

Abstract
Failure to clear amyloid-β (Aβ) from the brain is in part responsible for Aβ brain accumulation in Alzheimer's disease (AD). A critical protein for clearing Aβ across the blood–brain barrier is the efflux transporter P-glycoprotein (P-gp) in the luminal plasma membrane of the brain capillary endothelium. P-gp is reduced at the blood–brain barrier in AD, which has been shown to be associated with Aβ brain accumulation. However, the mechanism responsible for P-gp reduction in AD is not well understood. Here we focused on identifying critical mechanistic steps involved in reducing P-gp in AD. We exposed isolated rat brain capillaries to 100 nm Aβ40, Aβ40, aggregated Aβ40, and Aβ42. We observed that only Aβ40 triggered reduction of P-gp protein expression and transport activity levels; this occurred in a dose- and time-dependent manner. To identify the steps involved in Aβ-mediated P-gp reduction, we inhibited protein ubiquitination, protein trafficking, and the ubiquitin–proteasome system, and monitored P-gp protein expression, transport activity, and P-gp-ubiquitin levels. Thus, exposing brain capillaries to Aβ40 triggers ubiquitination, internalization, and proteasomal degradation of P-gp. These findings may provide potential therapeutic targets within the blood–brain barrier to limit P-gp degradation in AD and improve Aβ brain clearance.
SIGNIFICANCE STATEMENT The mechanism reducing blood–brain barrier P-glycoprotein (P-gp) in Alzheimer's disease is poorly understood. In the present study, we focused on defining this mechanism. We demonstrate that Aβ40 drives P-gp ubiquitination, internalization, and proteasome-dependent degradation, reducing P-gp protein expression and transport activity in isolated brain capillaries. These findings may provide potential therapeutic avenues within the blood–brain barrier to limit P-gp degradation in Alzheimer's disease and improve Aβ brain clearance.
Keywords: Alzheimer's disease, blood–brain barrier, P-glycoprotein, transporter, ubiquitin–proteasome system
Introduction
The etiology of excess amyloid-β (Aβ) brain accumulation in Alzheimer's disease (AD) is poorly understood. One contributing factor to Aβ brain accumulation is reduced Aβ clearance from the brain (Zlokovic and Frangione, 2003; Zlokovic, 2005). A neurovascular component in Aβ clearance has been implicated, suggesting that the blood–brain barrier is key in this process (Zlokovic and Frangione, 2003; Zlokovic, 2005). Studies show that P-glycoprotein (P-gp), an efflux transporter in the luminal plasma membrane of brain capillaries, is critical for clearing Aβ from brain into blood (Lam et al., 2001; Cirrito et al., 2005; Kuhnke et al., 2007; Hartz et al., 2010). We and others showed that P-gp protein expression and transport activity levels are reduced in brain capillaries from AD mouse models, suggesting a link between high Aβ levels and reduced levels of brain capillary P-gp (Hartz et al., 2010; Mehta et al., 2013).
Indeed, an inverse relationship between Aβ and P-gp protein levels has been observed in postmortem human brain samples (Wijesuriya et al., 2010; Jeynes and Provias, 2011), and capillary P-gp levels are significantly reduced and even undetectable in brain regions with high Aβ levels (Vogelgesang et al., 2002, 2004). Carrano et al. (2014) demonstrated that P-gp protein levels are reduced in human brain microvessels affected by high microvascular Aβ load in cerebral amyloid angiopathy. This was not observed in healthy individuals and AD patients without vascular amyloid deposits. Finally, PET studies in patients with mild and advanced AD confirmed that P-gp-mediated transport of [11C]-verapamil at the blood–brain barrier is lower than that in age-matched healthy individuals (van Assema et al., 2012; Deo et al., 2014). Collectively, these findings demonstrate an association between Aβ brain levels and blood–brain barrier P-gp. They suggest that P-gp is critical for Aβ clearance and that reduced P-gp protein expression and transport activity result in excessive Aβ brain accumulation.
The mechanism reducing blood–brain barrier P-gp in AD is poorly understood. In the present study, we focused on defining this mechanism. We demonstrate that Aβ40 drives P-gp ubiquitination-, internalization-, and proteasome-dependent degradation, reducing P-gp protein expression and transport activity in isolated brain capillaries. These findings may provide potential therapeutic avenues within the blood–brain barrier to limit P-gp degradation in AD and improve Aβ brain clearance.
Materials and Methods
Chemicals.
A11 antibody against Aβ40 oligomers, C219 antibody against P-gp, and antibodies against RAGE and β-actin were purchased from Abcam. Antibodies against LRP and ubiquitin, and purified IgG control antibody were obtained from Calbiochem-Novabiochem. OC antibody against Aβ40 was purchased from EMD Millipore, and antibodies 6E10 and 4G8, which recognize both Aβ40 and Aβ42, were obtained from Covance. Pierce Protein A/G Plus beaded agarose resin was obtained from Thermo Scientific, and the 20 S Proteasome Assay kit was purchased from Cayman Chemical. PYR-41 and bortezomib were from Selleckchem. [N-ε (4-Nitrobenzofurazan-7-yl)-d-Lys8]-cyclosporin A (NBD-CSA) was custom-synthesized by R. Wenger (Basel, Switzerland) (Schramm et al., 1995). PSC833 was a kind gift from Novartis. AggreSure β-Amyloid (1–40) (#AS-72215) was obtained from AnaSpec. Human Aβ1–40 (Aβ40), Aβ1–42 (Aβ42), and Aβ40–1reverse (Aβ40reverse) and all other chemicals were purchased from Sigma-Aldrich.
Animals.
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Kentucky and University of Minnesota (UK protocol #2014–1233, principal investigator A.M.S.H.; UMN protocol #0710A17842 and #1110A05865, principal investigator A.M.S.H.) and performed in strict accordance with Association for Assessment and Accreditation of Laboratory Animal Care regulations, the Department of Agriculture Animal Welfare Act, and the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Male Sprague Dawley rats (CD IGS rats; 275–300 g; 8–10 weeks) were purchased from Charles River. After shipping, rats were allowed to adapt to the new environment for a minimum of 5 d before they were used for experiments. Rats were kept under controlled environmental conditions (23°C, 35% relative humidity, 12 h dark-light cycle) with free access to tap water and standard rodent chow.
Aβ ELISA.
Total and oligomer-specific Aβ measurements were performed by ELISA (LeVine, 2004). Capture antibodies were immobilized on ELISA plates (Costar 9018). Total (monomeric + oligomeric) Aβ40 was determined by capturing with monoclonal antibody 6E10 (amino acids 3–8) and detecting with biotinylated monoclonal antibody 4G8 (amino acids 17–24). Oligomeric hAβ40 was determined by capturing with nonbiotinylated 4G8 and detecting with biotinylated 4G8 antibody. Capturing and detecting with the 4G8 antibodies to the same epitope recognize only oligomeric Aβ40 because the 4G8 epitope in the Aβ monomer is sequestered by the capture antibody and is not accessible to the biotinylated 4G8. The bound biotinylated 4G8 is quantified by streptavidin-HRP (Rockland Immunochemicals) and H2O2/tetramethylbenzidine peroxidase substrate.
Brain capillary isolation.
Rat brain capillaries were isolated as previously described (Hartz et al., 2004, 2008, 2010; Bauer et al., 2006). Briefly, rats were killed by CO2 inhalation and decapitated. Brains were removed, cleaned, dissected, and homogenized in ice-cold PBS (2.7 mm KCl, 1.46 mm KH2PO4, 136.9 mm NaCl, 8.1 mm Na2HPO4, supplemented with 5 mm d-glucose and 1 mm sodium pyruvate, pH 7.4). Ficoll PM400 was added to the homogenate to a final concentration of 15%, and the Ficoll-brain homogenate mix was centrifuged at 5800 × g for 20 min at 4°C. After resuspending the pellet in PBS containing 1% BSA (w/v), the capillary suspension was passed over a 40 ml glass bead column (0.4–0.6 mm; Sartorius StedimBiotech). Capillaries adhering to the glass beads were washed off the beads and collected in 1% BSA. Capillaries were washed with PBS (BSA-free) and used for experiments.
P-gp transport assay.
To determine P-gp transport activity, freshly isolated brain capillaries were incubated for 1 h at room temperature with the fluorescent P-gp-specific substrate NBD-CSA (2 μm in PBS buffer) (Hartz et al., 2004, 2008, 2010). For each treatment, images of 10 capillaries were acquired by confocal microscopy using a Zeiss LSM 710 inverted confocal microscope with a 40× 1.2 NA water-immersion objective and using the 488 nm line of an argon laser (Carl Zeiss). Images were analyzed by quantitating NBD-CSA fluorescence in the capillary lumen using ImageJ version 1.45s (Wayne Rasband, National Institutes of Health). Specific, luminal NBD-CSA fluorescence was taken as the difference between total luminal fluorescence and fluorescence in the presence of the P-gp-specific inhibitor PSC833 (5 μm) (Hartz et al., 2004, 2008, 2010).
Western blotting.
Protein expression levels in brain capillaries were analyzed by Western blotting as previously described (Hartz et al., 2004, 2008, 2010). Briefly, brain capillaries were homogenized in CelLytic MT cell lysis buffer (Sigma) containing Complete protease inhibitor (Roche). Homogenized brain capillary samples were centrifuged at 10,000 × g for 15 min at 4°C to remove nuclei, followed by a centrifugation of the denucleated supernatants at 100,000 × g for 90 min at 4°C to obtain brain capillary crude membranes. Brain capillary membranes were resuspended in buffer containing protease inhibitor and stored at −80°C until use.
Western blotting was done using the Invitrogen NuPage Bis-Tris electrophoresis and blotting system. After electrophoresis and protein transfer (30 V, 2 h), PVDF membranes (0.45 μm pore size) were blocked with protein-free T20 blocking buffer (Pierce) and incubated overnight with the primary antibody diluted in blocking buffer as indicated (C219, β-actin, ubiquitin, LRP, RAGE: all at 1 μg/ml). PVDF blotting membranes were washed and incubated with HRP-conjugated ImmunoPure secondary IgG (1:15,000; Pierce) for 1 h. Proteins were detected using SuperSignal West Pico Chemoluminescent Substrate (Pierce), and protein bands were visualized and imaged using a Bio-Rad Gel Doc 2000 gel documentation system with Quantity One software (Bio-Rad).
Dot blotting.
Dot blots were performed using the Whatman Minifold I 96-well system on Whatman Protran BA79 nitrocellulose membranes (pore size 0.1 μm, GE Healthcare). Blots were blocked, incubated, washed, and imaged as described in Western blotting.
Immunoprecipitation.
For immunoprecipitations, identical protein amounts (determined by Bradford assay) of Protein A/G bead-precleared capillary lysates were incubated with 5 μg P-gp antibody overnight at 4°C. The immune complexes were collected with Protein A/G agarose beads (2 h, room temperature), washed four times with RIPA buffer (150 mm NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0; Sigma-Aldrich) followed by a PBS wash. For ubiquitin immunoprecipitations, a ubiquitin enrichment kit from Pierce/Thermo Scientific was used according to the manufacturer's protocol. Immunoprecipitated proteins were eluted from agarose beads (IP: P-gp) or the ubiquitin affinity resin (IP: ubiquitin) with NuPAGE LDS sample buffer or Wes sample buffer and heated at 70°C for 10 min. IP samples eluted with NuPAGE LDS sample buffer were resolved by SDS-PAGE and analyzed by Western blotting as described above. Samples eluted with Wes sample buffer (see Fig. 5D; IP Ubiquitin) were analyzed and quantitated with a Simple Western assay using the Wes instrument (ProteinSimple). The Simple Western assay is a capillary electrophoresis technique that automates protein loading, separation, immunoprobing, washing, and detection, and allows absolute protein quantitation (O'Neill et al., 2006; Beccano-Kelly et al., 2014). For the assay, reagents of the Wes Master Kit (ProteinSimple) were used, and all steps of the assay were performed according to the manufacturer's protocol. A Wes Master Kit capillary cartridge and the prepared microplate were placed into the Wes instrument, which processed all assay steps automatically using default settings. Briefly, capillaries were loaded with both stacking and separation matrices followed by sample loading. During capillary electrophoresis, proteins were separated by size and then immobilized to the capillary wall. P-gp and ubiquitin were identified with primary antibodies against P-gp (C219, Thermo Scientific, 1:150) and ubiquitin (ubiquitin antibody, Abcam, 1:20), respectively, followed by immunodetection using Wes Master Kit HRP-conjugated anti-mouse secondary antibody and chemiluminescent substrate. Protein signals were automatically reported by the Compass software (version 2.6.5; ProteinSimple). Using Compass software, electropherograms were generated for each capillary (treatment group) and each protein (P-gp, ubiquitin), and the area under the curve was analyzed for P-gp. The area under the curve represents the signal intensity of the chemiluminescent reaction and is proportional to the amount of target protein in a respective capillary.
Figure 5.

Aβ40 triggers ubiquitination of P-gp. A, Isolated capillaries were exposed to 100 nm Aβ40. After 6 h, P-gp was immunoprecipitated and examined for P-gp and ubiquitin by Western blotting. Capillaries exposed to Aβ40 showed increased ubiquitination. IgG control: capillary lysate sample plus IgG antibody; negative control: capillary lysate sample but no primary antibody. B, Cross-experiment showing immunoprecipitation of ubiquitin followed by Western blot analysis of P-gp and ubiquitin. Increased ubiquitination of P-gp was found in capillaries exposed to Aβ40. C, The ubiquitin ligase inhibitor PYR-41 (10 μm) prevented Aβ40-mediated reduction of P-gp expression (Western blot, β-actin was used as protein loading control) and transport activity (luminal NBD-CSA fluorescence). D, P-gp and ubiquitin were immunoprecipitated and examined for P-gp and ubiquitin by Western blotting and simple Western assay using the Wes instrument. PYR-41 prevented P-gp ubiquitination in capillaries exposed to 100 nm Aβ40. Data are mean ± SEM (n = 10 capillaries per treatment group from one brain capillary isolation; pooled tissue from 10 rats). Units are arbitrary fluorescence units (scale, 0–255). ***p < 0.001, significantly lower than control.
20S proteasome activity assay.
We used the 20S proteasome assay kit from Cayman Chemical and followed the protocol provided by the manufacturer. Isolated capillaries were lysed in 20S proteasomal lysis buffer. Capillary lysate was incubated with 10 μl assay buffer and 10 μl proteasome substrate (SUC-LLVY-AMC) for 1 h at 37°C. Fluorescence intensity (ex 360 nm, em 480 nm) was measured using a microplate reader.
Statistical analysis.
Data are mean ± SEM. ANOVA or a two-tailed unpaired Student's t test was used to evaluate differences between controls and treated groups using Microsoft Excel 2010 and GraphPad Prism (version 6.01); differences were considered to be statistically significant when p < 0.05.
Results
Identification of Aβ forms
For the experiments described in the present study, we used 100 nm human Aβ40 dissolved in PBS supplemented with 5 mm d-glucose and 1 mm sodium pyruvate (pH 7.4, 25°C ± 0.5°C). We used several methods to identify the form in which Aβ40 was present in this solution. These involved measurements of Aβ40 monomer versus oligomer 0, 1, 6, and 24 h after preparation of the Aβ40 working solution. With Western blotting (LDS-PAGE under reducing conditions), we detected a band at 4 kDa for Aβ40 monomer with 4G8 antibody (recognizes Aβ17–24) and a weaker signal with 6E10 antibody (recognizes Aβ1–17; Fig. 1A). However, by Western blotting, we detected no Aβ40 oligomer bands using A11 antibody (detects amyloid oligomers) and faint bands using OC antibody (detects amyloid fibrils). Dot blot analysis using the same antibodies indicated that Aβ40 monomer and low levels of Aβ40 oligomer were present in the solution, but the amount of Aβ40 decreased over 24 h (Fig. 1B). By sandwich ELISA, we found a decreasing amount of total Aβ40 over time (Fig. 1C), which is likely due to nonspecific binding of the Aβ40 monomer to the test tube. By an oligomer-specific configuration sandwich ELISA, we observed a very low Aβ40 oligomer signal. The total amount of all Aβ40 species detectable in solution decreased dramatically over time. The lack of oligomeric Aβ40 under these conditions is not surprising as synthetic Aβ40 oligomerizes weakly in vitro (LeVine, 2004). Thus, the 100 nm Aβ40 solution used for our experiments primarily consisted of monomeric Aβ40. In an attempt to determine the size distribution of the various Aβ40 forms in the solution/dispersion, we performed nanoparticle tracking analysis using a NanoSight LM10 (Malvern Instruments). However, the Aβ40 concentration (100 nm) was too low, and the analysis was inconclusive (data not shown). Overall, these results are consistent with other findings showing that Aβ40 remains predominantly monomeric in aqueous buffer solutions, such as PBS (Stine et al., 2011).
Figure 1.
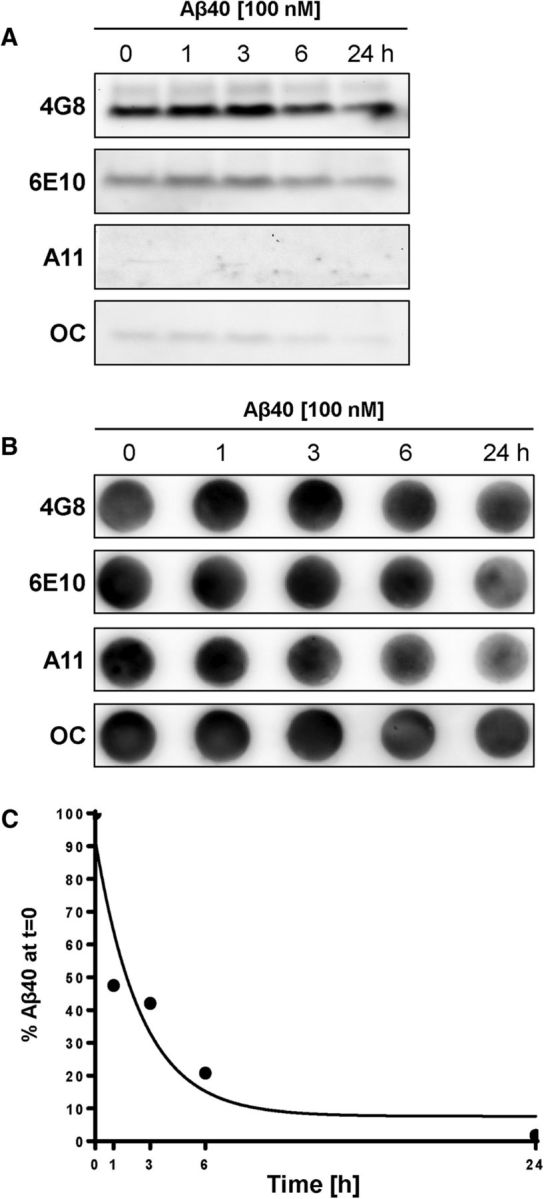
Determination of Aβ40 forms. A, Western blot showing bands for Aβ40 at 4 kDa with 4G8 antibody (recognizes Aβ17–24), weak bands at 4 kDa with 6E10 antibody (recognizes Aβ1–17), and no bands with A11 antibody (detects amyloid oligomers) and faint bands with OC antibody (detects amyloid fibrils) 0, 1, 6, and 24 h after making the 100 nm Aβ40 working solution in PBS. B, Dot blots showing low Aβ40 levels with 4G8, 6E10, A11, and OC antibodies that are decreasing over the course of 24 h. C, Oligomer-specific configuration ELISA shows that the total amount of Aβ40 in solution decreases over time.
Aβ40 reduces P-gp protein expression and transport activity
We measured P-gp protein expression and transport activity levels in freshly isolated rat brain capillaries that were exposed to 100 nm human Aβ40 for 1 or 6 h. After removing Aβ40, P-gp transport activity was determined by incubating brain capillaries with the P-gp-specific fluorescent substrate NBD-CSA (2 μm) and by measuring fluorescence of this dye in the capillary lumens by confocal microscopy and quantitative image analysis as described previously (Hartz et al., 2008, 2010). Figure 2 depicts representative images of a control brain capillary (no Aβ40) and a brain capillary exposed to Aβ40 for 6 h. Luminal NBD-CSA fluorescence was substantially decreased in capillaries exposed to Aβ40 compared with control capillaries (Fig. 2A,B). Image analysis revealed a reduction of 69.4 ± 6.2% (average of 10 independent experiments) in luminal NBD-CSA in brain capillaries exposed to Aβ40, indicating that Aβ40 reduced P-gp transport activity (Fig. 2C). This result is consistent with our previous data in isolated brain capillaries from wild-type and transgenic hAPP mice, where we observed a comparable reduction of P-gp transport activity levels with Aβ40 exposure (Hartz et al., 2010; their Fig. 2A,B). In addition to reduced P-gp transport activity, we also found a reduction of 40.3 ± 6.2% (average of 10 independent experiments) in P-gp protein expression in brain capillary membranes isolated from Aβ40-treated brain capillaries (6 h treatment). Aβ40 exposure did not affect protein expression levels of LRP1 (100 ± 4% compared with control, average of three independent experiments) and RAGE (95 ± 5.6% compared with control, average of three independent experiments; Fig. 2D). Dose–response experiments showed that both 1 and 6 h exposure of brain capillaries to 10, 50, and 100 nm Aβ40 reduced P-gp transport activity in a concentration-dependent manner, whereas only the 6 h exposure of capillaries to Aβ40 reduced P-gp protein expression levels (Fig. 2E,F). It is unlikely that Aβ40 acts as P-gp inhibitor because Aβ40-mediated reduction the effect can be abolished by inhibitors of the ubiquitin–proteasome pathway (data not shown). Thus, nanomolar concentrations of human Aβ40 trigger reduction of both P-gp expression and transport activity levels in isolated rat brain capillaries, but the time courses of loss of transport activity and loss of transporter protein expression levels differed.
Figure 2.
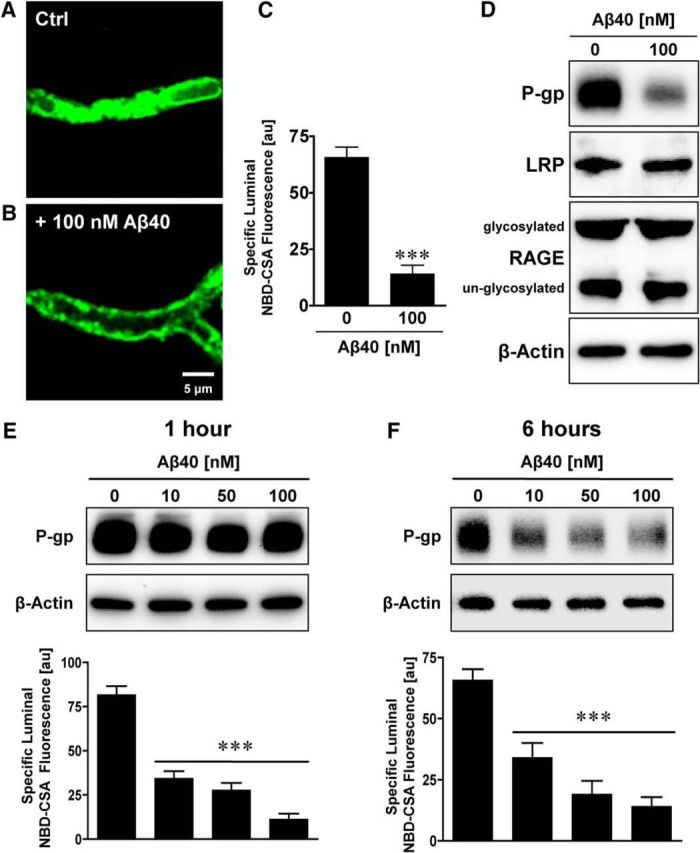
Aβ40 reduces P-gp protein expression and transport function. Representative confocal microscopy images of isolated brain capillaries that were exposed to 100 nm Aβ40 for 6 h and then exposed to the fluorescent P-gp substrate NBD-CSA (marker for P-gp transport activity). A, Representative image of an isolated brain capillary after 1 h of exposure to 2 μm NBD-CSA, showing steady-state NBD-CSA fluorescence. B, NBD-CSA fluorescence in the capillary lumen is reduced in capillaries that were exposed to Aβ40 for 6 h, indicating a decrease in P-gp transport activity. C, Specific NBD-CSA fluorescence in the capillary lumen was obtained through analysis of the confocal images and shows decreased luminal fluorescence in capillaries exposed to 100 nm Aβ40. D, Western blot showing that 100 nm Aβ40 reduces P-gp protein expression but has no effect on LRP or RAGE protein expression levels. E, Western blot showing that P-gp protein levels in capillaries exposed to 10–100 nm Aβ40 for 1 h remain at control levels. In contrast, P-gp transport activity levels decreased. F, Western blot showing that P-gp protein and transport activity levels decreased in a concentration-dependent manner after 6 h of Aβ40 exposure. β-Actin was used as protein loading control for Western blots. C, E, F, Data were obtained through analysis of the confocal images. Specific NBD-CSA fluorescence is the difference between total luminal fluorescence and fluorescence in the presence of the specific P-gp inhibitor PSC833, representing specific P-gp transport activity. Data are mean ± SEM (n = 10–15 capillaries per treatment group from one brain capillary isolation; pooled tissue from 10 rats). Units are arbitrary fluorescence units (scale, 0–255). ***p < 0.001, significantly lower than control.
To determine the effect of 100 nm Aβ40 on P-gp transport activity over time, we preincubated isolated rat brain capillaries with 2 μm NBD-CSA for 1 h to a steady state before adding Aβ40, and then monitored brain capillaries over 6 h. Figure 3A shows that luminal NBD-CSA fluorescence in control capillaries (no Aβ40) did not change over the 6 h time course (Hartz et al., 2010). In contrast, addition of Aβ40 reduced P-gp transport activity levels within 15 min. After 45 min, P-gp transporter activity was substantially decreased to a level that was comparable with that seen when P-gp was inhibited with PSC833; P-gp transport activity remained at this low level for the entire 6 h of Aβ40 exposure. Removing Aβ40 from the buffer medium after 45 min restored P-gp transport activity to control levels over the course of 6 h.
Figure 3.

Time course of Aβ40-mediated P-gp reduction. A, The effect of Aβ40 on P-gp transport activity is reversible. Capillaries were loaded for 60 min to steady state with 2 μm NBD-CSA. When 100 nm Aβ40 was added to the buffer (time 0 on graph), P-gp transport activity decreased rapidly; P-gp activity stayed decreased in capillaries exposed to Aβ40 for 6 h but recovered completely when Aβ40 was removed at time point 45 min. Western blot showing (B) no change of P-gp protein expression after 45 min exposure to Aβ40 and (C) no change of P-gp expression in capillaries that were first exposed to Aβ40 for 45 min followed by 5¼ h in Aβ40-free buffer (total experiment duration of 6 h). D, Consistent with Figure 2, Western blot showing reduced P-gp protein expression after 6 h exposure to Aβ40. β-Actin was used as a protein loading control for all Western blots. Data are mean ± SEM (n = 10–15 capillaries per treatment group from one brain capillary isolation; pooled tissue from 10 rats). Units are arbitrary fluorescence units (scale, 0–255). *p < 0.05, significantly lower than control. **p < 0.01, significantly lower than control. ***p < 0.001, significantly lower than control.
P-gp protein expression levels were unchanged after a 45 min Aβ40 exposure (Fig. 3B) and remained unchanged when Aβ40 was removed from the medium after 45 min and incubation in Aβ40-free medium continued for an additional 5¼ h (Fig. 3C). In contrast, brain capillaries that were exposed to Aβ40 for a total of 6 h exhibited reduced P-gp protein expression levels (Fig. 3D). The Aβ-mediated reduction of P-gp expression and transport activity levels was not reversible when Aβ40 was not removed after 45 min. Thus, Aβ40-mediated reduction of P-gp transport activity levels occurs within 15 min of initial Aβ40 exposure followed by a reduction in P-gp protein expression levels. The initial reduction in P-gp expression and transport activity levels is reversible when Aβ40 exposure of brain capillaries is limited to ≤45 min but becomes irreversible when Aβ40 remains in the medium for >45 min. This indicates that the transporter is degraded after 45 min of Aβ40 exposure, after which P-gp expression and transport activity cannot be restored.
In a series of control experiments, we exposed isolated rat brain capillaries to the aggregated form of Aβ40, to a peptide with the reverse sequence of that for Aβ40 (Aβ40reverse), and to Aβ42. In contrast to monomeric Aβ40, 6 h exposure of isolated rat brain capillaries to aggregated Aβ40 did not affect P-gp protein expression and transport activity (Fig. 4A,B). LRP and RAGE protein expression levels also remained unchanged by the aggregated form of Aβ40. When we exposed isolated rat brain capillaries to Aβ40reverse (Fig. 4C) or to human Aβ42 (Fig. 4D) for 6 h, P-gp protein expression and transport activity did not change either. These data indicate that, under these experimental conditions (100 nm of respective human Aβ species for 6 h), reduction in P-gp protein expression and transport activity levels in isolated rat brain capillaries is specific to Aβ40, whereas aggregated Aβ40, Aβ40reverse, or Aβ42 had no effect.
Figure 4.

Aggregated Aβ40, reverse Aβ40, and Aβ42 have no effect on P-gp. Exposing capillaries to 100 nm Aβ40 decreases P-gp protein expression (Western blot) and transport activity (luminal NBD-CSA fluorescence). Aggregated Aβ40 (A, B), Aβ40reverse (reverse amino acid sequence of Aβ40) (C), and Aβ42 (D) had no effect on P-gp expression or activity. β-Actin was used as protein loading control for Western blots. Data are mean ± SEM (n = 10 capillaries per treatment group from one brain capillary isolation; pooled tissue from 10 rats). Units are arbitrary fluorescence units (scale, 0–255). ***p < 0.001, significantly lower than control.
Aβ40 activates the ubiquitin–proteasome pathway
To determine whether exposure to human Aβ40 triggers the reduction in P-gp protein expression and transport activity levels by activating the ubiquitin–proteasome pathway, we exposed isolated rat brain capillaries to 100 nm Aβ40 for 6 h, immunoprecipitated P-gp or ubiquitin from capillary lysates, and separated protein samples by SDS-PAGE and Western blotting. The Western blot in Figure 5A (P-gp immunoprecipitation) shows increased ubiquitin levels in brain capillaries treated with Aβ40. In the cross-experiment (Fig. 5B) where we immunoprecipitated ubiquitin, we observed increased P-gp protein levels associated with ubiquitin after Aβ40 exposure. We detected no signal in the IgG controls (capillary lysate sample plus IgG antibody) and negative controls (capillary lysate sample but no primary antibody; Fig. 5A), indicating that the observed signals are specific for P-gp and ubiquitin. Consistent with increased P-gp ubiquitination, PYR-41, a cell-permeable inhibitor of ubiquitin ligase E1, abolished the Aβ40-mediated reduction of P-gp protein expression and transport activity levels (Fig. 5C), as well as ubiquitination of P-gp (Fig. 5D). Using the Simple Western assay system (ProteinSimple), we determined the amount of P-gp that was immunoprecipitated by ubiquitin: control samples contained 4.3 ng P-gp protein, samples from capillaries exposed to Aβ40 contained 6.9 ng P-gp protein, and samples treated with both Aβ40 and PYR-41 contained 3.3 ng P-gp protein based on a P-gp protein standard curve. Data from six independent immunoprecipitation experiments indicate that Aβ40 increased the amount of ubiquitinated P-gp by 53 ± 9.3%. Together, these data demonstrate that Aβ40 exposure increases P-gp ubiquitination in isolated brain capillaries.
Next, we investigated whether Aβ40 triggers internalization of P-gp, which is the step following ubiquitination in the proteasome pathway. We exposed isolated rat brain capillaries to Aβ40 for 6 h with or without the microtubule inhibitors, brefeldin A and nocodazole, to stop intracellular trafficking. Figure 6 shows that both inhibitors blocked Aβ40-mediated reduction of P-gp protein expression and transport activity levels. These data suggest that blocking intracellular trafficking prevents P-gp internalization from the plasma membrane into the cell.
Figure 6.

Microtubule inhibitors prevent Aβ40-mediated P-gp reduction. The microtubule inhibitors brefeldin A (1 μg/ml) (A) and nocodazole (100 nm) (B) prevent Aβ40-mediated reduction of P-gp protein expression (Western blot) and transport activity (luminal NBD-CSA fluorescence). β-Actin was used as protein loading control for Western blots. Data are mean ± SEM (n = 10 capillaries per treatment group from one brain capillary isolation; pooled tissue from 10 rats). Units are arbitrary fluorescence units (scale, 0–255). ***p < 0.001, significantly lower than control.
To verify that the final step of the ubiquitin–proteasome pathway was involved in P-gp reduction, we determined whether P-gp was subject to proteasomal or lysosomal degradation. We exposed isolated rat brain capillaries to Aβ40 with or without the proteasome inhibitors lactacystin, MG-132, and bortzomib, or the lysosomal vacuolar type H+-ATPase inhibitor bafilomycin A1. All three proteasome inhibitors prevented Aβ40-mediated reduction of P-gp protein expression and transport activity levels (Fig. 7A–C), whereas the lysosomal inhibitor bafilomycin A1 had no such effect (Fig. 7D). Consistent with this, we found increased 20S proteasomal activity in isolated rat brain capillaries that were exposed to Aβ40 for 6 h (Fig. 7E). The inhibitors themselves had no effect on P-gp protein expression and transport function (Fig. 7F). This suggests that, following ubiquitination and internalization, P-gp is directed to the proteasome for degradation.
Figure 7.

Inhibition of the proteasome prevents Aβ40-mediated P-gp reduction. The proteasome inhibitors lactacystin (10 μm) (A), MG-132 (150 nm) (B), and bortezomib (100 nm) (C) abolish Aβ40-mediated reduction of P-gp expression (Western blot) and transport activity (luminal NBD-CSA fluorescence). D, The lysosomal inhibitor bafilomycin A1 (10 nm) did not prevent P-gp reduction triggered by Aβ40. E, The 20S proteasome activity was increased in isolated rat brain capillaries exposed to Aβ40 for 6 h. β-Actin was used as protein loading control for Western blots. F, The inhibitors PYR-41, nocodazole, and bortzomib had no effect on P-gp protein expression and transport activity. Data are mean ± SEM (n = 10 capillaries per treatment group from one brain capillary isolation; pooled tissue from 10 rats). Units are arbitrary fluorescence units (scale, 0–255). ***p < 0.001, significantly lower or higher than control.
Together, the present data demonstrate that exposing brain capillaries to Aβ40 triggers ubiquitination, internalization, and proteasomal degradation of P-gp at the blood–brain barrier (Fig. 8).
Figure 8.

Proposed signaling pathway. Based on the data presented here, we propose the following signaling pathway: Aβ40 triggers: (1) ubiquitination, (2) internalization, and (3) proteasomal degradation of blood–brain barrier P-gp, which results in reduced P-gp protein expression and transport function.
Discussion
We and others previously showed that P-gp protein expression and transport activity are reduced in human Aβ-overexpressing AD mouse models (Hartz et al., 2010; Mehta et al., 2013). These data are supported by studies with postmortem human brain tissue that demonstrate P-gp expression and activity levels are lower in the neurovasculature from patients with mild cognitive impairment and AD compared with those in age-matched, healthy individuals (Wijesuriya et al., 2010; Jeynes and Provias, 2011; van Assema et al., 2012; Deo et al., 2014). These studies suggest a link between high Aβ levels and reduced brain capillary P-gp in AD. However, the cellular mechanism that drives P-gp reduction in the blood–brain barrier in AD has not been unraveled.
In the present study, we demonstrate that nanomolar concentrations of Aβ40 decrease P-gp expression and transport activity in rat brain capillaries and we begin to describe the mechanism through which this occurs. Our data show that exposing brain capillaries to monomeric Aβ40 decreases P-gp protein expression and transport activity levels in a time- and concentration-dependent manner (Figs. 1–3). At 100 nm, neither aggregated Aβ40, nor Aβ40reverse, nor Aβ42 had any effect (Fig. 4). Aβ40 concentrations measured in postmortem brain tissue from AD patients are in the mid to high nanomolar range, similar to the Aβ40 concentrations used in the present study (Gravina et al., 1995; Hardy and Selkoe, 2002). Immunoprecipitation demonstrated that Aβ40 triggers P-gp ubiquitination, and experiments with microtubule and proteasomal inhibitors indicate that P-gp is internalized and degraded by the proteasome (Figs. 6, 7). Consistent with this, 20S proteasomal activity in capillaries was increased by Aβ40. Based on these data, we propose that Aβ40 activates the ubiquitin–proteasome system at the blood–brain barrier, resulting in P-gp degradation and, thus, reduction in P-gp protein expression and activity levels (Fig. 8). These data for intact brain capillaries demonstrate a cause-and-effect relationship between exposure to nanomolar concentrations of Aβ40 and reduced P-gp expression and transport activity.
Recent reports suggest that Aβ42 could also be involved in the reduction of blood–brain barrier P-gp. Brenn et al. (2011) implanted an osmotic pump subcutaneously into FVB mice for 24 h peripheral administration of Aβ40 and Aβ42, and showed that Aβ42, but not Aβ40, slightly reduced P-gp mRNA levels in the brain, whereas P-gp protein expression levels remained unchanged. Carrano et al. (2014) exposed a human cerebral microvascular endothelial cell line (hCMEC/D3) to various Aβ forms for 24 h and found that oligomeric Aβ42, but not fibrillary Aβ42 or Aβ40, reduced P-gp mRNA levels. Park et al. (2014) showed that Aβ42-mediated reduction in P-gp protein expression in bEnd.3 cells depended on RAGE and that a yet to-be-identified astrocyte-derived factor attenuated the Aβ-induced decrease in P-gp expression in a bEnd.3-astrocyte coculture. However, the effects observed in these studies may not be a realistic reflection of the situation in the AD brain because they may be artifacts of administering Aβ42 into the periphery (Brenn et al., 2011), of using immortalized cultured cells (Carrano et al., 2014), and of applying excessive Aβ42 concentrations (Park et al., 2014). In addition, it is possible that the Aβ42 effect on blood–brain barrier P-gp is time-, concentration-, and context-dependent, which could explain some of the discrepancies between our data and those by other groups described above.
Ubiquitination followed by lysosomal or proteasomal degradation plays a crucial role in regulating expression and function of membrane proteins (Ciechanover, 1994; Ciechanover and Iwai, 2004). P-gp is a 180 kDa membrane protein that undergoes endocytosis and recycling, and its half-life has been determined in multidrug-resistant cancer cell lines to be ∼14–17 h (Muller et al., 1995; Kim et al., 1997). P-gp trafficking, localization, stability, and function are, in part, regulated by the ubiquitin–proteasome pathway (Loo and Clarke, 1999). One protease-cleavage site of P-gp is in the first extracellular loop near the glycosylation site (Arg113); two more sites (Arg680 and Leu682) are located in the linker region joining the two halves of P-gp (Loo and Clarke, 1998; Nuti and Rao, 2002). Zhang et al. (2004) were the first to show that P-gp stability is regulated by the ubiquitin–proteasome pathway. These authors found that inhibition of N-glycosylation enhances P-gp ubiquitination and degradation, thereby reducing transporter activity (Zhang et al., 2004). In a recent study, Katayama et al. (2013) showed that the ubiquitin E3 ligase subunit FBXO15 and the ubiquitin-conjugating enzyme E2 R1 regulate P-gp expression in human colorectal cancer cell lines. The authors demonstrated that FBXO15 knockdown increases cell-surface P-gp expression and enhances P-gp-mediated efflux, suggesting that the ubiquitin–proteasome system regulates P-gp expression and activity levels. Our study indicates that Aβ40 triggers P-gp ubiquitination, internalization, and proteasome-dependent degradation leading to reduction of P-gp expression and activity levels in brain capillaries (Figs. 5–7). We show that the lysosomal inhibitor bafilomycin A does not attenuate Aβ40-mediated P-gp reduction, suggesting that proteasomal degradation, rather than lysosomal degradation, reduces P-gp levels (Fig. 7). Recently, we demonstrated that P-gp is a substrate for the ubiquitin ligase NEDD4-1, which is increased in brain capillaries from 9-month-old hAPP mice (Tg2576 model) and may be involved in Aβ-mediated P-gp reduction (Akkaya et al., 2015). A recent study by Aida et al. (2014) on the ABC transporters BSEP and MRP2 suggests that transporter degradation is multifaceted and complex. The authors propose that, in addition to the ubiquitin–proteasome and lysosomal pathways, proteins destined for degradation can also be routed to a ubiquitination-independent, and bafilomycin-insensitive pathway. Although our data are conclusive, several mechanistic details remain to be determined. For example, it is unknown whether P-gp is mono- or poly-ubiquitinated, which ubiquitin ligases catalyze this step, and which signals drive Aβ40 activation of the ubiquitin–proteasome pathway.
The ubiquitin–proteasome system is critical for normal brain function because it regulates proper protein turnover, development, and differentiation, signals transduction, and is responsible for the disposal of unneeded waste proteins (Yi and Ehlers, 2007). In AD, however, the ubiquitin–proteasome system is dysfunctional and is suspected to contribute to the pathogenesis of the disease (Oddo, 2008; Hong et al., 2014). In this regard, the ubiquitin protein ligases, UBE2 and UBE3, are upregulated in AD, and elevated levels of ubiquitinated proteins have been detected in neurofibrillary tangles and Aβ plaques in postmortem AD brains (Mori et al., 1987; Perry et al., 1987; Keck et al., 2003). Overall, the majority of available studies suggest that the ubiquitin–proteasome system is impaired and defective in neurons in the late-stage AD brain from deceased patients. In contrast, little is known about changes in the subset of the ubiquitin–proteasome system at the blood–brain barrier. In this regard, Deane et al. (2004) showed Aβ-mediated proteasomal degradation of LRP in brain capillaries from 6- to 9-month-old, transgenic hAPP mice that were cognitively impaired. In isolated brain capillaries from nontransgenic wild-type mice, we found that Aβ40 increases proteasome activity and ubiquitinylated P-gp levels, resulting in reduced P-gp expression and activity levels (Figs. 5–7). Thus, our data support a possible role for the ubiquitin–proteasome system in reducing P-gp in AD at the level of the blood–brain barrier.
The present experiments show that the time courses of Aβ40-induced loss of P-gp transport activity and protein expression do not coincide. Transport activity decreased rapidly with Aβ40 exposure, whereas the reduction in transporter expression was delayed (at least past 45 min of exposure). Removal of Aβ40 early in the time course fully restored transport activity and did not alter transporter expression in the long term. In 6 h experiments, pharmacologically inhibiting P-gp ubiquitination, trafficking, or degradation at the proteasome abolished the loss of transporter activity and transporter protein expression. One would expect this result when ubiquitination was inhibited, but the lack of effect of Aβ40 exposure when trafficking and proteasomal degradation were inhibited suggests a more complex scenario than that depicted in Figure 8. One possibility for P-gp is that both ubiquitination and trafficking are reversible when degradation does not occur over a period of hours. These aspects of the mechanism require further study, as does the mechanism by which Aβ40 signals P-gp ubiquitination, internalization, and proteasome activation.
Collectively, our findings and those from other groups indicate that P-gp in the blood–brain barrier is part of the Aβ brain clearance system and that P-gp protein expression and transport activity levels are reduced in AD (Lam et al., 2001; Cirrito et al., 2005; Kuhnke et al., 2007; Hartz et al., 2010; Mehta et al., 2013). We also demonstrate in rat brain capillaries that Aβ40 triggers reduction in P-gp expression and activity levels through the ubiquitin–proteasome system, which, for the first time, provides a plausible mechanism how P-gp is reduced in AD. This implies a pernicious positive feedback loop where reduced P-gp levels lead to increased Aβ brain accumulation, which in turn drives further P-gp degradation, leading to even greater increases in Aβ brain levels and eventually AD pathology. Clearly, this scenario could contribute to the progressive nature of AD. In this regard, our data imply that the ubiquitin–proteasome system may potentially serve as an early therapeutic target to protect P-gp from degradation and limit Aβ from accumulation in the brain, thereby delaying the onset and slowing the progression of AD. Current studies are underway to test the effect of PYR-41, nocodazole, and bortezomib in an AD mouse model in vivo.
Footnotes
This work was supported by National Institute on Aging Grant 1R01AG039621 to A.M.S.H., 3M Science and Technology Doctoral Fellowship to A.W., 3M Grant P01-AG005119-20 to H.L., and the Division of Intramural Research at National Institute of Environmental Health Sciences/National Institutes of Health to D.S.M. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health. We thank Stephanie Edelmann, Ralf Rempe, Yu Zhong, and Tal Ashkenazi-Frolinger for proofreading the manuscript and editorial assistance; Britt Johnson, Kevin Viken, and Emma Soldner for technical assistance; and Lynne Bemis and Evan Odean for the NanoSight analysis.
The authors declare no competing financial interests.
References
- Aida K, Hayashi H, Inamura K, Mizuno T, Sugiyama Y. Differential roles of ubiquitination in the degradation mechanism of cell surface-resident bile salt export pump and multidrug resistance-associated protein 2. Mol Pharmacol. 2014;85:482–491. doi: 10.1124/mol.113.091090. [DOI] [PubMed] [Google Scholar]
- Akkaya BG, Zolnerciks JK, Ritchie TK, Bauer B, Hartz AMS, Sullivan JA, Linton KJ. The multidrug resistance pump ABCB1 is a substrate for the ubiquitin ligase Nedd4–1. Mol Membr Biol. 2015;32:39–45. doi: 10.3109/09687688.2015.1023378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer B, Yang X, Hartz AM, Olson ER, Zhao R, Kalvass JC, Pollack GM, Miller DS. In vivo activation of human pregnane X receptor tightens the blood–brain barrier to methadone through P-glycoprotein up-regulation. Mol Pharmacol. 2006;70:1212–1219. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- Beccano-Kelly DA, Kuhlmann N, Tatarnikov I, Volta M, Munsie LN, Chou P, Cao LP, Han H, Tapia L, Farrer MJ, Milnerwood AJ. Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front Cell Neurosci. 2014;8:301. doi: 10.3389/fncel.2014.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenn A, Grube M, Peters M, Fischer A, Jedlitschky G, Kroemer HK, Warzok RW, Vogelgesang S. Beta-amyloid downregulates MDR1-P-glycoprotein (Abcb1) expression at the blood–brain barrier in mice. Int J Alzheimers Dis. 2011;2011:690121. doi: 10.4061/2011/690121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A, Snkhchyan H, Kooij G, van der Pol S, van Horssen J, Veerhuis R, Hoozemans J, Rozemuller A, de Vries HE. ATP-binding cassette transporters P-glycoprotein and breast cancer related protein are reduced in capillary cerebral amyloid angiopathy. Neurobiol Aging. 2014;35:565–575. doi: 10.1016/j.neurobiolaging.2013.09.015. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin–proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Iwai K. The ubiquitin system: from basic mechanisms to the patient bed. IUBMB Life. 2004;56:193–201. doi: 10.1080/1521654042000223616. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM. P-glycoprotein deficiency at the blood–brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Deo AK, Borson S, Link JM, Domino K, Eary JF, Ke B, Richards TL, Mankoff DA, Minoshima S, O'Sullivan F, Eyal S, Hsiao P, Maravilla K, Unadkat JD. Activity of P-glycoprotein, a beta-amyloid transporter at the blood–brain barrier, is compromised in patients with mild Alzheimer disease. J Nucl Med. 2014;55:1106–1111. doi: 10.2967/jnumed.113.130161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, Suzuki N, Younkin SG. Amyloid beta protein (A beta) in Alzheimer's disease brain: biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43) J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Fricker G, Miller DS. Rapid regulation of P-glycoprotein at the blood–brain barrier by endothelin-1. Mol Pharmacol. 2004;66:387–394. doi: 10.1124/mol.104.001503. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Block ML, Hong JS, Miller DS. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood–brain barrier. FASEB J. 2008;22:2723–2733. doi: 10.1096/fj.08-106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Miller DS, Bauer B. Restoring blood–brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer's disease. Mol Pharmacol. 2010;77:715–723. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L, Huang HC, Jiang ZF. Relationship between amyloid-beta and the ubiquitin–proteasome system in Alzheimer's disease. Neurol Res. 2014;36:276–282. doi: 10.1179/1743132813Y.0000000288. [DOI] [PubMed] [Google Scholar]
- Jeynes B, Provias J. An investigation into the role of P-glycoprotein in Alzheimer's disease lesion pathogenesis. Neurosci Lett. 2011;487:389–393. doi: 10.1016/j.neulet.2010.10.063. [DOI] [PubMed] [Google Scholar]
- Katayama K, Noguchi K, Sugimoto Y. FBXO15 regulates P-glycoprotein/ABCB1 expression through the ubiquitin–proteasome pathway in cancer cells. Cancer Sci. 2013;104:694–702. doi: 10.1111/cas.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- Kim H, Barroso M, Samanta R, Greenberger L, Sztul E. Experimentally induced changes in the endocytic traffic of P-glycoprotein alter drug resistance of cancer cells. J Physiol. 1997;273:C687–C702. doi: 10.1152/ajpcell.1997.273.2.C687. [DOI] [PubMed] [Google Scholar]
- Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I, Cascorbi I, Walker LC, Kroemer HK, Warzok RW, Vogelgesang S. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer's amyloid-beta peptides: implications for the mechanisms of Abeta clearance at the blood–brain barrier. Brain Pathol. 2007;17:347–353. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB. beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001;76:1121–1128. doi: 10.1046/j.1471-4159.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- LeVine H., 3rd Alzheimer's beta-peptide oligomer formation at physiologic concentrations. Anal Biochem. 2004;335:81–90. doi: 10.1016/j.ab.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. Quality control by proteases in the endoplasmic reticulum: removal of a protease-sensitive site enhances expression of human P-glycoprotein. J Biol Chem. 1998;273:32373–32376. doi: 10.1074/jbc.273.49.32373. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J Biol Chem. 1999;274:24759–24765. doi: 10.1074/jbc.274.35.24759. [DOI] [PubMed] [Google Scholar]
- Mehta DC, Short JL, Nicolazzo JA. Altered brain uptake of therapeutics in a triple transgenic mouse model of Alzheimer's disease. Pharm Res. 2013;30:2868–2879. doi: 10.1007/s11095-013-1116-2. [DOI] [PubMed] [Google Scholar]
- Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- Muller C, Laurent G, Ling V. P-glycoprotein stability is affected by serum deprivation and high cell density in multidrug-resistant cells. J Cell Physiol. 1995;163:538–544. doi: 10.1002/jcp.1041630314. [DOI] [PubMed] [Google Scholar]
- Nuti SL, Rao US. Proteolytic cleavage of the linker region of the human P-glycoprotein modulates its ATPase function. J Biol Chem. 2002;277:29417–29423. doi: 10.1074/jbc.M204054200. [DOI] [PubMed] [Google Scholar]
- Oddo S. The ubiquitin–proteasome system in Alzheimer's disease. J Cell Mol Med. 2008;12:363–373. doi: 10.1111/j.1582-4934.2008.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill RA, Bhamidipati A, Bi X, Deb-Basu D, Cahill L, Ferrante J, Gentalen E, Glazer M, Gossett J, Hacker K, Kirby C, Knittle J, Loder R, Mastroieni C, Maclaren M, Mills T, Nguyen U, Parker N, Rice A, Roach D, et al. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proc Natl Acad Sci U S A. 2006;103:16153–16158. doi: 10.1073/pnas.0607973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park R, Kook SY, Park JC, Mook-Jung I. Abeta1–42 reduces P-glycoprotein in the blood–brain barrier through RAGE-NF-kappaB signaling. Cell Death Dis. 2014;5:e1299. doi: 10.1038/cddis.2014.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci U S A. 1987;84:3033–3036. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm U, Fricker G, Wenger R, Miller DS. P-glycoprotein-mediated secretion of a fluorescent cyclosporin analogue by teleost renal proximal tubules. J Physiol. 1995;268:F46–F52. doi: 10.1152/ajprenal.1995.268.1.F46. [DOI] [PubMed] [Google Scholar]
- Stine WB, Jungbauer L, Yu C, LaDu MJ. Preparing synthetic Abeta in different aggregation states. Methods Mol Biol. 2011;670:13–32. doi: 10.1007/978-1-60761-744-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Assema DM, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, Comans EF, Hoetjes NJ, Tolboom N, Langer O, Müller M, Scheltens P, Lammertsma AA, van Berckel BN. Blood–brain barrier P-glycoprotein function in Alzheimer's disease. Brain. 2012;135:181–189. doi: 10.1093/brain/awr298. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW. Deposition of Alzheimer's beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics. 2002;12:535–541. doi: 10.1097/00008571-200210000-00005. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, Siegmund W, Walker LC, Pahnke J. The role of P-glycoprotein in cerebral amyloid angiopathy: implications for the early pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2004;1:121–125. doi: 10.2174/1567205043332225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesuriya HC, Bullock JY, Faull RL, Hladky SB, Barrand MA. ABC efflux transporters in brain vasculature of Alzheimer's subjects. Brain Res. 2010;1358:228–238. doi: 10.1016/j.brainres.2010.08.034. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wu JY, Hait WN, Yang JM. Regulation of the stability of P-glycoprotein by ubiquitination. Mol Pharmacol. 2004;66:395–403. doi: 10.1124/mol.104.001966. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Frangione B. Transport-clearance hypothesis for Alzheimer's disease and potential therapeutic implications in Abeta metabolism in Alzheimer's disease. Austin, TX: Landes Bioscience; 2003. [Google Scholar]