

Graphical abstract
A number of triazolo-benzodiazepines including drugs such as alprazolam have been developed as protein interaction inhibitors that target bromodomains of the BET family.
Keywords: BRD4, Bromodomains, Benzodiazepines, Alprazolam, Benzotriazepines
Abstract
Benzodiazepines are psychoactive drugs with anxiolytic, sedative, skeletal muscle relaxant and amnestic properties. Recently triazolo-benzodiazepines have been also described as potent and highly selective protein interaction inhibitors of bromodomain and extra-terminal (BET) proteins, a family of transcriptional co-regulators that play a key role in cancer cell survival and proliferation, but the requirements for high affinity interaction of this compound class with bromodomains has not been described. Here we provide insight into the structure–activity relationship (SAR) and selectivity of this versatile scaffold. In addition, using high resolution crystal structures we compared the binding mode of a series of benzodiazepine (BzD) and related triazolo-benzotriazepines (BzT) derivatives including clinically approved drugs such as alprazolam and midazolam. Our analysis revealed the importance of the 1-methyl triazolo ring system for BET binding and suggests modifications for the development of further high affinity bromodomain inhibitors.
1. Introduction
Benzodiazepines (BzDs) are drug-like small molecules that led to the development of a large number of approved drugs that modulate the function of the GABA (gamma-aminobutyric acid) receptor. BzDs have generally sedative, anxiolytic, amnesic and muscle relaxing properties and have been approved for the treatment of sleeping disorders, seizures, muscle spasms and anxiety.1 For instance, alprazolam (8-chloro-1-methyl-6-phenyl-4H-[1,2,4]triazolo[4,3-a][1,4]benzodiazepine) is a potent short acting BzD used for the treatment of anxiety disorders2 whereas midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine) is prescribed for the treatment of acute seizures, insomnia as well as a sedative drug.3 L-655,708 (ethyl (13aS)-7-methoxy-9-oxo-11,12,13,13a-tetrahydro-9H-imidazo[1,5-a]pyrrolo[2,1-c][1,4]benzodiazepine-1-carboxylate) was the first subtype-selective inverse agonist at the BzD binding site that has been discovered. This BzD binds preferentially to the α5 subtype of the GABAA receptor.4 The high interest in this compound class in medicinal chemistry and medicine made many BzD analogues synthetically accessible and established a rich body of literature on their pharmacological properties.5, 6, 7 A variety of biological activities and synthetic routes have also been established for the related benzotriazepines (BzT).8
Recently, we and others described BzDs as potent protein interaction inhibitors that selectively bind to acetyl lysine (Kac) recognition modules of the BET (bromodomain and extra-terminal) family of transcriptional co-regulators.9, 10, 11 Importantly, the discovered inhibitor JQ1 has no significant GABA receptor activity most likely due to its bulky substitution at the 2 position of the benzodiazepine ring system.9
Bromodomains constitute a highly diverse family of interaction domains that comprise 61 members in humans. All bromodomains share a conserved fold that comprises a left-handed bundle of four alpha helices (αZ, αA, αB, αC) linked by diverse loop regions (ZA and BC loops) that flank the substrate binding site. The helical bromodomain bundle creates a deep central hydrophobic cavity that specifically recognizes sequences that contain ε-N-acetylated lysine residues.12 Several crystal structures with peptidic substrates revealed a common anchor point that recognizes the acetyl moiety: In all bromodomain substrate complexes a conserved asparagine residue forms a hydrogen bond with the carbonyl oxygen of the N-acetyl group of the substrate peptide providing a starting point for the design of acetyl lysine mimetic and competitive ligands.13 Even though it has not been demonstrated structurally, in some bromodomains the asparagine is substituted by a threonine or tyrosine residue providing an alternative hydrogen donor to the acetyl lysine carbonyl.
Bromodomain-containing proteins are reader domains of epigenetic marks that play key roles in transcription control and chromatin remodeling.14 In histones, ε-N-acetylation of lysine residues has been associated with open chromatin architecture and transcriptional activation and the recent discovery of selective and potent BzD inhibitors suggested that these protein interaction modules can be efficiently targeted by drug-like molecules.9, 10, 11 The therapeutic potential of bromodomains has recently been reviewed.15
The BET family of bromodomain containing proteins is represented by four proteins that express several splice isoforms in mammals (BRD2, BRD3, BRD4 and BRDT). Proteins of this family share a common domain architecture that comprises two N-terminal bromodomains and an ET (extra terminal) domain but each family member contains diverse C-termini.16 The discovery of the selective BET inhibitor iBET11 provided compelling evidence for targeting BET bromodomains in inflammatory diseases and reports have linked BET proteins to cancer. Both BRD2 and BRD3 are over-expressed in nasopharyngeal carcinoma17 and high expression levels for the testis specific isoform BRDT have been detected in lung cancer.18 In addition, BRD4 recruits the positive transcription elongation factor complex (P-TEFb) to transcriptional start sites, a key regulatory event controlling transcriptional elongation of many growth promoting genes.19, 20
Modulation of P-TEFb activity developed into a promising strategy for the treatment of chronic lymphocytic leukemia—a core component of the P-TEFb complex is cyclin dependent kinase-9 (CDK9) which has been successfully targeted by the ATP competitive inhibitor flavopiridol.21, 22 Indeed a recent RNAi screen has identified BRD4 as a key target required for the survival and maintenance of acute myeloid leukemia (AML) cells.23 Importantly, the bromodomains of both BRD3 and BRD4 have been identified in recurrent chromosomal translocations with NUT (Nuclear protein in testis), giving rise to an extremely aggressive untreatable subtype of squamous carcinoma termed NUT midline carcinoma (NMC).24, 25, 26 The recent development of the potent and selective inhibitor JQ1 and its evaluation in mouse models of patient derived cancer cells provided a compelling case for targeting BET bromodomains in NMC.9
Here we describe the structural requirements for high affinity interactions of BzDs and BzTs with BET bromodomains and discuss an initial SAR (structure–activity relationship) for these compound classes. We discovered that the clinically approved BzDs alprazolam and midazolam bound with low μM affinities to BET bromodomain and determined several high resolution co-crystal structures to further guide structure based design efforts. In addition, the BzTs are presented as an alternative versatile scaffold that specifically binds with nM potency to the acetyl lysine binding site of BET bromodomains.
2. Results and discussion
The recent disclosure of the triazolo-benzodiazepine iBET as a bromodomain inhibitor11 prompted us to investigate the interaction of a number of clinically approved BzDs with these protein interaction modules (Fig. 1). The temperature shift binding assay has emerged as a rapid screening technology for detection of protein ligands and has been shown to correlate well with binding constants determined by direct biophysical methods such as isothermal titration calorimetry and enzymatic assays.9, 27 Using this methodology we identified alprazolam and surprisingly midazolam as compounds that interact with BET bromodomains (Fig. 2A). Selectivity screening against the BET family of bromodomains and five diverse bromodomains that belong to different families that constitute the bromodomain phylogenetic tree revealed that both alprazolam and midazolam maintained their selectivity for BET BRDs. The array of Tm data also suggested a slight preference of midazolam for the second bromodomain of BET family members (Fig. 2A and B). In contrast, the related BzDs estazolam and triazolam showed only very weak interaction suggesting that the methyl group at the triazolo and imidazolo ring is a required component for the interaction of the scaffold with BET bromodomains. Comparison of alprazolam with triazolam revealed that the chloro substitution at the 2 position of the phenyl ring is not tolerated. The GABAA receptor α5 specific inhibitor L655708 showed no significant interaction with BET bromodomains. To obtain better insight into the SAR of this compound class we initiated a synthetic effort on the benzodiazepine scaffold. For studying the role of the 6-aryl substitutent in alprazolam, 6-ethyl analogues were prepared, furthermore we modified the substituents in the triazolo ring (C-1) and investigated related triazolo-benzotriazepines (BzTs) in which the chiral carbon atom present in JQ1, iBET and related compounds has been replaced by nitrogen. This greatly simplifies synthesis by abolishing the need for a stereoselective route or separation of enantiomers.
Figure 1.

Chemical structures of studied clinical BzDs and published BET bromodomain inhibitors.
Figure 2.

(A) Temperature shift data measured on synthesized inhibitors as well as clinical BzDs. Synthesized compounds have been initially screened at 100 μM compound concentration and interacting compounds were re-evaluated at 10 μM compound concentration. Temperature shift data are color coded as indicated in the figure. (B) Example of a raw data trace of a temperature shift experiment. Shown are data measured on BRD4(1) alone (black line) and BRD4(1) in the presence of 10 mM alprazolam (red line). (C) Isothermal titration calorimetry data. Shown are data measured on alprazolam (red) and BzD-7 (black). The panel shows raw binding heats of 8 μL injections of BRD4(1) into a solution of each of the two inhibitors. The first injection (2 μL) was not included into the data analysis. The insert shows normalized binding heats and non-linear least squares fit to the experimental data (solid lines). The derived thermodynamic data are compiled in Table 1.
3. Chemical synthesis
The 6-ethyl-triazolobenzodiazepines were prepared starting from known28 benzodiazepinone 1 (Scheme 1). Conversion of 1 to the thiolactam 2 could not be accomplished under standard conditions29 (P2S5 in pyridine or other high boiling solvents), but was successfully realized by using Lawesson’s reagent in anhydrous THF under nitrogen atmosphere. Target compounds 4a–e were obtained by condensation of thiolactam 2 with carboxylic acid hydrazides 3a–e. Residual hydrazides 3a–e were found to be poorly separable from the products 4a–e by column chromatography. This problem was solved by converting the hydrazides into water-soluble condensation products upon stirring with glucose solution prior to extraction with an organic solvent. The triazolo-benzotriazepines were prepared according to published procedures8a (Scheme 2).
Scheme 1.

Synthesis outline of target compounds 4a–e. Reagents and conditions: (i) Lawesson′s reagent (1.1 equiv), THF, rt, 24 h, 53%; (ii) hydrazides 3a–e (2 equiv), n-butanol, 130 °C, 24 h, 17–67%.
Scheme 2.

Studied benzotriazepines (BzT) prepared according to Richter et al. 8a
As expected the (thio)lactams BzD-2 and BzD-3 showed no significant interaction with bromodomains confirming the importance of the triazolo ring system. This observation has been also confirmed with the BzT intermediates Bz-T2 and Bz-T4. The binding pocket of BET BRDs harbors a number of conserved water molecules. With the BzD series we explored the possibility of displacing these waters by ligand atoms employing larger substituents than the methyl group at the triazolo ring that would protrude deeper into the acetyl lysine pocket. However, all bulkier groups resulted in Tm shifts not larger than 2° at high (100 μM) compound concentration indicating very weak interaction with BET bromodomains. In support of this observation also the 6-ethyl analogue BzD-5 of alprazolam showed strongly reduced binding activity. Even though the methyl substituent was not extensively tested in the BzT series it appeared to be optimal. Substitution of the methyl moiety (BzT-7) with a primary amine (BzT-8) led to significantly weaker interactions and a carbonyl group at this position (BzT-9) abolished interaction with all screened bromodomains (Fig. 2). The open-chain synthetic precursor BzT-4 of BzT-9 showed only very poor activity.
We used isothermal titration calorimetry (ITC) to precisely determine the binding constants of alprazolam and the best hit of the BzT series (BzT-7) in solution. Titration of the first bromodomain of BRD4 (BRD4(1)) into alprazolam resulted in large exothermic binding heats (ΔH: −6.96 kcal/mol). We determined a dissociation constant (KD) of 2.5 μM for this interaction (Fig. 2C). Comparison with ITC data that have been collected on JQ1 indicated that the loss of the tertiary butyl ester at position 2 in the diazepam ring leads to a significant reduction of binding affinity probably due to the loss of a second hydrogen bond to the acetyl lysine anchoring residue N140. ITC titrations carried out on BzT-7 revealed a binding constant of 0.64 μM indicating an excellent ligand efficacy of this compound. The thermodynamic data are compiled in Table 1.
Table 1.
Thermodynamic parameters of BzD and BzT interaction with BRD4(1)
| Protein | ΔTm (°C) | Kd (μM) | ΔHobs (kcal/mol) | N | TΔS (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|---|---|---|---|
| (+)JQ1a | 9.3 | 0.049 ± 0.02 | −8.42 ± 0.019 | 1.00 ± 0.001 | 1.22 | −9.64 |
| Alprazolam | 4.7 | 2.46 ± 0.11 | −6.96 ± 0.05 | 1.10 ± 0.006 | 0.44 | −7.40 |
| BzT-7 | 4.2 | 0.64 ± 0.03 | −6.16 ± 0.03 | 0.98 ± 0.003 | 2.00 | −8.16 |
According to Filippakopoulos et al. 9 Titrations were carried out in 50 mM HEPES pH 7.4 (at 25 °C), 150 mM NaCl and 15 °C while stirring at 295 rpm. In all cases the protein was titrated into the ligand solution (reverse titration).
4. Binding mode of tested BzDs and BzTs
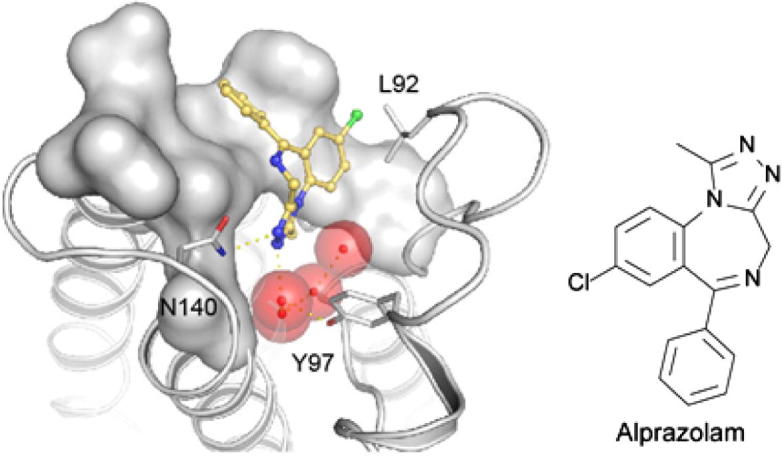
In order to obtain structural insight into the binding mode of clinically approved BzDs we determined the co-crystal structures of the first bromodomain of BRD4 with alprazolam and midazolam and compared them to structures of related ligands as well as the apo-structure. As expected, the co-crystal structures revealed the canonical alpha helical fold that is typical for bromodomains (Fig. 3A). Comparison with the apo-structure (PDB-ID: 2oss)9 revealed only minor backbone re-arrangements. However, in some cases different conformation of side chains were observed. In particular I146 assumed a different rotamer conformation compared to the apo-structure (Fig. 3B). In apo-BRD4(1) four structural water molecules were observed in the acetyl lysine cavity which were coordinated either directly or indirectly (through interaction with directly bound water molecules) by the conserved tyrosine residue Y97. In all BzD and BzT complexes the inhibitors assumed similar binding modes with good shape complementarity with the BRD4(1) acetyl lysine binding site. All structures were refined to high resolution with low R/Rfree-values (Table 2). The ligands were well defined by electron density (Fig. 3C–E).
Figure 3.

Structure of BzD and BzT BRD4 complexes. (A) Structural overview of the BRD4(1) structure. The main structural elements are labeled and the acetyl lysine mimetic inhibitor alprazolam is shown in ball and stick representation. (B) Superimposition of the apo structure of BRD4(1) (green) and the alprazolam ligand complex (blue). The figure shows main interaction residues and details of the acetyl lysine binding site as well as conserved water molecules (shown as red balls). (C–E) Detailed view of the co-crystallized ligands. The acetyl lysine binding site is shown as a transparent white surface. Residues interacting with the inhibitors are labeled in C and the orientation has been maintained in C–E. The conserved acetyl lysine anchoring residue N140 is labeled in blue. A 2FoFc electron density map that has been contoured at 2σ is shown in the lower panel for each ligand.
Table 2.
Data collection and refinement statistics
| Data collection | |||
|---|---|---|---|
| PDB ID | 3U5J | 3U5K | 3U5L |
| BRD4 ligand | Alprazolam | Midazolam | BzT-7 |
| Space group | P2(1)2(1)2(1) | P3(1) | P2(1)2(1)2(1) |
| Cell dimensions: a, b, c (Å) | 37.30, 44.21, 78.36 | 89.55, 89.55, 64.41 | 37.36, 43.13, 78.38 |
| α, β, γ (deg) | 90.00, 90.00, 90.00 | 90.00, 90.00, 120.00 | 90.00, 90.00, 90.00 |
| Resolutiona (Å) | 1.60 (1.69–1.60) | 1.80 (1.90–1.80) | 1.39 (1.46–1.39) |
| Unique observationsa | 17,472 (2392) | 51,230 (6551) | 25,967 (3709) |
| Completenessa (%) | 98.6 (95.3) | 95.7 (83.9) | 96.9 (96.9) |
| Redundancy⁎ | 6.3 (5.3) | 4.1 (3.4) | 6.7 (6.9) |
| Rmergea | 0.056 (0.225) | 0.102 (0.522) | 0.063 (0.239) |
| I/σIa | 20.7 (6.4) | 7.8 (2.1) | 18.4 (7.4) |
| Refinement | |||
| Resolution (Å) | 1.60 | 1.80 | 1.39 |
| Rwork/Rfree (%) | 14.6/17.7 | 20.9/25.9 | 11.1/14.2 |
| Number of atoms (p/o/w)b | 1098/38/216 | 4055/92/231 | 1125/27/218 |
| B-factors (Å2) (p/o/w)b | 11.57/15.77/26.53 | 20.34/31.67/19.97 | 8.67/6.76/25.16 |
| r.m.s.d bonds (Å) | 0.015 | 0.016 | 0.013 |
| r.m.s.d angles (o) | 1.538 | 1.640 | 1.700 |
| Ramachadran Favoured (%) | 97.52 | 96.71 | 97.30 |
| Allowed (%) | 2.48 | 3.09 | 2.70 |
| Disallowed (%) | 0.00 | 0.21 | 0.00 |
Values in parentheses correspond to the highest resolution shell.
(p/o/w): Protein atoms, other (ligand) atoms, water.
The water network observed in apo-BRD4(1) was slightly rearranged in structures that bound the triazolo-benzodiazepine ring system (Fig. 4). The missing second hydrogen bond to N140 in the alprazolam complex results most likely in the lower affinity of this ligand compared to iBET (Fig. 4B). The complex with midazolam (Fig. 4C) revealed the influence of the lack of the nitrogen in position 3 of the ring system (annulated imidazole instead of 1,2,4-triazole). The inability of midazolam to form a hydrogen bond with the acetyl lysine anchoring residue N140 leads to an outward shift of this inhibitor. In addition, 2 of the 4 structural water molecules are not present in the midazolam complex. The 2-fluorophenyl ring system is rotated by about 30°. This is most likely a consequence of the fluoro substitution that requires reorientation of this aromatic ring system. Larger halogens in that position will rotate the ring further explaining the observed inactivity of triazolam that contains a 2-chlorophenyl moiety.
Figure 4.

Details of the BzD and BzT interaction with BRD4(1). Shown is (A) iBET, (B) alprazolam, (C) midazolam and (D) BzT-7. Conserved water molecules are shown as red spheres.
Triazolo-benzotriazepines were discovered as a new versatile scaffold with strong BRD4 binding affinity. The binding mode of BzT-7 was found to be reminiscent of the related BzD alprazolam with identical conservation of the four water molecules (Fig. 4D).
5. Conclusions
Here we describe structural requirements for the development of benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. We found that the clinical inhibitor alprazolam binds with low μM affinity to the acetyl lysine binding site of BRD4(1). The co-crystal structure with BRD4(1) showed that the triazolo ring acts as an acetyl lysine mimetic scaffold forming a hydrogen bond with the conserved residue N140 that acts as a hydrogen bond anchor point for acetylated substrates in most bromodomains. Surprisingly, also midazolam, that lacks the hydrogen bond forming nitrogen in the triazolo ring bound to BRD4(1). The binding mode of this BzD is characterized by a reorientation of midazolam in the acetyl lysine binding pocket and in an altered network of structural water molecules. However, FDA-approved sedatives inhibit BRD4 at high concentrations making it unlikely that this activity will cause side effects due to inhibition of BRD4 mediated transcription control during therapy. This unexpected binding mode may be explored for the development of more potent inhibitors. A small expansion of the triazolo-bezodiazepine scaffold and comparison with estazolam underline the importance of the 3-methyl group in the triazolo ring system. Furthermore, we report compounds of the triazolo-benzotriazepine class as submicromolar inhibitors of BET bromodomains with similar binding mode as BzD and excellent ligand efficiency.
6. Experimental
Alprazolam, estazolam, midazolam, L-655,708 and triazolam were purchased from Sigma.
General Analysis of chemical compounds: Melting points were determined on a Büchi Melting Point B-540 apparatus and are uncorrected. NMR spectra were recorded on a Jeol GSX 400 (1H NMR: 400 MHz, 13C NMR: 100 MHz) and a Jeol JNMR-GX 500 (1H NMR: 500 MHz, 13C NMR: 125 MHz) spectrometer, respectively. Chemical shifts are reported in δ (ppm) units relative to the internal standard tetramethylsilane (TMS). Infrared spectroscopy was done on a Perkin–Elmer FT-IR Paragon 1000. Mass spectra were obtained on a Hewlett Packard 5989 A Mass Spectrometer by either chemical ionization (CI) or electron ionization (EI). High resolution mass spectra (HRMS) were measured on a Jeol JMS GCmate II. All chemicals and solvents used were of analytical grade and no further purification was needed. Flash column chromatography was performed on silica gel Si 60 (40–63 μm).
6.1. 7-Chloro-5-ethyl-1H-benzo[e][1,4]diazepine-2(3H)-thione (2)
7-Chloro-5-ethyl-1H-benzo[e][1,4]diazepin-2(3H)-one (1; 2.0 g, 9.1 mmol, 1.0 equiv) and Lawesson’s reagent (4.0 g, 10 mmol, 1.1 equiv) were suspended in 19 mL of anhydrous tetrahydrofuran and stirred for 24 h at room temperature under nitrogen atmosphere. The crude product was extracted three times in a separating funnel with a dichloromethane/water mixture. The combined organic phases were washed with water, dried over sodium sulfate, filtered and evaporated. The purification was done by flash column chromatography (ethyl acetate/isohexane, 1:2), yielding thiolactam 2 (1.1 g, 4.8 mmol, 53 %) as a yellow solid. mp: 168.7 °C; 1H NMR (400 MHz, CD2Cl2): δ (ppm) 1.13 (t, 3JHH = 7.4 Hz, 3H, CH3), 2.75 (q, 3JHH = 7.3 Hz, 2H, CH2–CH3), 4.49 (s, 2H, H-3), 7.12 (d, 3JHH = 8.6 Hz, 1H, H-9), 7.45 (dd, 4JHH = 2.3 Hz, 3JHH = 8.6 Hz, 1H, H-8), 7.58 (d, 4JHH = 2.3 Hz, 1H, H-6), 10.19 (bs s, 1H, H-1); 13C NMR (100 MHz, CD2Cl2): δ (ppm) 11.4 (CH3), 31.8 (CH2–CH3), 62.9 (C-3), 122.9 (C-9), 128.7 (C-6), 131.1 (C-7), 131.5 (C-5a), 131.6 (C-8), 136.9 (C-9a), 172.4 (C-5), 201.6 (C-2); IR [cm−1]: v = 3113, 3070, 2973, 2899, 2853, 2731, 2675, 1635, 1576, 1520, 1474, 1358, 1163, 1009, 838; MS (EI): m/z (%) = 238 (100) [M+•]; HR-MS (EI+): calcd for C11H11ClN2S [M+•] 238.0331; found 238.0296.
6.2. General procedure for the preparation of triazolo-benzodiazepines 4a–e
Thiolactam 2 (1.0 equiv) and carboxylic acid hydrazide 3a–e (2.0 equiv) were dissolved in n-butanol and heated to 130 °C in a sealed vial under nitrogen atmosphere for 24 h. The reaction mixture was cooled to room temperature and stirred with an aqueous glucose solution for 2 h. The crude product was extracted several times with dichloromethane. The combined organic layers were washed with water, dried over sodium sulfate, filtered and evaporated. Purification was done by flash column chromatography (dichloromethane/methanol, 9:1), giving the triazolo-benzodiazepines 4a–e in 17–67% yield.
6.2.1. 8-Chloro-6-ethyl-1-methyl-4H-benzo[f][1,2,4]triazolo[4,3-a][1,4]diazepine (4a)
Yield: 40%; yellow solid; mp: 173.5–174.9 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 1.09 (t, 3JHH = 7.3 Hz, 3H, CH2–CH3), 2.60 (s, 3H, 1-CH3), 2.56–2.65 (m, 1H, CHH–CH3), 2.80–2.89 (m, 1H, CHH–CH3), 3.93 (d, 2JHH = 13.0 Hz, 1H, 4-CHH), 5.28 (d, 2JHH = 13.0 Hz, 1H, 4-CHH), 7.35 (d, 3JHH = 8.6 Hz, 1H, H-10), 7.60 (dd, 4JHH = 2.4 Hz, 3JHH = 8.6 Hz, 1H, H-9), 7.68 (d, 4JHH = 2.3 Hz, 1H, H-7); 13C NMR (125 MHz, CDCl3): δ (ppm) 11.0 (CH2–CH3), 12.4 (1-CH3), 32.3 (CH2–CH3), 45.7 (C-4), 124.6 (C-10), 128.9 (C-7), 130.7 (C-10a), 131.1 (C-9), 131.9 (C-6a), 133.9 (C-8), 150.2 (C-1), 154.9 (C-3a), 170.9 (C-6); IR [cm−1]: v = 3066, 2971, 2933, 2852, 2360, 1635, 1542, 1485, 1426, 1378, 1111, 831; MS (CI): m/z (%) = 261 (100) [MH+]; MS (EI): m/z (%) = 260 (30) [M+•], 225 (100) [M+•—Cl]; HR-MS (EI+): calcd for C13H13ClN4 [M+•] 260.0829; found 260.0813.
6.2.2. 8-Chloro-1,6-diethyl-4H-benzo[f][1,2,4]triazolo[4,3-a] [1,4]diazepine (4b)
Yield: 45%; yellow solid; mp: 203.6–205.9 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 1.07 (t, 3JHH = 7.4 Hz, 3H, 6-CH2–CH3), 1.34 (t, 3JHH = 7.5 Hz, 3H, 1-CH2–CH3), 2.60–2.70 (m, 1H, 6-CHH–CH3), 2.77–2.89 (m, 2H, 1-CHH–CH3, 6-CHH–CH3), 3.00–3.11 (m, 1H, 1-CHH–CH3), 3.92 (d, 2JHH = 13.0 Hz, 1H, 4-CHH), 5.28 (d, 2JHH = 13.0 Hz, 1H, 4-CHH), 7.37 (d, 3JHH = 8.6 Hz, 1H, H-10), 7.59 (dd, 4JHH = 2.4 Hz, 3JHH = 8.6 Hz, 1H, H-9), 7.67 (d, 4JHH = 2.3 Hz, 1H, H-7); 13C NMR (125 MHz, CDCl3): δ (ppm) 11.1 (6-CH2–CH3), 11.5 (1-CH2–CH3), 19.8 (1-CH2-CH3), 32.4 (6-CH2-CH3), 45.7 (C-4), 124.6 (C-10), 128.8 (C-7), 130.9 (C-10a), 131.1 (C-9), 131.8 (C-6a), 133.8 (C-8), 154.8 (C-1), 155.1 (C-3a), 171.0 (C-6); IR [cm−1]: v = 3060, 2986, 2934, 2873, 2362, 2343, 1630, 1540, 1485, 1430, 1261, 1108, 1027, 818, 799; MS (CI): m/z (%) = 275 (100) [MH+]; MS (EI): m/z (%) = 274 (40) [M+•], 245 (40) [M+•—Et], 239 (100) [M+•—Cl]; HR-MS (EI+): calcd for C14H15ClN4 [M+•] 274.0985; found 274.0987.
6.2.3. 8-Chloro-6-ethyl-1-phenyl-4H-benzo[f][1,2,4]triazolo[4,3-a][1,4]diazepine (4c)
Yield: 61%; light yellow solid; mp: 176.8 °C; 1H NMR (400 MHz, CD2Cl2): δ (ppm) 1.13 (t, 3JHH = 7.3 Hz, 3H, CH3), 2.67–2.80 (m, 1H, CHH–CH3), 2.82–2.96 (m, 1H, CHH–CH3), 4.00 (d, 2JHH = 13.0 Hz, 1H, 4-CHH), 5.21 (d, 2JHH = 13.1 Hz, 1H, 4-CHH), 6.87 (d, 3JHH = 8.7 Hz, 1H, H-10), 7.30 (dd, 4JHH = 2.4 Hz, 3JHH = 8.7 Hz, 1H, H-9), 7.37–7.52 (m, 5 H, H-2′, H-3′, H-4′), 7.69 (d, 4JHH = 2.4 Hz, 1H, H-7); 13C NMR (100 MHz, CD2Cl2): δ (ppm) 11.2 (CH3), 32.8 (CH2–CH3), 46.2 (C-4), 126.8 (C-10), 127.2 (C-1′), 128.8 (C-2′), 128.9 (C-7), 129.3 (C-3′), 130.7 (C-4′), 131.1 (C-9), 132.0 (C-10a), 132.2 (C-6a), 133.9 (C-8), 153.6 (C-1), 157.3 (C-3a), 171.9 (C-6); IR [cm−1]: v = 3057, 2970, 2930, 2854, 2365, 2345, 1631, 1534, 1485, 1472, 1422, 1287, 1107, 977, 821, 768, 696; MS (CI): m/z (%) = 323 (100) [MH+]; MS (EI): m/z (%) = 322 (40) [M+•], 293 (40) [M+•—Et], 287 (100) [M+•—Cl]; HR-MS (EI+): calcd for C18H15ClN4 [M+•] 322.0985; found 322.0963; Elemental analysis calcd (%) for C18H15ClN4: C 66.98, H 4.68, N 17.36; found C 65.57, H 4.78, N 17.01.
6.2.4. 8-Chloro-6-ethyl-1-(pyridin-4-yl)-4H-benzo[f][1,2,4]triazolo[4,3-a][1,4]diazepine (4d)
Yield: 17%; yellow solid; mp: 157.3–159.9 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 1.16 (t, 3JHH = 7.4 Hz, 3H, CH3), 2.72–2.82 (m, 1H, CHH–CH3), 2.88–2.98 (m, 1H, CHH–CH3), 4.01 (d, 2JHH = 13.3 Hz, 1H, 4-CHH), 5.36 (d, 2JHH = 13.3 Hz, 1H, 4-CHH), 6.90 (d, 3JHH = 8.7 Hz, 1H, H-10), 7.39 (dd, 4JHH = 2.3 Hz, 3JHH = 8.7 Hz, 1H, H-9), 7.41 (dd, 2H, 3JHH = 6.2 Hz, H-2′), 7.72 (d, 4JHH = 2.3 Hz, 1H, H-7), 8.72 (d, 3JHH = 5.7 Hz, 2H, H-3′); 13C NMR (125 MHz, CDCl3): δ (ppm) 11.3 (CH3), 32.7 (CH2–CH3), 45.9 (C-4), 122.3 (C-2′), 126.4 (C-10), 129.0 (C-7), 131.1 (C-10a), 131.3 (C-9), 131.9 (C-6a), 134.3 (C-1′), 134.7 (C-8), 150.9 (C-3′), 151.2 (C-1), 158.0 (C-3a), 171.7 (C-6); IR [cm−1]: v = 3036, 2971, 2927, 2853, 2366, 2344, 2225, 1633, 1604, 1531, 1484, 1467, 1434, 1290, 1110, 985, 827, 728, 576; MS (CI): m/z (%) = 324 (100) [MH+]; MS (EI): m/z (%) = 323 (30) [M+•], 294 (50) [M+•—Et], 288 (100) [M+•—Cl]; HR-MS (EI+): calcd for C17H14ClN5 [M+•] 323.0938; found 323.0934.
6.2.5. 8-Chloro-6-ethyl-1-(3-methoxyphenyl)-4H-benzo[f][1,2,4] triazolo[4,3-a][1,4]diazepine (4e)
Yield: 67%; yellow solid; mp: 132.6–133.7 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) 1.14 (t, 3JHH = 7.4 Hz, 3H, CH2–CH3), 2.70–2.82 (m, 1H, CHH–CH3), 2.83–2.96 (m, 1H, CHH–CH3), 3.81 (s, 3H, OCH3), 4.00 (d, 2JHH = 13.4 Hz, 1H, 4-CHH), 5.33 (d, 2JHH = 13.2 Hz, 1H, 4-CHH), 6.88 (ddd, 4JHH = 1.0 Hz, 4JHH = 1.3 Hz, 3JHH = 7.7 Hz, 1H, H-6′), 6.91 (d, 3JHH = 8.7 Hz, 1H, H-10), 7.00 (ddd, 4JHH = 1.0 Hz, 4JHH = 2.5 Hz, 3JHH = 8.4 Hz, 1H, H-4′), 7.14 (dd, 4JHH = 1.7 Hz, 4JHH = 2.3 Hz, 1H, H-2′), 7.30 (t, 3JHH = 7.8 Hz, 1H, H-5′), 7.32 (dd, 1H, 4JHH = 2.4 Hz, 3JHH = 8.7 Hz, H-9), 7.66 (d, 4JHH = 2.4 Hz, 1H, H-7); 13C NMR (100 MHz, CDCl3): δ (ppm) 11.2 (CH2–CH3), 32.6 (CH2–CH3), 45.9 (C-4), 55.4 (OCH3), 113.7 (C-2′), 116.4 (C-4′), 120.6 (C-6′), 126.4 (C-10), 127.6 (C-1′), 128.4 (C-7), 130.0 (C-5′), 130.8 (C-9), 131.4 (C-10a), 131.5 (C-6a), 133.7 (C-8), 153.2 (C-1), 156.9 (C-3a), 159.9 (C-3′), 171.7 (C-6); IR [cm−1]: v = 3072, 2970, 2935, 1632, 1583, 1535, 1485, 1466, 1435, 1319, 1287, 1238, 1108, 1045, 994, 753; MS (CI): m/z (%) = 353 (100) [MH+]; MS (EI): m/z (%) = 352 (80) [M+•], 323 (50) [M+•—Et], 317 (100) [M+•—Cl]; HR-MS (EI+): calcd for C19H17ClN4O [M+•] 352.1091; found 352.1098.
The NMR spectra of all synthesized compounds are shown in the Supplementary data of this manuscript.
6.3. Protein stability shift assay
Thermal melting experiments were carried out using an Mx3005p Real Time PCR machine (Stratagene). Proteins were buffered in 10 mM HEPES pH 7.5, 500 mM NaCl and assayed in a 96-well plate at a final concentration of 2 μM in 20 μL volume. Compounds were added at a final concentration of 10 μM or 100 μM in order to probe weaker interactions. SYPRO Orange (Molecular Probes) was added as a fluorescence probe at a dilution of 1:1000. Excitation and emission filters for the SYPRO-Orange dye were set to 465 nm and 590 nm, respectively. The temperature was raised with a step of 3 °C per minute from 25 to 96 °C and fluorescence readings were taken at each interval. The temperature dependence of the fluorescence during the protein denaturation process was approximated by the equation
| (1) |
where ΔuG(T) is the difference in unfolding free energy between the folded and unfolded state, R is the gas constant and yF and yU are the fluorescence intensity of the probe in the presence of completely folded and unfolded protein respectively. The baselines of the denatured and native states were approximated by a linear fit. The observed temperature shifts, , were recorded as the difference between the transition midpoints of sample and reference wells containing protein without ligand in the same plate and determined by non-linear least squares fit.
6.4. Isothermal titration calorimetry
Experiments were carried out on a VP-ITC titration microcalorimeter from MicroCal™, LLC (Northampton, MA). All experiments were carried out at 15 °C while stirring at 295 rpm, in ITC buffer (50 mM HEPES pH 7.4 at 25 °C, 150 mM NaCl). The microsyringe was loaded with a solution of the protein sample (150 μM BRD4(1) in ITC buffer). All titrations were conducted using an initial control injection of 2 μL followed by 34 identical injections of 8 μL with a duration of 16 s (per injection) and a spacing of 250 s between injections. The heat of dilution was determined by independent titrations (protein into buffer) and was subtracted from the experimental data. The collected data were evaluated using a single binding site model and the MicroCal™ Origin software. Thermodynamic parameters were calculated (ΔG = ΔH−TΔS = −RTlnKB, where ΔG, ΔH and ΔS are the changes in free energy, enthalpy and entropy of binding respectively).
6.5. Protein expression and purification
Proteins were cloned, expressed and purified as previously described.9
6.6. Crystallization
Aliquots of the purified proteins were set up for crystallization using a mosquito™ crystallization robot (TTP Labtech, Royston UK). Coarse screens were typically setup onto Greiner 3-well plates using three different drop ratios of precipitant to protein per condition (100 + 50 nL, 75 + 75 nL and 50 + 100 nL). Initial hits were optimized further scaling up the drop sizes. All crystallizations were carried out using the sitting drop vapor diffusion method at 4 °C. BRD4(1) crystals with alprazolam were grown by mixing 200 nL of the protein (9.5 mg/mL and 5 mM final ligand concentration) with 100 nL of reservoir solution containing 0.20 M sodium sulfate, 0.1 M BT-Propane pH 6.5, 20% PEG3350 and 10% ethylene glycol. BRD4(1) crystals with midazolam were grown by mixing 200 nL of protein (9.36 mg/mL and 5 mM final ligand concentration) with 100 nL of reservoir solution containing 0.1 M magnesium chloride, 0.1 M MES pH 6.5, 15% PEG6000 and 10% ethylene glycol. BRD4(1) crystals with BzT-7 were grown by mixing 200 nL of protein (9 mg/mL and 5 mM final ligand concentration) with 200 nL of reservoir solution containing 0.1 M MES pH 6.5, 10% PEG3350 and 10% ethylene glycol. In all cases diffraction quality crystals grew within a few days.
6.7. Data collection and structure solution
All crystals were cryo-protected using the well solution supplemented with additional ethylene glycol and were flash frozen in liquid nitrogen. Data were collected in-house on a Rigaku FRE rotating anode system equipped with a RAXIS-IV detector (alprazolam and midazolam complexes) or at the Diamond beamline I04.1 (BzT-7 complex). Indexing and integration was carried out using MOSFLM30 and scaling was performed with SCALA31 or XDS.32 Initial phases were calculated by molecular replacement with PHASER33 using the known models of BRD4(1) (PDB ID 2OSS). Initial models were built by ARP/wARP34 followed by manual building in COOT.35 Refinement was carried out in REFMAC5.36 In all cases thermal motions were analyzed using TLSMD37 and hydrogen atoms were included in late refinement cycles. Data collection and refinement statistics can be compiled in Table 2. The models and structure factors have been deposited with PDB accession codes: 3U5J (BRD4(1)/alprazolam), 3U5K (BRD4(1)/midazolam), 3U5L (BRD4(1)/BzT-7), respectively.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2011.10.080.
Contributor Information
Franz Bracher, Email: franz.bracher@cup.uni-muenchen.de.
Stefan Knapp, Email: stefan.knapp@sgc.ox.ac.uk.
Supplementary data
Supplementary Figures.
References and notes
- 1.Olkkola K.T., Ahonen J. Handb. Exp. Pharmacol. 2008;182:335. doi: 10.1007/978-3-540-74806-9_16. [DOI] [PubMed] [Google Scholar]
- 2.Verster J.C., Volkerts E.R. CNS Drug Rev. 2004;10:45. doi: 10.1111/j.1527-3458.2004.tb00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sofou K., Kristjansdottir R., Papachatzakis N.E., Ahmadzadeh A., Uvebrant P. J. Child Neurol. 2009;24:918. doi: 10.1177/0883073809332768. [DOI] [PubMed] [Google Scholar]
- 4.(a) Quirk K., Blurton P., Fletcher S., Leeson P., Tang F., Mellilo D., Ragan C.I., McKernan R.M. J. Neurochem. 2001;77:445. doi: 10.1016/s0028-3908(96)00061-5. [DOI] [PubMed] [Google Scholar]
- 5.Atack J.R. Expert Opin. Investig. Drugs. 2005;14:601. doi: 10.1517/13543784.14.5.601. [DOI] [PubMed] [Google Scholar]
- 6.Korpi E.R., Mattila M.J., Wisden W., Luddens H. Ann. Med. 1997;29:275. doi: 10.3109/07853899708999348. [DOI] [PubMed] [Google Scholar]
- 7.Dubnick B., Lippa A.S., Klepner C.A., Coupet J., Greenblatt E.N., Beer B. Pharmacol. Biochem. Behav. 1983;18:311. doi: 10.1016/0091-3057(83)90385-4. [DOI] [PubMed] [Google Scholar]
- 8.(a) Richter P.H., Scheefeldt U. Pharmazie. 1991;46:701. [PubMed] [Google Scholar]; (b) McDonald I.M., Austin C., Buck I.M., Dunstone D.J., Griffin E., Harper E.A., Hull R.A., Kalindjian S.B., Linney I.D., Low C.M., Pether M.J., Spencer J., Wright P.T., Adatia T., Bashall A. J. Med. Chem. 2006;49:2253. doi: 10.1021/jm051219x. [DOI] [PubMed] [Google Scholar]; (c) Fernandez P., Guillen M.I., Ubeda A., Lopez-Cremades P., Aller E., Lorenzo A., Molina P., Alcaraz M.J. Naunyn Schmiedebergs Arch. Pharmacol. 2003;368:26. doi: 10.1007/s00210-003-0768-6. [DOI] [PubMed] [Google Scholar]
- 9.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I., Philpott M., Munro S., McKeown M.R., Wang Y., Christie A.L., West N., Cameron M.J., Schwartz B., Heightman T.D., La Thangue N., French C.A., Wiest O., Kung A.L., Knapp S., Bradner J.E. Nature. 2010;468:1067. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung C.W., Coste H., White J.H., Mirguet O., Wilde J., Gosmini R.L., Delves C., Magny S.M., Woodward R., Hughes S.A., Boursier E.V., Flynn H., Bouillot A.M., Bamborough P., Brusq J.M., Gellibert F.J., Jones E.J., Riou A.M., Homes P., Martin S.L., Uings I.J., Toum J., Clement C.A., Boullay A.B., Grimley R.L., Blandel F.M., Prinjha R.K., Lee K., Kirilovsky J., Nicodeme E. J. Med. Chem. 2011;54:3827. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 11.Nicodeme E., Jeffrey K.L., Schaefer U., Beinke S., Dewell S., Chung C.W., Chandwani R., Marazzi I., Wilson P., Coste H., White J., Kirilovsky J., Rice C.M., Lora J.M., Prinjha R.K., Lee K., Tarakhovsky A. Nature. 2010;468:1119. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng L., Zhou M.M. FEBS Lett. 2002;513:124. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 13.Owen D.J., Ornaghi P., Yang J.C., Lowe N., Evans P.R., Ballario P., Neuhaus D., Filetici P., Travers A.A. EMBO J. 2000;19:6141. doi: 10.1093/emboj/19.22.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Kouzarides T. Cell. 2007;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]; (b) Dey A., Nishiyama A., Karpova T., McNally J., Ozato K. Mol. Biol. Cell. 2009;20:4899. doi: 10.1091/mbc.E09-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller S., Filippakopoulos P., Knapp S. Expert Rev. Mol. Med. 2011;13:e29. doi: 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu S.Y., Chiang C.M. J. Biol. Chem. 2007;282:13141. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 17.Zhou M., Peng C., Nie X.M., Zhang B.C., Zhu S.G., Yu Y., Li X.L., Li G.Y. Ai Zheng. 2003;22:123. [PubMed] [Google Scholar]
- 18.Scanlan M.J., Altorki N.K., Gure A.O., Williamson B., Jungbluth A., Chen Y.T., Old L.J. Cancer Lett. 2000;150:155. doi: 10.1016/s0304-3835(99)00385-7. [DOI] [PubMed] [Google Scholar]
- 19.Yang Z., He N., Zhou Q. Mol. Cell Biol. 2008;28:967. doi: 10.1128/MCB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahl P.B., Lin C.Y., Seila A.C., Flynn R.A., McCuine S., Burge C.B., Sharp P.A., Young R.A. Cell. 2010;141:432. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phelps M.A., Lin T.S., Johnson A.J., Hurh E., Rozewski D.M., Farley K.L., Wu D., Blum K.A., Fischer B., Mitchell S.M., Moran M.E., Brooker-McEldowney M., Heerema N.A., Jarjoura D., Schaaf L.J., Byrd J.C., Grever M.R., Dalton J.T. Blood. 2009;113:2637. doi: 10.1182/blood-2008-07-168583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson D.M., Joseph B., Hillion J., Segal J., Karp J.E., Resar L.M. Leuk. Lymphoma. 2011;52:1999. doi: 10.3109/10428194.2011.591012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuber J., Shi J., Wang E., Rappaport A.R., Herrmann H., Sison E.A., Magoon D., Qi J., Blatt K., Wunderlich M., Taylor M.J., Johns C., Chicas A., Mulloy J.C., Kogan S.C., Brown P., Valent P., Bradner J.E., Lowe S.W., Vakoc C.R. Nature. 2011;478:524. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.French C.A. Cancer Genet. Cytogenet. 2010;203:16. doi: 10.1016/j.cancergencyto.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haack H., Johnson L.A., Fry C.J., Crosby K., Polakiewicz R.D., Stelow E.B., Hong S.M., Schwartz B.E., Cameron M.J., Rubin M.A., Chang M.C., Aster J.C., French C.A. Am. J. Surg. Pathol. 2009;33:984. doi: 10.1097/PAS.0b013e318198d666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.French C.A., Miyoshi I., Kubonishi I., Grier H.E., Perez-Atayde A.R., Fletcher J.A. Cancer Res. 2003;63:304. [PubMed] [Google Scholar]
- 27.(a) Bullock A.N., Debreczeni J.E., Fedorov O.Y., Nelson A., Marsden B.D., Knapp S. J. Med. Chem. 2005;48:7604. doi: 10.1021/jm0504858. [DOI] [PubMed] [Google Scholar]; (b) Fedorov O., Huber K., Eisenreich A., Filippakopoulos P., King O., Bullock A.N., Szklarczyk D., Jensen L.J., Fabbro D., Trappe J., Rauch U., Bracher F., Knapp S. Chem. Biol. 2011;18:67. doi: 10.1016/j.chembiol.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell, S. C.; Childress, S. J., United States Patent 3714145 (1973).
- 29.Hester J.B., Jr., Rudzik A.D., Kamdar B.V. J. Med. Chem. 1971;14:1078. doi: 10.1021/jm00293a015. [DOI] [PubMed] [Google Scholar]
- 30.Leslie, A. G. W.; Powell, H. MOSFLM, 7.01; MRC Laboratory of Molecular Biology: Cambridge, 2007.
- 31.Evans, P. SCALA - scale together multiple observations of reflections, 3.3.0; MRC Laboratory of Molecular Biology: Cambridge, 2007.
- 32.Kabsch W., Xds Acta Crystallogr. D Biol. Crystallogr. 2010;66:125. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy A.J., Grosse-Kunstleve R.W., Storoni L.C., Read R.J. Acta Crystallogr. D Biol. Crystallogr. 2005;61:458. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 34.Perrakis A., Morris R., Lamzin V.S. Nat. Struct. Biol. 1999;6:458. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P., Cowtan K. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Murshudov G.N., Vagin A.A., Dodson E.J. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Painter J., Merritt E.A. Acta Crystallogr. D Biol. Crystallogr. 2006;62:439. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures.