286D GALCs with or without treatment with 2 mm of 4-PBA. 4-PBA significantly enhanced the GALC activities in cells expressing WT, G286D, and T529M GALCs. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). n.s., Not significant. C, The addition of 4-PBA for 24 h increased the trafficking to the lysosome (LAMP2; green) of WT and all mutant GALCs (red) in transfected HEK-293T cells, possibly due to the overall increase in GALC precursor by 4-PBA. Orthogonal views (XY, XZ, and YZ) of a z-stack. White lines indicate section positions. Scale bar: C, 5 μm.
Collectively, these data suggest that 4-PBA may only affect the precursor level of GALC, not its processing. Because of the increased total GALC level, the addition of 4-PBA enhanced the activities of WT, G286D, and T529M significantly in total cells (Fig. 8B). The effect on Y567S was not statistically significant. Interestingly, GALC activity was also increased in the empty vector control by 4-PBA treatment, indicating that there could be a basal level of GALC expression in HEK-293T cells that is negligible in normal conditions. Trafficking analysis using anti-GALC and anti-LAMP2 antibodies showed that 4-PBA increased the localization of WT and all mutants GALCs to the lysosome (Fig. 8C), possibly due to the overall increase in GALC precursor by 4-PBA.
Discussion
In this study, we showed that measuring GALC activity in lysosomal fractions provides a better correlation between the GALC mutation and disease severity. We further show that common polymorphisms modulate GALC activity in ways that are predicted by disease severity and that may be exploited for improving the clinical outcome. These novel findings are important for the diagnosis, prognosis, and design of novel therapies for GLD.
Our study confirms and significantly extends upon the previous study (Lee et al., 2010). They showed alterations in processing, trafficking, and enzymatic activity of one infantile- and two later-onset GALC mutants. We have extended that analysis to three more infantile- and three later-onset mutants, in combination with various common cis-polymorphisms, to show that lack of arrival to lysosomes correlates with lack of processing of GALC and the severity of disease. Second, cis-polymorphisms can have important effects on trafficking, processing, and activity of GALC mutants. Furthermore, we confirmed that the differential trafficking of GALC mutants is conserved among cell types, including myelinating glia (oligodendrocytes and Schwann cells) and HEK-293T embryonic kidney cells. Finally, we follow up on these observations to show that direct measure of GALC activity from lysosomal fractions in transfected cells, as well as from patient fibroblasts, better distinguishes the GALC activities of infantile GLD from the later-onset GLD mutants.
Current diagnosis of GLD in newborn screening is based on reduced GALC activity in total cell lysates from blood and DNA analysis (http://www.wadsworth.org/). However, the mutation–phenotype relationship often does not correlate with clinical outcomes, such as age of onset and survival. The reason for the variability in the genotype–phenotype relationship for GLD was previously unknown (Wenger, 2011b). In the lysosome, GALC catabolizes galactolipids, such as galactosylceramide and psychosine (Suzuki, 2004). Because clinical laboratories use total cell lysates from blood cells or skin fibroblasts to measure GALC activity, it is possible that GALC present in other subcellular organelles could falsely skew the enzymatic activity measured in total cells after lysis but would not reflect the useful activity of GALC in vivo in the lysosome.
In this study, we postulated that this confounding error is one reason why GALC activity from total cell lysates may not always be a good prognostic predictor. Our data support this hypothesis. We showed that infantile-onset mutation in GALC results in alterations in trafficking and processing of GALC associated with severely reduced lysosomal GALC activity. In contrast, a later-onset mutation retains partial ability to traffic GALC to lysosomes where it is processed at lower levels and retains a low, but existent, GALC activity. Furthermore, we show that cis-polymorphisms can modify trafficking and processing of a mutant GALC, including its insertion into the lysosome, and access to its substrates. These data provide the first evidence for how cis-polymorphisms may modulate the function of a mutant GALC and produce genotype/phenotype variability in patients.
At first glance, analysis of lysosomal extracts appears to be designed to eliminate GALC-specific activities that are falsely high because this approach would remove other confounding cellular sources of GALC (Fig. 9). However, falsely high values are not the problem in presymptomatic or newborn screening. In the latter, the problem is to distinguish very low infantile-onset GLD GALC activities from low, later-onset GLD GALC activities and from falsely low, not disease-causing GALC activities (e.g., carriers and other false positives). Nonetheless, comparing Figure 1F (lysosomal) with Figure 1E (whole cell) shows how lysosomal extracts actually succeed. The specific activities of WT or later-onset G286D increase by 2.5- to 3.5-fold in the lysosomal fraction, largely because GALC per unit protein values are much higher in the lysosome, whereas the specific activities of infantile-onset T529M or Y567S values fall slightly; here there is only the small amount of GALC in the lysosome in either extract. As a result, the somewhat low levels of later-onset G286D are now easily distinguished from very low infantile-onset levels. In addition, we note that the range of GALC activities are expanded over a larger range (from 3 to 55 in the lysosome instead of from 2.5 to 20 nmol/h/mg in the whole cell) potentially helping to distinguish all true GALC levels from background noise and to better separate lower GALC levels due to carrier or polymorphism effects from GLD-causing levels.
Figure 9.

Model for how measured GALC activities may not correlate with disease severity in GLD; 5% of normal GALC activity is regarded as the general cutoff for diagnosis of GLD. The diagram represents two different possibilities for the localization of GALC that can produce the same whole-cell lysate GALC activity when measured in patient blood. A, If the majority of remaining GALC is <5% but is found concentrated in the lysosome, then the patient will have milder phenotype and present with a later-onset form of the disease. B, If, however, most of the remaining GALC is localized to the ER or Golgi, due to defective trafficking, then the patient will have a more severe phenotype and may manifest with the infantile-onset form of the disease.
A total of 5% of normal GALC activity is considered to be the general cutoff for diagnosis of GLD. We propose that, if the majority of this GALC activity (<5%) is present in the lysosome, then the patient will have a milder phenotype and will present with the later-onset form of the disease (Fig. 9A). In contrast, if this GALC activity is localized aberrantly to the ER or Golgi, due to a defect in trafficking of GALC protein, then the patient will have a more severe phenotype and will present with the infantile-onset form of the disease (Fig. 9B). Indeed, it has been recently reported that the GALC precursor protein undergoes more effective processing in the later-onset form of the disease than in the infantile-onset form (Hossain et al., 2014), indicating that the factor most responsible for the pathogenesis of the disease may indeed be the ability to process the precursor form of GALC to the mature form. Therefore, we think that it is necessary to measure GALC-specific activity in various subcellular organelles, including the lysosome, to better understand the different sources that may contribute to overall GALC activity in whole-cell lysates, currently measured in the clinic.
In addition to being trafficked directly to the lysosome, the GALC precursor is also secreted into the extracellular space and then taken up either by the secreting cell itself or by adjacent cells, at which point it is finally delivered to lysosomes (Nagano et al., 1998). GLD mutations also lead to impaired GALC secretion and/or reduced cellular reuptake (Lee et al., 2010), indicating that the mutations may prevent cross-correction of adjacent cells with significant GALC enzyme. The cis-polymorphisms could also impact this secretion/uptake process by affecting GALC trafficking. In a future study, we plan to measure the effects of these mutations and cis-polymorphisms on GALC secretion and cellular reuptake.
The use of the chemical chaperone strategy is theoretically attractive because it is applicable to a wide range of pathologic conditions. Interestingly, we identified 4-PBA as an enhancer of the GALC precursor level, which is a low-molecular weight fatty acid that increases protein stability by the binding to and masking of exposed hydrophobic regions of misfolded proteins. 4-PBA is already used clinically in children with urea cycle disorders (Maestri et al., 1996), as a chemotherapeutic agent (Chen WY et al., 1997), and in cystic fibrosis, where it partially facilitates trafficking of the mutant cystic fibrosis transmembrane conductance regulator ΔF508 (Rubenstein and Zeitlin, 2000). Partial effects of chaperones for the treatment of GLD may be sufficient because it is generally accepted that a residual enzyme activity of only ∼10% provides clinical efficacy in lysosomal storage diseases (Parenti, 2009). Also, because of their small size, chaperones have the potential to be taken orally with broad bioavailability, including to the brain. Indeed, α-lobeline, a pharmacological chaperone and weak inhibitor of GALC, was found to rescue function of the D544N infantile-onset GLD mutant in transfected cells (Lee et al., 2010). Another recent study has also demonstrated that N-octyl-4-epi-β-valienamine significantly increases the enzyme activity and the processing of later-onset GALC mutants (Hossain et al., 2015). Additionally, a cellular high-throughput screening assay using GLD fibroblasts has been developed for small molecules that enhance the residual mutant GALC activity (Ribbens et al., 2013). Unfortunately, in many contexts (Fig. 8A), 4-PBA affects mainly the level of precursor GALC and not its processed fragments, thus limiting the therapeutic application. In addition, accumulation of precursor proteins has the potential to cause other side effects, including an unfolded protein response (D'Antonio et al., 2013). It will be necessary to screen for other chemical chaperones that facilitate trafficking of mutant GALCs to the lysosome.
In conclusion, we found that cis-polymorphisms that are associated with specific mutations explain, in part, the clinical variability associated with a given genetic mutation. We suggest that measurement of GALC activity in a subcellular organelle-specific manner may better predict which infants with low GALC activity are at higher risk for the infantile phenotype and are therefore more suitable as potential candidates for HCT. This method will also help to distinguish other children with low GALC activity that will eventually develop later-onset phenotypes, perhaps many years before the initial onset of symptoms. Although the majority of this study has been performed in transfected cells on exogenously expressed GALC, we did confirm the improved discrimination of infantile- and later-onset GALC levels in lysosomal fractions prepared from patient fibroblasts. In a future study, we will extend this analysis to total cell lysates versus lysosomal fractions in induced pluripotent stem cells and induced pluripotent stem cell-derived oligodendrocytes from GLD patients (Tappino et al., 2010) to confirm the difference between infantile- and later-onset forms, and the effect of cis-polymorphisms, on trafficking, lysosomal processing, and GALC activity.
Footnotes
This work was supported by National Institutes of Health Grant R03-NS087359 to D.S., Western New York Stem Cell Pilot Studies to D.S., Empire State Development Corporation for Research Foundation-Krabbe Disease Research Working Capital Grant W753 to L.W. and M.L.F., Empire State Development Corporation for Krabbe Disease Research Capital Equipment Grant U446 to L.W. and M.L.F., and Hunter's Hope Foundation. We thank Mirella Filocamo for information regarding Krabbe dermal fibroblast records; Christopher B. Eckman for providing the polyclonal GALC antibody; Franca Cambi for providing the oli-neu cells; Edward Hurley for technical support; and Nadav I. Weinstock for useful comments on the manuscript.
The authors declare no competing financial interests.
References
- Chen WY, Bailey EC, McCune SL, Dong JY, Townes TM. Reactivation of silenced, virally transduced genes by inhibitors of histone deacetylase. Proc Natl Acad Sci U S A. 1997;94:5798–5803. doi: 10.1073/pnas.94.11.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Rafi MA, de Gala G, Wenger DA. Cloning and expression cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum Mol Genet. 1993;2:1841–1845. doi: 10.1093/hmg/2.11.1841. [DOI] [PubMed] [Google Scholar]
- D'Antonio M, Musner N, Scapin C, Ungaro D, Del Carro U, Ron D, Feltri ML, Wrabetz L. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J Exp Med. 2013;210:821–838. doi: 10.1084/jem.20122005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane JE, Graham SC, Kim NN, Stein PE, McNair R, Cachón-González MB, Cox TM, Read RJ. Insights into Krabbe disease from structures of galactocerebrosidase. Proc Natl Acad Sci U S A. 2011;108:15169–15173. doi: 10.1073/pnas.1105639108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gasperi R, Gama Sosa MA, Sartorato EL, Battistini S, MacFarlane H, Gusella JF, Krivit W, Kolodny EH. Molecular heterogeneity of late-onset forms of globoid-cell leukodystrophy. Am J Hum Genet. 1996;59:1233–1242. [PMC free article] [PubMed] [Google Scholar]
- Duffner PK, Barczykowski A, Kay DM, Jalal K, Yan L, Abdelhalim A, Gill S, Gill AL, Carter R. Later onset phenotypes of Krabbe disease: results of the World-Wide Registry. Pediatr Neurol. 2012;46:298–306. doi: 10.1016/j.pediatrneurol.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci. 2006;26:10967–10983. doi: 10.1523/JNEUROSCI.2572-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, Scherer SS, Wrabetz L, Kamholz J, Shy ME. Mitogen-expanded Schwann cells retain the capacity to myelinate regenerating axons after transplantation into rat sciatic nerve. Proc Natl Acad Sci U S A. 1992;89:8827–8831. doi: 10.1073/pnas.89.18.8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya H, Kukita Y, Nagano S, Sakai Y, Yamashita Y, Fukuyama H, Inatomi Y, Saito Y, Koike R, Tsuji S, Fukumaki Y, Hayashi K, Kobayashi T. Adult onset globoid cell leukodystrophy (Krabbe disease): analysis of galactosylceramidase cDNA from four Japanese patients. Hum Genet. 1997;100:450–456. doi: 10.1007/s004390050532. [DOI] [PubMed] [Google Scholar]
- Harzer K, Knoblich R, Rolfs A, Bauer P, Eggers J. Residual galactosylsphingosine (psychosine) b-galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clin Chim Acta. 2002;317:77–84. doi: 10.1016/S0009-8981(01)00791-4. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Otomo T, Saito S, Ohno K, Sakuraba H, Hamada Y, Ozono K, Sakai N. Late-onset Krabbe disease is predominant in Japan and its mutant precursor protein undergoes more effective processing than the infantile-onset form. Gene. 2014;534:144–154. doi: 10.1016/j.gene.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Higaki K, Saito S, Ohno K, Sakuraba H, Nanba E, Suzuki Y, Ozono K, Sakai N. Chaperone therapy for Krabbe disease: potential for late-onset GALC mutations. J Hum Genet. 2015;60:539–545. doi: 10.1038/jhg.2015.61. [DOI] [PubMed] [Google Scholar]
- Jung M, Kramer E, Grzenkowski M, Tang K, Biakemore W, Aguzzi A, Khazaie K, Chiichlia K, Blankenfeld Gv, Kettenmann H, Trotter J. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. EurJ Neurosci. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Kuehnel W. Color atlas of cytology, histology, and microscopic anatomy. Ed 4. New York: Thieme; 2003. [Google Scholar]
- Lattanzi A, Neri M, Maderna C, di Girolamo I, Martino S, Orlacchio A, Amendola M, Naldini L, Gritti A. Widespread enzymatic correction of CNS tissues by a single intracerebral injection of therapeutic lentiviral vector in leukodystrophy mouse models. Hum Mol Genet. 2010;19:2208–2227. doi: 10.1093/hmg/ddq099. [DOI] [PubMed] [Google Scholar]
- Lee WC, Tsoi YK, Dickey CA, Delucia MW, Dickson DW, Eckman CB. Suppression of galactosylceramidase (GALC) expression in the twitcher mouse model of globoid cell leukodystrophy (GLD) is caused by nonsense-mediated mRNA decay (NMD) Neurobiol Dis. 2006;23:273–280. doi: 10.1016/j.nbd.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Lee WC, Kang D, Causevic E, Herdt AR, Eckman EA, Eckman CB. Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J Neurosci. 2010;30:5489–5497. doi: 10.1523/JNEUROSCI.6383-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissens W, Arena A, Seneca S, Rafi M, Sorge G, Liebaers I, Wenger D, Fiumara A. A single mutation in the GALC gene is responsible for the majority of late onset Krabbe disease patients in the Catania (Sicily, Italy) region. Hum Mutat. 2007;28:742. doi: 10.1002/humu.9500. [DOI] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Wenger DA. Multiple mutations in the GALC gene in a patient with adult-onset Krabbe disease. Ann Neurol. 1996;40:116–119. doi: 10.1002/ana.410400119. [DOI] [PubMed] [Google Scholar]
- Maestri NE, Brusilow SW, Clissold DB, Bassett SS. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med. 1996;335:855–859. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- Martino S, Tiribuzi R, Tortori A, Conti D, Visigalli I, Lattanzi A, Biffi A, Gritti A, Orlacchio A. Specific determination of beta-galactocerebrosidase activity via competitive inhibition of beta-galactosidase. Clin Chem. 2009;55:541–548. doi: 10.1373/clinchem.2008.115873. [DOI] [PubMed] [Google Scholar]
- Nagano S, Yamada T, Shinnoh N, Furuya H, Taniwaki T, Kira J. Expression and processing of recombinant human galactosylceramidase. Clin Chim Acta. 1998;276:53–61. doi: 10.1016/S0009-8981(98)00095-3. [DOI] [PubMed] [Google Scholar]
- Pannuzzo G, Cardile V, Costantino-Ceccarini E, Alvares E, Mazzone D, Perciavalle V. A galactose-free diet enriched in soy isoflavones and antioxidants results in delayed onset of symptoms of Krabbe disease in twitcher mice. Mol Genet Metab. 2010;100:234–240. doi: 10.1016/j.ymgme.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafi MA, Luzi P, Zlotogora J, Wenger DA. Two different mutations are responsible for Krabbe disease in the Druze and Moslem Arab populations in Israel. Hum Genet. 1996;97:304–308. doi: 10.1007/BF02185759. [DOI] [PubMed] [Google Scholar]
- Raghavan S, Zeng B, Torres PA, Pastores GM, Kolodny EH, Kurtzberg J, Krivit W. Globoid cell leukodystrophy (Krabbe disease): normal umbilical cord blood galactocerebrosidase activity and polymorphic mutations. J Inherit Metab Dis. 2005;28:1005–1009. doi: 10.1007/s10545-005-4138-z. [DOI] [PubMed] [Google Scholar]
- Ribbens J, Whiteley G, Furuya H, Southall N, Hu X, Marugan J, Ferrer M, Maegawa GH. A high-throughput screening assay using Krabbe disease patient cells. Anal Biochem. 2013;434:15–25. doi: 10.1016/j.ab.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of ΔF508-CFTR. Am J Physiol Cell Physiol. 2000;278:C259–C267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- Satoh JI, Tokumoto H, Kurohara K, Yukitake M, Matsui M, Kuroda Y, Yamamoto T, Furuya H, Shinnoh N, Kobayashi T, Kukita Y, Hayashi K. Adult-onset Krabbe disease with homozygous T1853C mutation in the galactocerebrosidase gene: unusual MRI findings of corticospinal tract demyelination. Neurology. 1997;49:1392–1399. doi: 10.1212/WNL.49.5.1392. [DOI] [PubMed] [Google Scholar]
- Shin D, Shin JY, McManus MT, Ptácek LJ, Fu YH. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann Neurol. 2009;66:843–857. doi: 10.1002/ana.21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Howng SY, Ptáèek LJ, Fu YH. miR-32 and its target SLC45A3 regulate the lipid metabolism of oligodendrocytes and myelin. Neuroscience. 2012;213:29–37. doi: 10.1016/j.neuroscience.2012.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K. Myelin biology and disorders. New York: Elsevier; 2004. Krabbe disease. [Google Scholar]
- Tappino B, Biancheri R, Mort M, Regis S, Corsolini F, Rossi A, Stroppiano M, Lualdi S, Fiumara A, Bembi B, Di Rocco M, Cooper DN, Filocamo M. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum Mutat. 2010;31:E1894–E1914. doi: 10.1002/humu.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA. NCBI Bookshelf. Bethesda, MD: NCBI; 2011a. Krabbe disease. [Google Scholar]
- Wenger DA. Leukodystrophies (International Review of Child Neurology Series) London: Mac Keith; 2011b. Krabbe disease (globoid cell leukodystrophy) pp. 90–105. [Google Scholar]
- Xu C, Sakai N, Taniike M, Inui K, Ozono K. Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correlation. J Hum Genet. 2006;51:548–554. doi: 10.1007/s10038-006-0396-3. [DOI] [PubMed] [Google Scholar]
- Yagi T, Matsuda J, Takikita S, Mohri I, Suzuki K, Suzuko K. Comparative clinico-pathological study of saposin-A-deficient (SAP-A−/−) and twitcher mice. J Neuropathol Exp Neurol. 2004;63:721–734. doi: 10.1093/jnen/63.7.721. [DOI] [PubMed] [Google Scholar]