

Abstract
LRSAM1 mutations have been found in recessive and dominant forms of Charcot–Marie–Tooth disease. Within one generation of the original Dutch family in which the dominant LRSAM1 mutation was identified, three of the five affected family members have developed Parkinson's disease between ages 50 and 65 years, many years after neuropathy onset. We speculate that this late‐onset parkinsonism is part of the LRSAM1 phenotype, thus associating a hitherto peripheral nerve disease with a central nervous system phenotype. How the mutated Lrsam1 protein, which normally has E3 ubiquitin ligase activity and is expressed in the nervous system, impacts on substantia nigra neurons is unclear.
Introduction
The hereditary neuropathies are clinically, but even more so genetically, a very heterogeneous group of diseases. Recently, members of our group identified a frameshift mutation in LRSAM1 as the cause of dominantly inherited, axonal sensorimotor neuropathy in a Dutch family (Charcot–Marie–Tooth, CMT, type 2P).1 This finding was confirmed by others,2, 3 and another homozygous mutation in this gene was previously found in a recessive CMT family (AR‐CMT2P).4 In the original paper on the Dutch family, the clinical description incidentally mentioned that two of the affected members showed signs of Parkinson's disease (PD). This observation was not highlighted, and this possible association does not come up through the usual search engines in the medical literature. As we have recently seen a third family member with signs of PD, we believe that this is not just a chance association, and that the LRSAM1 mutation might be related to the co‐occurring PD. Here, we detail the parkinsonian phenotype of three affected family members.
Case Descriptions
Figure 1 shows the pedigree of the family. Three of the five affected family members in the second generation suffer from parkinsonism, on top of the axonal sensorimotor neuropathy.
Figure 1.
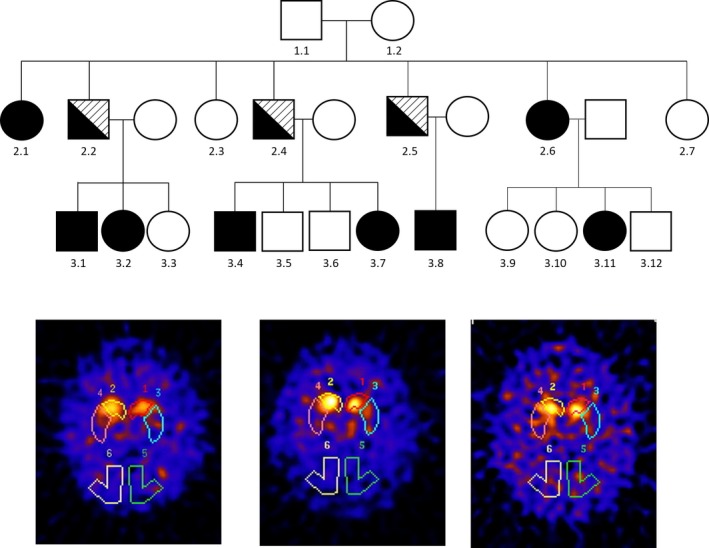
The top shows the family pedigree. Squares: males, circles: females. Black filled symbols indicate those affected by Charcot–Marie–Tooth (CMT). Symbols that are half black and half lined indicate those affected by both CMT and Parkinson's disease. The bottom shows the abnormal DaT SPECT scans of the three males with both CMT and Parkinson's disease. Note the asymmetrically reduced uptake and pattern of more putamen than caudate involvement.
Case 1 (2.2)
This man, now 72 years old, diagnosed earlier with CMT2, was referred to us at 66 years because of reduced right leg agility since 2 years. He mentioned more difficulties with walking and festination. The right leg was getting weaker. He could previously walk with a cane, but now had to use a stroller. His right more than left leg occasionally glued to the floor. He mentioned difficulty turning in bed. He experienced reduced dexterity on the right with a hand tremor. Balance was impaired and he had one fall. Smell was normal. There was mild urinary urgency with erectile dysfunction since 2 years. Cognition was unchanged. Upon examination at that time, we saw a masked face, slightly monotonous speech, jerky pursuit eye movements, rigidity and bradykinesia of all limbs but more pronounced on the right, dragging of the right leg and a foot drop on the left, rest and action tremor of the right hand, weakness and wasting of distal more than proximal leg muscles, sensory disturbances in a sock‐and‐glove distribution, and lower limb areflexia. A dopamine transporter (DaT) SPECT scan (Fig. 1) demonstrated bilaterally reduced uptake, putamen more than caudate and more pronounced on the left. He was started on levodopa and noticed benefit with levodopa/carbidopa CR 125 mg tid (but no extra improvement with a higher dose).
Case 2 (2.4)
This man, now 68 years old, was seen at the age of 66 years because of functional decline. He was already carrying the diagnosis of CMT2. Since 1 year, he was feeling more stiff and slow. Increased walking difficulties had led to wheelchair dependency. He had noticed more difficulty dual tasking and loss of initiative. He mentioned the presence of a hand tremor, left more than right. There was hyposmia and some urinary urgency. Upon examination, we found wasting and weakness of distal more than proximal leg muscles, and to a lesser extent also of distal more than proximal arm muscles, combined with areflexia and hypesthesia of the hands and anesthesia of both legs. There was bradykinesia, more pronounced on the left, and rigidity that was more pronounced on the right with cogwheeling. MRI showed some focal white matter hyperintensities and slight cortical atrophy. A DaT scan demonstrated profoundly reduced basal ganglia uptake (putamen bilaterally and left caudate; Fig. 1). Levodopa and later ropinirol were started, with moderate benefit. An attempt to stop the levodopa led to worsening of motor signs, which again improved when levodopa was re‐installed (to now 1000 mg per day). The CMT features continue to dominate the clinical picture and to have the greatest impact on his level of functioning.
Case 3 (2.5)
The youngest brother, now aged 67 years, was diagnosed with PD at the age of 52 years, when he was already known to have CMT2. He had noticed reduced arm swing, with stiffness and reduced dexterity of his left arm for about one and a half year. His sense of smell had diminished. On examination, in addition to the known CMT stigmata, there was a slightly monotonous speech, absent arm swing on the left, an action tremor of the left more than right hand, and left‐sided rigidity. He was initially started on pergolide, but a switch to levodopa with entacapone provided good relief of his mainly left‐sided parkinsonism. After 4 years of treatment, peak‐dose dyskinesias developed and he started to freezing and some falls; he needed a stroller because of this. A DaT scan at the age of 60 years showed severely reduced uptake in left more than right putamen, and to a lesser extent in left more than right caudate (Fig. 1). Examination at the age of 61 years in an on‐phase showed masked face, weak and monotonous voice, no tremor, mild bradykinesia and rigidity left more than right, dyskinesias of all extremities but left more than right, severely impaired walking (with support) with freezing episodes, and reduced balance. Over the following years, while being on levodopa and amantadine, there was progression of dyskinesias, unpredictable on–off fluctuations arose, and a further progression of gait and balance difficulties, as well as of his lower limb sensory disturbances, were apparent. He underwent deep brain stimulation of the subthalamic nucleus at age 64. The procedure was complicated by a subdural hemorrhage and he did not gain much benefit over the following months. At present, there is severe dysarthria, lateral flexion of the trunk to the right, severe bradykinesia, no dyskinesias, and he can only walk with a stroller during which freezing and poor balance are observed.
Genetic Findings
As described in the original paper,1 all family members affected by CMT2, including the three with concomitant PD, were carrying a frameshift mutation (c.2121_2122insGC; p.Leu708Argfx28) in the LRSAM1 gene. In addition, we excluded mutations in other genes that are associated with genetic forms of parkinsonism in subject 2.4 (SNCA, parkin, PINK1, DJ‐1, GBA, POLG, VPS35, DNAJC13, LRRK2, ATXN2, ATXN3, CACNA1A, TBP, and ATN1). For detailed methodology, see Data S1.
Discussion
We here share the observation that three members from a CMT2P family with LRSAM1 mutations have developed dopa‐responsive parkinsonism and speculate that parkinsonism is part of the LRSAM1 phenotype. The parkinsonian features developed at ages 50–65 years, many years after the onset of peripheral neuropathy, and were asymmetric. Two patients had reduced smell. All showed an effect of levodopa, although the actual functional benefit was modest because of the coexisting neuropathy. One patient also showed the long‐term complications of dopaminergic treatment. Nuclear imaging proved that the parkinsonian features were due to degeneration of the nigrostriatal pathway. Therefore, at the clinical and imaging levels, the parkinsonian component of the phenotype in these LRSAM1 mutation carriers seems in keeping with classic PD. We have no neuropathological data to answer the question of whether there is Lewy body pathology.
The other family members did not show any signs of PD, but we did not systematically do DaT scanning or smell testing to look for preclinical nigra degeneration or premotor signs, respectively.
The combination of parkinsonism and neuropathy has a differential diagnosis, but we believe that the specific context of this family rules out most alternative explanations. There is a possibility of a second genetic condition in the family that explains the parkinsonism. We were able to exclude mutations in most of the currently known PARK genes, but a mutation in another gene – yet unknown to be related to genetic forms of parkinsonism – remains possible.
Interestingly, there is an older report on seven cases, of which five were familial, with CMT type 2 and dopamine‐responsive parkinsonism that developed years later.5 We have contacted the first author, who indicated that there was no genetic follow‐up and DNA samples were not available for testing (C. Tranchant, pers. comm.).
LRSAM1 encodes a multifunctional protein and is expressed in the fetal and adult nervous system.1 Its ubiquitylating capacity was shown to be perturbed as a result of the Dutch mutation. The only known target for LRSAM1 ubiquitylation is TSG101, a component of the ESCRT machinery (endosomal‐sorting complex required for transport). Ubiquitylation has mostly been associated with protein degradation, however, other nonproteolytic pathways have been described, such as internalization and modulation of protein interactions, cellular localization, transcription, and signaling. The fate of the ubiquitylated substrate largely depends on the type of ubiquitin modification and other (opposing) factors such as the deubiquitylating enzymes. Little is known about the specificity of noncanonical polyubiquitylation that LRSAM1 has been described to favor in vitro. It is interesting to note that parkin also mediates formation of Lys‐27‐ubiquitin chains.6 Mutations in PINK1 and parkin mediate degradation of mitochondria through mitophagy, and lead to failure of mitochondrial translocation.7 Recently, LRSAM1 was shown to be crucial for autophagy of intracellular Salmonella of infected HeLa cells. This raises intriguing questions regarding the endogenous LRSAM1 targets and seems to link processes of mitochondrial dynamics and autophagy. Animal models have proven that LRSAM1 mutations cause neurological disease in mouse and zebrafish.1, 8 In this respect, it is interesting to note that lrsam1−/− mice when challenged with acrylamide showed poor coordination and tremor.8 Additionally, LRSAM1 has been linked to Huntington's disease (HD), suggesting that LRSAM1 also plays a role in CNS neurons.9 It is yet unclear what this role is and how mutations in this gene lead to substantia nigra pathology.
Further studies are encouraged, such as carefully checking for signs of PD in other LRSAM1 pedigrees, considering to test for LRSAM1 mutations in patients with this particular combination, and investigating whether mutations in this gene are involved in genetic forms of parkinsonism in the absence of neuropathy.
Author Contributions
M. B. A. carried out the clinical evaluation and wrote the first draft; M. A. J. W., H. J. S., and F. B. were involved in the original gene identification and critical review of the paper; R. A. E. and B. R. B. contributed to the clinical evaluation and critical review of the paper; M. Q. and V. B. did the genetic analyses and critical review of the paper; and B. P. W. contributed to the clinical evaluation, writing of the paper, and general supervision.
Conflict of Interest
Dr. Bloem reports grants from Netherlands Organization for Scientific Research, Prinses Beatrix Foundation, Stichting Parkinson Fonds, Michael J Fox Foundation, Parkinson Vereniging, National Parkinson Foundation, personal fees from Danone, Zambon, Abbvie, Teva, outside the submitted work. Dr. Bonifati reports personal fees from Elsevier Ltd, Springer Science + Business Media, LLC, outside the submitted work. Dr. van de Warrenburg reports grants from Radboud university medical center, BBMRI‐NL, Gossweiler Foundation, ‘Wetenschapsfonds’ Dutch dystonia society, outside the submitted work.
Supporting information
Data S1. Mutation Analysis of Genes That Can Lead to Genetic Forms of Parkinsonism
References
- 1. Weterman MA, Sorrentino V, Kasher PR, et al. A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum Mol Genet 2012;21:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nicolaou P, Cianchetti C, Minaidou A, et al. A novel LRSAM1 mutation is associated with autosomal dominant axonal Charcot‐Marie‐Tooth disease. Eur J Hum Genet 2013;21:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Engeholm M, Sekler J, Schöndorf DC, et al. A novel mutation in LRSAM1 causes axonal Charcot‐Marie‐Tooth disease with dominant inheritance. BMC Neurol 2014;14:118. doi:10.1186/1471‐2377‐14‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guernsey DL, Jiang H, Bedard K, et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot‐Marie‐Tooth disease. PLoS Genet 2010;6: e1001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tranchant C, Ruh D, Warter JM. Type II Charcot‐Marie‐Tooth and dopa‐sensitive Parkinson disease. Rev Neurol (Paris) 1994;150:72–74. [PubMed] [Google Scholar]
- 6. Huett A, Heath RJ, Begun J, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin‐dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe 2012;12:778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010;12:119–131. [DOI] [PubMed] [Google Scholar]
- 8. Bogdanik LP, Sleigh JN, Tian C, et al. Loss of the E3 ubiquitin ligase LRSAM1 sensitizes peripheral axons to degeneration in a mouse model of Charcot‐Marie‐Tooth disease. Dis Model Mech 2013;6:780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang B, Seredenina T, Coppola G, et al. Gene expression profiling of R6/2 transgenic mice with different CAG repeat lengths reveals genes associated with disease onset and progression in Huntington's disease. Neurobiol Dis 2011;42:459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Mutation Analysis of Genes That Can Lead to Genetic Forms of Parkinsonism