

Abstract
The protein SF3B1 is a core component of the spliceosome, the large ribonucleoprotein complex responsible for pre-mRNA splicing. Interest in SF3B1 intensified when tumor exome sequencing revealed frequent specific SF3B1 mutations in a variety of neoplasia and when SF3B1 was identified as the target of three different cancer cell growth inhibitors. A better mechanistic understanding of SF3B1's role in splicing is required to capitalize on these discoveries. Using the inhibitor compounds, we probed SF3B1 function in the spliceosome in an in vitro splicing system. Formerly, the inhibitors were shown to block early steps of spliceosome assembly, consistent with a previously determined role of SF3B1 in intron recognition. We now report that SF3B1 inhibitors also interfere with later events in the spliceosome cycle, including exon ligation. These observations are consistent with a requirement for SF3B1 throughout the splicing process. Additional experiments aimed at understanding how three structurally distinct molecules produce nearly identical effects on splicing revealed that inactive analogs of each compound interchangeably compete with the active inhibitors to restore splicing. The competition indicates that all three types of compounds interact with the same site on SF3B1 and likely interfere with its function by the same mechanism, supporting a shared pharmacophore model. It also suggests that SF3B1 inhibition does not result from binding alone, but is consistent with a model in which the compounds affect a conformational change in the protein. Together, our studies reveal new mechanistic insight into SF3B1 as a principal player in the spliceosome and as a target of inhibitor compounds.
Keywords: SF3B1, spliceosome, pre-mRNA splicing, inhibitor
INTRODUCTION
The spliceosome is the extremely complicated ribonucleoprotein (RNP) machine responsible for pre-mRNA splicing, an essential step in eukaryotic gene expression. The complexity of the spliceosome regularly impedes functional characterization of its over 100 components. One of these components is SF3B1, which with six other proteins forms the SF3B subunit of the U2 small nuclear RNP (snRNP). SF3B1 has garnered recent attention in cancer biology. It was identified as the molecular target of three potent cytotoxic agents: spliceostatin A (SSA, 1), pladienolide B (PB, 2), and herboxidiene (HB, 3) (Kaida et al. 2007; Kotake et al. 2007; Hasegawa et al. 2011), which are being investigated as potential chemotherapeutic leads (Eskens et al. 2013). Additionally, a specific set of SF3B1 mutations is prevalent in a variety of cancers (Yoshida and Ogawa 2014). However, how SF3B1 and its mutations contribute to cancer is murky, in large part because we do not know the details of SF3B1 function in the spliceosome.
When SF3B1 is depleted or targeted by inhibitors, in vitro spliceosome assembly halts early at the point in which U2 snRNP recognizes a sequence in introns called the branch point (Brosi et al. 1993; Roybal and Jurica 2010; Corrionero et al. 2011; Folco et al. 2011; Effenberger et al. 2014). Yet there are hints that SF3B1 may function at multiple stages because the stability of its association with the spliceosome appears to be regulated (Coltri et al. 2010; Lardelli et al. 2010; Ilagan et al. 2013). For each splicing event, five snRNPs (U1, U2, U4, U5, and U6) and dozens of additional proteins associate, rearrange, and leave to create a catalytically competent spliceosome. This “spliceosome cycle” is often represented as a dynamic series of intermediate complexes (H/E → A → B → C) based on the components present and stage of splicing catalysis. As noted above, SF3B1 joins early with U2 snRNP during A complex formation, but, unlike most other spliceosome components that play an early role in intron recognition and then leave, SF3B1 remains associated with the spliceosome after full assembly to B complex and through activation and catalysis (Agafonov et al. 2011; Ilagan et al. 2013). Until now, SF3B1 function at these later stages has not been directly examined.
Another open question regarding SF3B1 inhibition is how three compounds with very different chemical structures exhibit nearly identical effects on spliceosome assembly and in cells. Structure–activity relationship (SAR) data for the three compounds and related molecules have been steadily emerging (Sakai et al. 2002; Mizui et al. 2004; Lagisetti et al. 2008, 2013, 2014; Albert et al. 2009; Fan et al. 2011; Gundluru et al. 2011; Muller et al. 2011; Villa et al. 2012, 2013; Gao et al. 2013; Ghosh and Chen 2013; Arai et al. 2014; Effenberger et al. 2014; Ghosh et al. 2014a,b,c; He et al. 2014). SSA (1), which is similar to FR901464 (Nakajima et al. 1996b), meayamycin (Albert et al. 2009), thailanstatins (Liu et al. 2013), and sudemycins (Fan et al. 2011), differs in structure relative to the other two compounds. PB (2), which is related to E7107 and FD-895 (Kotake et al. 2007; Villa et al. 2012), and HB (3), a member of GEX1 family (Sakai et al. 2002), share a similar side chain, but have different ring structures (macrolide vs. tetrahydropyran). A common pharmacophore for the three types of inhibitors has been suggested (Lagisetti et al. 2008, 2014), but no shared feature has been shown to be critical for activity. It is important to know how these compounds individually interact with SF3B1 and how these interactions interfere with its activity. This information is critical for comparing the effects of the different compounds and identification of the best candidate for drug development.
In cells, treatment with SF3B1 inhibitors leads to changes in alternative splicing of genes involved in apoptosis and the cell cycle, among others (Kaida et al. 2007; Kotake et al. 2007; Lagisetti et al. 2009; Corrionero et al. 2011; Hasegawa et al. 2011; Convertini et al. 2014; Effenberger et al. 2014). Microarray analysis of 2000 splicing events from nearly 500 genes suggested that SSA affects alternative splicing of introns with weak branch points (Corrionero et al. 2011). The selective effect of SF3B1 inhibitors on a limited set of gene transcripts presumably results in tumor cells showing higher sensitivity than healthy cells (Lagisetti et al. 2008), but what triggers changes in particular splicing events as a result of SF3B1 inhibition remains to be determined. At some level, pre-mRNA sequence must play a role, but how that sequence information is communicated to the spliceosome is poorly understood. By directly examining the relationship between intron sequence and SF3B1 activity, we may be able to understand how alternative splicing is modulated by the drugs, and why tumor cells exhibit higher sensitivity.
In this paper, we present data that address questions of SF3B1 function and its interactions with inhibitors. Our results suggest that SF3B1 inhibitors share a common interaction site and we propose that they interfere with a conformational change in the protein to affect its activity. By bypassing the earlier stages of inhibition, we find that the compounds interfere with additional stages of spliceosome assembly and catalysis, suggesting that SF3B1 activity is required at multiple steps. We also show that weakening branch point and polypyrimidine sequence does not necessarily confer sensitivity to SF3B1 splicing inhibition in vitro, indicating that in cells additional factors likely contribute to which genes the drugs will target. Together, our results expand the role of SF3B1 function in the spliceosome and show that SF3B1 inhibitors share the same mechanism, which is fundamental to understanding what happens when SF3B1 is mutated or modulated in cells.
RESULTS
Inactive analogs compete with SF3B1 inhibitors
It is well established that SSA (1), PB (2), and HB (3) bind to the core splicing protein SF3B1 and inhibit splicing in the in vitro system. We previously identified inactive analogs of the three compounds [Fig. 1; iSSA (SSE), 4; inactive pladienolide (iPB), 5; inactive herboxidiene (iHB), 6] that revealed different structural features crucial for splicing inhibition (Roybal and Jurica 2010; Effenberger et al. 2013, 2014; Ghosh et al. 2014b,c). However, we do not know whether the compounds are inactive because they cannot interact with SF3B1 any longer, or because they interact but do not interfere with the function of SF3B1. To distinguish between the two scenarios, we set up competition assays in which we incubated a pre-mRNA substrate under splicing conditions with different ratios of active to inactive compounds (Fig. 2A). If an inactive compound still interacts with SF3B1, then it will outcompete the active version and, when in excess, restore splicing. On the other hand, if inactive compounds do not interact, then even large excess will not be able to outcompete the active version and splicing will not be restored. As previously shown, SSA (1), PB (2), and HB (3) each completely inhibit in vitro splicing at 1 µM, whereas the corresponding inactive derivatives have no effect (Fig. 2A, lanes 3–5 versus 6–7). Surprisingly, addition of the corresponding inactive compound at increasing concentrations restores splicing completely for PB (2) and HB (3), and partially for SSA (1) (Fig. 2A, lanes 9–17). For example, 10 µM iHB (6) restores ∼70% of splicing in the presence of 1 µM HB (3), and 100 µM iHB (6) restores essentially all splicing. iSSA (SSE, 4) also restores splicing in the presence of SSA (1), but only partially, even at the highest concentration tested. This result is consistent with the inactive compounds interacting with SF3B1 in competition with their active counterparts, although with different potencies. In that scenario, it would also mean that interaction with SF3B1 is not sufficient for splicing inhibition, because the inactive analogs do not interfere with in vitro splicing. Instead, the compounds could impair SF3B1 by modulating a conformational change in the protein or a binding partner, and different small changes of chemical features (i.e., the separate features altered in the inactive compounds) affect how the compounds interfere with the structural shift. Alternatively, the active compounds could sterically occlude an interaction between SF3B1 and another spliceosome protein or RNA. However, it is more difficult to imagine how small changes in the inactive compound would maintain binding, but no longer sterically interfere with an SF3B1 interaction.
FIGURE 1.

Chemical structures of compounds used in this study. SSA (1), spliceostatin A; PB (2), pladienolide B; HB (3), herboxidiene; iSSA (4), inactive spliceostatin A (also known as SSE, spliceostatin E); iPB (5), inactive pladienolide B; iHB (6), inactive herboxidiene. IC50 refers to the concentration required to reduce in vitro splicing by half compared to DMSO-treated control reactions. Inactive compounds do not interfere with in vitro splicing up to a concentration of 200 µM.
FIGURE 2.
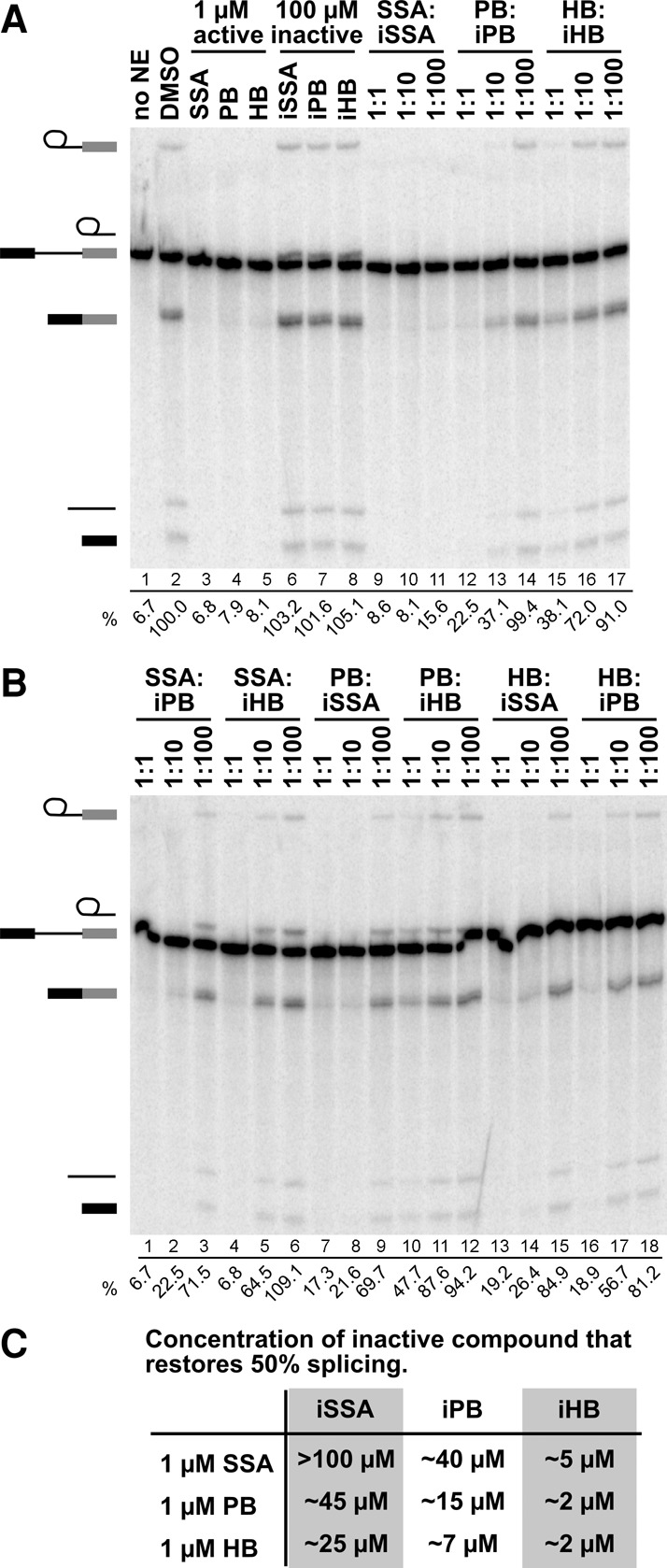
SSA, PB, and HB bind to the same site on SF3B1. (A) Denaturing gel analysis of RNA isolated from splicing reactions incubated with no nuclear extract (NE), DMSO, or the indicated compounds. In lanes 3–8, compounds were added at the specified concentration; in lanes 9–17, splicing reactions contained 1 µM active compound and 1, 10, or 100 µM inactive compound. Identities of bands are schematized to the left as (from top to bottom) lariat intermediate, free lariat, pre-mRNA, mRNA, free intron, and 5′ exon intermediate. Splicing efficiency, shown below each lane, is determined as the percentage of mRNA relative to the sum of pre-mRNA, intermediates, and mRNA. (B) Same as panel A. (C) Columns list the concentration of inactive compounds that restores 50% splicing in the presence of 1 µM active compound (rows). Values are estimated from dosage response plots of the splicing reactions shown in A and B.
Three distinct SF3B1 inhibitors bind to the same site
Given the similar effects that the three SF3B1 inhibitors have on cell growth and morphology, and the potential of a common pharmacophore, we hypothesized that SSA (1), PB (2), and HB (3) interact with the same site on SF3B1. We tested this hypothesis using the same competition assay with an active inhibitor and increasing concentrations of inactive analogs of the two structurally distinct inhibitors [e.g., SSA (1) with increasing concentrations of iPB (5) and iHB (6)]. Strikingly, all three inactive analogs are able to outcompete any other active compound (Fig. 2B). This result provides the first evidence that all three inhibitors interact with the same site in SF3B1.
Based on plots of splicing efficiency in the presence of 1 µM active compound vs. increasing active compound, we estimated effective concentration of inactive compound that restores splicing by 50%. These data indicate that both active and inactive compounds differ in their affinity for SF3B1 (Fig. 2C). With all the inactive analogs, we found that less compound is required to compete for HB (3) inhibition relative to the amounts required to compete with PB (2) and SSA (1). Conversely, more of each inactive compound was required to compete with SSA (1) relative to HB (3) and PB (2). These data allow us to rank the active compounds relative to one another for SF3B1 affinity, with SSA (1) > PB (2) > HB (3). Similarly, the inactive analogs can also be ranked according to their affinities. Consistently, less iHB (6) was required to compete with all three active compounds relative to iPB (5) and iSSA (SSE, 4). In contrast, iSSA (SSE, 4) was the worst competitor of the inactive compounds for all three active molecules. If competition between active and inactive compounds is mediated by affinity, these results indicate that the feature that is modified to inactivate SSA (1) is more important for SF3B1 interaction than the features that are modified in PB (2) and HB (3), respectively.
SF3B1 inhibition can be independent of branch point sequence in vitro
Corrionero et al. (2011) reported that in cells alternative splicing changes caused by SSA (1) are more likely to occur with introns containing weak branch point sequences. They proposed that the drug allows nearby strong branch sequence decoys to compete with a weak branch point for U2 small nuclear RNA (snRNA) base pairing. To look at the interplay between pre-mRNA sequence and susceptibility to SF3B1 inhibition, we tested the hypothesis that splicing substrates with a weak branch point sequence will be more sensitive to SF3B1 inhibitors relative to those with a strong branch point sequence. If the function of SF3B1 targeted by the inhibitors can be compensated by more stable U2 snRNA base pairing, we expected to see an increase in IC50 value with branch sequence strength (i.e., more drug is required for inhibition). We generated pre-mRNA substrates for in vitro splicing with different branch point sequence strengths, including those from introns that show drug sensitivity in cells (Corrionero et al. 2011; Effenberger et al. 2014) and measured splicing efficiency of these pre-mRNAs with increasing concentration of the drug SSA (1) (Fig. 3A). As expected, we found that changes from consensus in branch point sequence result in a decreased splicing efficiency in the in vitro assay system (Supplemental Fig. S1A). However, the concentration of SSA that reduced splicing by 50% relative to the no-drug control was similar for each substrate. Consistent with a common interaction with SF3B1, we saw similar results with PB (Fig. 3B; Supplemental Fig. S1B). These data indicate that substrates with the different branch point sequences that we tested have the same dependence on SF3B1 in NE. It also implies that U2 snRNA/pre-mRNA base pairing alone may not explain the apparent different sensitivities of splicing events to SF3B1 inhibitors in cells.
FIGURE 3.
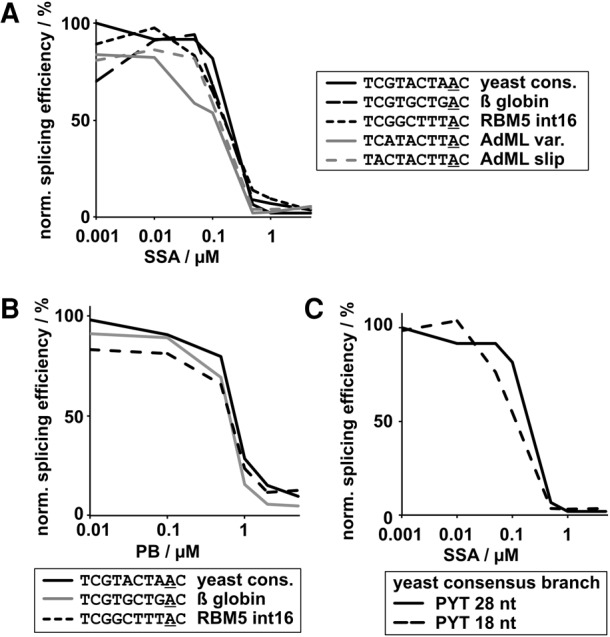
Branch point region and polypyrimidine tract (PYT) strength do not correlate with compound sensitivity. In vitro splicing efficiencies of pre-mRNA substrates with different branch point regions normalized to DMSO control reactions are shown plotted against (A) SSA (1) or (B) PB (2) concentration. The branch point A is underlined. (C) Same as A using pre-mRNA substrates with different PYT length.
Because SF3B1 interactions with the protein U2AF2 at the polypyrimidine tract (PYT) downstream from the branch point are also important for splicing (Gozani et al. 1998), we also examined the effect of PYT length on SF3B1 inhibition (Fig. 3C; Supplemental Fig. S1C). Again, the splicing efficiency is decreased with a shorter PYT, but the IC50 value for SSA (1) is not affected. This result shows that in an in vitro context, the function of SF3B1 that is targeted by the drug is not compensated by its interaction with U2AF2 bound to the PYT. Together, these results show that splicing sequence strength of these substrates alone does not confer increased sensitivity to SSA in vitro. It also suggests that other factors and/or additional sequence context play a role in mediating the differential splicing changes observed for transcripts with SF3B1 inhibitors in cells. Alternatively, or in addition, differences in branch point sequence could affect spliceosome assembly at a kinetic level, which is mediated by SF3B1. Such differences may not be apparent from our splicing assays, which are end-point assays. However, in the context of competing splice sites in cells, a difference in assembly rate could result in alternative splicing changes.
SF3B1 inhibitors interfere with spliceosome assembly after ATP-dependent stabilization of A complex
All three SF3B1 inhibitors have been shown to affect early spliceosome assembly in vitro by causing a stall at an A-like complex (Roybal and Jurica 2010; Corrionero et al. 2011; Folco et al. 2011; Effenberger et al. 2014). Additionally, Folco et al. (2011) reported that the PB analog E7107 inhibits binding of an oligonucleotide containing the branch point sequence to U2 snRNP in NE. Notably, when the extract was pre-treated with ATP, the compound had no effect, which led to a proposal that E7107 (and by extension, PB) blocks an ATP-dependent conformational change in U2 snRNP that exposes the branch point binding sequence and allows for stable U2 snRNP association with A complex. It is not clear whether the conformational change occurs prior to U2 snRNP association with pre-mRNA and whether it is the same ATP-dependent step associated with stabilization of the A complex. Arguing against the later possibility, base pairing between U2 snRNA and pre-mRNA occurring both before ATP addition and in SSA-inhibited spliceosomes has been shown (Wassarman and Steitz 1992; Corrionero et al. 2011).
To shed light onto these questions, we first tested the hypothesis that splicing inhibition by PB is due to its blocking the ATP-dependent conformational change in U2 snRNP, which is mediated by SF3B1 prior to A complex assembly. If true, spliceosome assembly would not be inhibited if U2 snRNP had already taken on this conformation. We pre-incubated NE with ATP before and after PB (2) addition, and then used those extracts for in vitro splicing. In both cases, we observe a loss of splicing and a block in assembly at the same A-like complex (Fig. 4A). We conclude that the role of SF3B1 in modulating an ATP-dependent conformational change in the isolated U2 snRNP previously described is not sufficient for spliceosome assembly, which suggests additional functions for SF3B1 in splicing.
FIGURE 4.

SF3B1 inhibitors interfere with spliceosome assembly after stable A complex formation. (A) (Top panel) Native gel analysis of spliceosome complexes assembled in HeLa NE incubated for indicated time (minutes). No ATP refers to ATP-depleted NE; +ATP indicates ATP-depleted extract with added ATP; in lanes 3–6, NE was pre-incubated with 1 µM PB (2) followed by addition of ATP and pre-mRNA substrate; in lanes 7–10, NE was pre-incubated with ATP followed by addition of 1 µM PB (2) and pre-mRNA substrate. The identities of spliceosome complexes are denoted with assembly occurring in the following order: H/E → A → B → C. (Lower panel) Denaturing gel analysis of the same in vitro splicing reactions shown in the top. Band identities are indicated on the left as in Figure 2. (B) Representative native gel analysis of spliceosome complexes assembled for 30 min at 30°C in the presence of PB (2) (lanes 1–5) or in NE depleted of U4/6 snRNAs (ΔU4/6), after which DMSO, 1 µM PB (2), or iPB (5) was added. The complexes were then challenged with increasing heparin (hep.) concentration in which 1x is 0.5 mg/mL. (C) Native gels as shown in B were quantified to calculate the percent of A complex relative to total splicing complexes with increasing heparin concentration. Values were normalized to the amount of A complex in 0.5 mg/mL (1x) heparin and represent averages of technical replicates of 2–4 independent assays. (D) Lanes 1–9 show native gel analysis of spliceosome complexes assembled for 30 min (unless otherwise indicated) at 30°C in: no NE, mock-depleted NE (mock), U2 or U4/U6 snRNA-depleted NE (ΔU2, ΔU4/U6), a mixture of ΔU2 and ΔU4/U6 (mix). For lanes 10–12, stable A complex was assembled in ΔU4/6 NE for 10 min, after which ΔU2 NE and indicated compounds were added and incubated for an additional 20 min. When included, HB and SSA are at 5 µM.
Previous research showed that SF3B1 inhibitors interfere with stabilization of A complex and the ATP-dependent formation of direct interactions between SF3B1 and pre-mRNA on either side of the branch point (Gozani et al. 1996, 1998; Corrionero et al. 2011; Folco et al. 2011). Those studies showed that in the presence of SSA and E7107, the A-like complex that forms lacks SF3B1/pre-mRNA interactions and is sensitive to heparin treatment (Corrionero et al. 2011; Folco et al. 2011). We also find that an A-like complex that forms in the presence of PB is unstable with increasing heparin concentrations, consistent with the proposed function of SF3B1 in the ATP-dependent stabilization of U2 snRNP at the branch point sequence (Fig. 4B, lanes 1–5; Fig. 4C). If the role of SF3B1 in splicing is limited to achieving stable A complex, SF3B1 inhibitors should not affect assembly following stable U2 snRNP incorporation into the spliceosome. To test this hypothesis, we bypassed the first block induced by the compounds by assembling stable A complex in NE depleted of U4/U6 snRNAs. We achieved depletion by using the endogenous RNase H present in splicing extracts in combination with short DNA oligonucleotides complementary to U4 and U6 snRNAs (Blencowe et al. 1989) (Supplemental Fig. S2A). As expected, the A complex that accumulates in this extract is much more stable to heparin concentration relative to the complexes that form in the presence of SF3B1 inhibitors (Fig. 4B, lanes 6–10; Fig. 4C). We then used these stable A complexes to test whether SF3B1 inhibitors would affect the formation of mature spliceosomes when chased with new extract containing U4/U6 snRNAs. To protect the U4/U6 snRNAs in the chase extract, we used DNase I digestion to destroy the RNase H oligos in the ΔU4/6 extracts. To prevent additional A complex assembly, we also depleted U2 snRNA in the chase extract by the same RNase H digestion followed by DNaseI digestion protocol (Supplemental Fig. S2A). As expected, no complexes form past E/H in the ΔU2 extract (Fig. 4D, lanes 5 and 7), and when ΔU4/6 and ΔU2 extracts are mixed, normal complex formation occurs (Fig. 4D, lane 3).
With DMSO in the ΔU2 chase extract, spliceosome assembly is rescued as evidenced by the appearance of a robust B/C complex band (Fig. 4D, lane 10). In contrast, little to no higher-order spliceosome complexes form in the presence of SF3B1 inhibitors HB (3) and SSA (1) (Fig. 4D, lanes 11 and 12); and although a small amount of B/C complex may still be forming, we do not detect splicing chemistry (Supplemental Fig. S2C). This result is not consistent with our original hypothesis, because ATP-dependent stabilization of A complex is not sufficient to bypass further sensitivity to SF3B1 inhibitors. Instead, the results suggest two possible scenarios, which are not mutually exclusive: (1) There is another requirement for SF3B1 after ATP-dependent stabilization of A complex, and/or (2) ATP-dependent stabilization of A complex is reversible and SF3B1 is required to maintain the stable conformation. To address the second possibility, we assembled A complex in ΔU4/U6 NE, then added DMSO, PB (2), or iPB (5), and then challenged the complexes with increasing heparin concentration. Native gel show that at high heparin concentration, ATP–stabilized A complex appears to partially convert to a faster migrating complex, but compared with DMSO control, both active and inactive drugs have no effect on the complex (Fig. 4B, lanes 6–20; Fig. 4C). This result does not support the second scenario, which predicts that A complex would be less stable in the presence of active drugs. It is consistent with SF3B1 playing a role in progression to B complex, in addition to its role in A complex assembly. There is, however, also a formal possibility that binding of the inhibitors to SF3B1 interferes with the activity of another component of the spliceosome required for this transition, for which SF3B1 is normally a passive bystander.
SF3B1 inhibitors block exon ligation
Given that SF3B1 is present throughout the spliceosome cycle (Agafonov et al. 2011; Ilagan et al. 2013), we hypothesized that in addition to its likely roles in transitioning into and out of A complex, it also functions at later stages of spliceosome assembly. We decided to examine this possibility in the context of the catalytic spliceosome, specifically between the two steps of splicing chemistry, using the bimolecular exon ligation reaction (Fig. 5A; Konforti and Konarska 1995; Anderson and Moore 1997). If SF3B1 also has a role in the spliceosome after 1st step chemistry, then exon ligation would be affected by SF3B1 inhibitors. To bypass the early spliceosome assembly blocks induced by PB (2), we incubated an unlabeled pre-mRNA 5′ substrate that contains a 5′ exon and an intron in NE. Spliceosome assemble on this substrate and complete 1st step chemistry to produce a free 5′ exon and lariat intron intermediate (Konforti and Konarska 1995; Anderson and Moore 1997). We then added a labeled 3′ substrate consisting of a 3′ exon preceded by a 3′ splice site with and without drug. In the assay, with just DMSO added, we detected a labeled mRNA band that is a result of exon ligation (Fig. 5B, lane 3). However, the mRNA band nearly disappears with increasing concentration of PB (2) (Fig. 5B, lanes 4–9). We repeated the assay with SSA (1), with the same result (Supplemental Fig. S3). The inhibition of exon ligation indicates that PB (2) and SSA (1) interact with their target, SF3B1, in the catalytic spliceosome. It is also consistent with a functional requirement for SF3B1 after 1st step chemistry. There is no data linking SF3B1 directly to splicing catalysis, so it is likely that the protein plays a structural role such as positioning RNA and/or protein, the conformation of which is modulated by the inhibitor. Again, however, we cannot rule out the possibility that binding of the inhibitors to SF3B1 interferes with the activity of another component of the spliceosome required for exon ligation and that SF3B1 simply is a spectator. We also note that relative to the earliest point of inhibition, a higher concentration of the drug is required to block exon ligation. We have not determined the basis for this difference, although one possibility may be that the interaction site in SF3B1 for inhibitors may be partially occluded in the context of the catalytic spliceosome.
FIGURE 5.
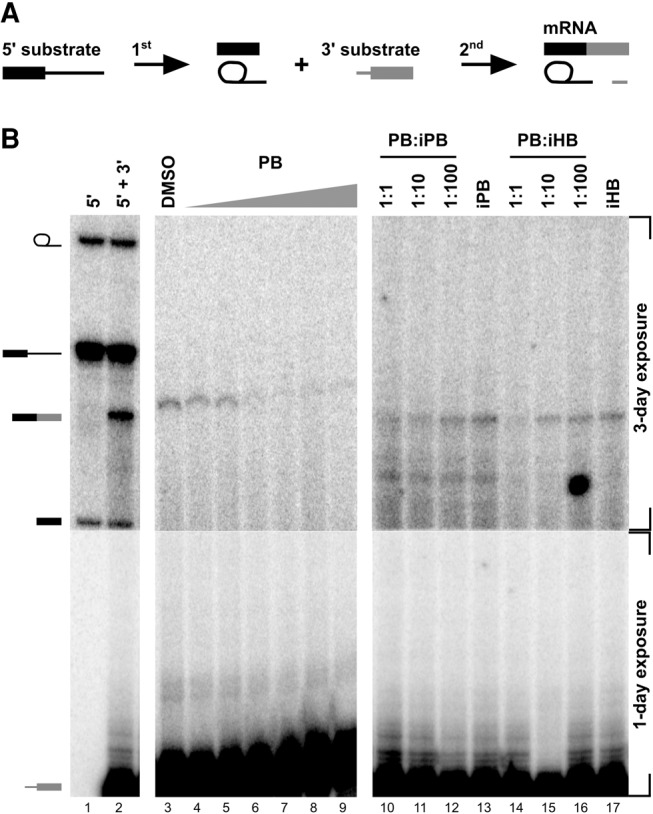
Bimolecular assay shows that PB inhibits exon ligation. (A) Schematic of the bimolecular exon ligation reaction. The 5′ substrate consists of a 5′ exon and intron containing the branch point and PYT and is capable of 1st step splicing chemistry to produce splicing intermediates. The 3′ substrate consists of a 3′ splice site and 3′ exon and is capable of 2nd step chemistry to produce mRNA when added in trans. To test for inhibition, PB is added with the 3′ substrate. (B) Denaturing gel analysis of in vitro bimolecular exon ligation reactions. Identities of bands are schematized to the left as (from top to bottom) lariat intermediate, 5′ substrate, mRNA, 5′ exon, and 3′ exon. A 3-d exposure is shown for the top region of the gel, while the bottom region is a 1-d exposure. In lanes 1 and 2, both 5′ and 3′ substrates are radiolabeled. In lanes 3–17, only the 3′ substrate is radiolabeled. In lanes 4–9, increasing concentrations of PB (2) (0.1, 1, 2, 5, 10, 100 µM) were included with the 3′ substrate. In lanes 10–12 and 14–16, active compound was added at 2 µM and increasing concentrations of inactive compounds were added at the indicated ratio. In lanes 13 and 17, inactive compound is at 200 µM.
We also tested whether inactive analogs could compete with PB (2) and SSA (1) inhibition of exon ligation. As with earlier inhibited steps, increasing concentrations of both inactive PB (5) and inactive HB (6) restore mRNA production (Fig. 5B, lanes 10–17; Supplemental Fig. S3). This result supports the idea that a similar SF3B1 function is required at multiple steps of the spliceosome cycle, including after 1st step chemistry.
DISCUSSION
Our studies shed new light on the role of SF3B1 in the spliceosome and the mechanisms of splicing inhibition by three structurally distinct SF3B1 inhibitors. In terms of inhibitor mechanism, we found that inactivating changes to SF3B1 inhibitors do not necessarily abrogate compound interactions with the protein. This result means that interaction alone is not sufficient to confer SF3B1 inhibition, and leads us to speculate that the inhibitors also interfere with a conformational change in SF3B1 that is required for its role in splicing (Fig. 6).
FIGURE 6.
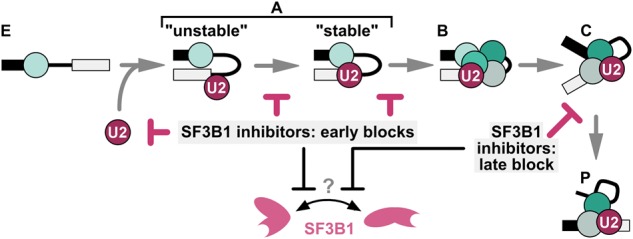
Model of SF3B1 function in the spliceosome cycle. Spliceosomes assemble on a pre-mRNA substrate through a series of intermediate splicing complexes (E → A → B → C → P). We propose that SF3B1 inhibitors affect assembly at multiple steps by interfering with a conformational change in SF3B1.
We also found that SSA (1), PB (2), and HB (3) very likely interact with the same site on SF3B1, which strongly supports the presence of a common pharmacophore between the compounds, as predicted by Webb and co-workers (Lagisetti et al. 2008, 2014), although the full features of the pharmacophore are still not fully apparent. PB (2) and HB (3) resemble each other with a side chain linked to a ring structure. The side chains are similar, and the positions that differ do not affect PB (2) activity as evidenced by analogs that also differ in those positions and still retain activity (Sakai et al. 2002; Mizui et al. 2004; Effenberger et al. 2014; Ghosh et al. 2014b). In our study, the modification that inactivates PB (5) is the loss of two methyl groups in the side chain, a change that has never been present in active HB analogs (Sakai et al. 2002; Effenberger et al. 2014). In terms of the rings, the PB (2) macrolide ring is more complex than the tetrahydropyran of HB (3), and the positions that are altered in inactive HB (6) do not have an obvious equivalent in PB (2). Previous SAR data for PB indicates that an acetyl group on the macrolide ring is important (Mizui et al. 2004). SSA (1) has a very different architecture, and the shared characteristics with PB (2) and HB (3) are not obvious. The inactivating change in SSA (4) is localized to the ring containing an epoxide (note that iSSA is equivalent to SSE) (Ghosh et al. 2014c). Our data indicate a shared interaction site, and it will be very interesting to determine how these three compounds with very different structures interact with SF3B1. It is also remarkable that changes to distinct chemical features inactivate them and yet do not abrogate binding. Nonetheless, these data represent an excellent starting point for SAR studies to differentiate the features of the common pharmacophore that confer binding from those that directly confer inhibition.
Even though these compounds behave similarly in vitro, their structural differences could affect their stability, permeability, etc. in cells. However, all three drugs have been tested in a cytological profiling assay in HeLa cells with nearly identical outcomes (Effenberger et al. 2014). The three compounds confer similar “mega-speckle” phenotypes in HeLa cells as determined by immunofluorescence staining with an antibody to the splicing factor SRSF2 (SC-35) (Kaida et al. 2007; Effenberger et al. 2014). The Koide laboratory showed that HB (3) and SSA (1) have similar IC50 values for splicing of a minigene reporter in HEK293-II cells, although meamycin B, another FR901464 derivative, is more potent purportedly because it is more stable (Gao et al. 2013). For splicing of endogenous genes, SSA (1) and HB (3) both cause retention of an intron in the p27 transcript (Kaida et al. 2007; Hasegawa et al. 2011), and SSA (1) and PB (2) both promote exon 16 skipping in the RBM5 transcript (Corrionero et al. 2011; Effenberger et al. 2014). The compounds have also each been assayed for cytotoxicity in a variety of cancer cell lines, however with little overlap (Nakajima et al. 1996a; Sakai et al. 2002, 2004; Mizui et al. 2004; Gao et al. 2013). The reported IC50 values are in the low nanomolar range for all three compounds, although in some cases, HB (3) seems to be a somewhat less potent growth inhibitor (Gao et al. 2013). Therefore, most indications suggest that as in vitro the compounds behave similarly in cells. Still, whether they all affect the same splicing events and can be used interchangeably needs to be tested directly.
In using the drugs to investigate SF3B1 function, we were surprised that in vitro the requirement for SF3B1 activity can be independent of branch point sequence and PYT strength. This result contrasts with the effect of SF3B1 inhibitors in cells, which appear to preferentially affect splicing of introns with weaker branch point sequences. We envision that in cells other factors, potentially including assembly kinetics, also play a role in specifying the alternative splicing changes induced by SF3B1 inhibitors. Identifying those factors will be key to understanding how cancer cells are sensitized to SF3B1 inhibitors.
The observation that SF3B1 inhibitors affect the spliceosome cycle from early assembly to splicing catalysis represents a new and important addition to our understanding of SF3B1 function in the spliceosome. Importantly, inhibition of SF3B1 at different stages is rescued by inactive analogs, suggesting that the same activity of SF3B1 is involved. We propose that SF3B1 cycles between conformational states to progress the spliceosome to the next stage, and that the inhibitors interfere with this change (Fig. 6). This model is consistent with observations from the Reed laboratory that a PB analog inhibits a conformational switch in SF3B1 that allows U2 snRNP to stably bind an oligonucleotide containing the branch point sequence (Folco et al. 2011). It is also consistent with the observation that SSA prevents a contact between SF3B1 and the pre-mRNA that normally forms during A complex stabilization (Gozani et al. 1996, 1998; Corrionero et al. 2011). We speculate that the changes in stability of the association of the SF3B complex with the spliceosome, which have been observed before catalytic activation and after exon ligation, result from the same conformational switch (Coltri et al. 2010; Lardelli et al. 2010; Ilagan et al. 2013). It is possible that changes in SF3B1 conformation are linked to the rearrangements in U2 snRNA structure that also cycle during spliceosome assembly and which clearly have roles in branch point recognition, A complex stabilization, and catalytic activation of the spliceosome. These rearrangements are in turn also linked to the activity of the RNA-dependent ATPases Prp5 and Prp16 (Perriman et al. 2003; Hilliker et al. 2007; Perriman and Ares 2007, 2010). Therefore, it may be significant that ATP treatment bypassed inhibition of U2 snRNP interactions with a branch point sequence oligo, and that ATP-stabilization renders A complex resistant to heparin in the presence of PB. It is possible that in both instances, the SF3B1 inhibitors interfere with the formation of an ATP-dependent interaction. However, once the interaction is in place, the compounds no longer impact that particular step. The SF3B1 inhibitors open the door to investigating these possibilities in the future.
MATERIALS AND METHODS
Synthesis of SF3B1 inhibitors and analogs
The synthesis of SF3B1 inhibitors and analogs as well as the determination of IC50 values is described elsewhere (Ghosh and Li 2011; Ghosh and Anderson 2012; Ghosh and Chen 2013; Effenberger et al. 2014; Ghosh et al. 2014b,c). iHB (6) synthesis will be reported in A Ghosh (in prep.).
In vitro splicing reactions
Pre-mRNA substrate was derived from the adenovirus major late (AdML) transcript. 32P-UTP body-labeled G(5′)ppp(5′)G-capped substrate was generated by T7 run-off transcription followed by gel purification. Where indicated, additional pre-mRNA substrates with varying branch point region or PYT length were used. NE was prepared as previously described (Dignam et al. 1983) from HeLa cells grown in DMEM/F-12 1:1 and 5% (v/v) newborn calf serum. For splicing reactions, 5 nM pre-mRNA substrate was incubated with 60 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg mL−1 tRNA, and 50% (v/v) HeLa NE at 30°C for 5 to 60 min. In some experiments, HeLa NEs were ATP-depleted by incubation at 30°C for 30 min. SF3B1 inhibitors, inactive analogs, and combinations thereof were added at the beginning of the splicing reactions unless otherwise noted.
Bimolecular exon ligation
Templates for 5′ and 3′ substrates were generated by PCR of the same AdML construct using primers that added a 5′ T7 promoter sequence. 32P-UTP body-labeled G(5′)ppp(5′)G-capped 5′ substrate and 32P-UTP body-labeled GMP-capped 3′ substrate were generated by T7 run-off transcription followed by gel purification. Unlabeled G(5′)ppp(5′)G-capped 5′ substrate was generated by T7 run-off transcription followed by size-exclusion chromatography. For bimolecular exon ligation reactions, 5 nM 5′ substrate was incubated under the in vitro splicing conditions described above. After 30 min, 3′ substrate with and without SF3B1 inhibitors was added to the reaction and incubated for an additional 60 min.
Denaturing gel analysis
RNA was extracted from in vitro splicing or bimolecular ligation reactions and separated on a 15% (v/v) denaturing polyacrylamide gel. 32P-labeled RNA species were visualized by phosphorimaging and quantified with ImageQuant software (Molecular Dynamics). Splicing efficiency was quantified as the amount of mRNA relative to total RNA and normalized to a dimethyl sulfoxide (DMSO) control reaction. IC50 values for inhibitors are defined as the concentration of inhibitor at which splicing efficiency is reduced by 50% and were derived from averaged plots of splicing efficiency versus compound concentration from 2–4 independent assays.
Native gel analysis
Splicing reactions were set up as described above and incubated at 30°C for 0–45 min. Time point samples were kept on ice until all samples were ready for analysis. For gels shown in Figure 4A,D, 10 µL of splicing reactions were mixed with 10 µL of 2× native gel loading buffer [20 mM Trizma base, 20 mM glycine, 25% (v/v) glycerol, 0.1% (w/v) cyan blue, 0.1% (w/v) bromphenol blue, 1 mg mL−1 heparin sulfate] and incubated at room temperature for 5 min before loading onto a 1.6% (w/v) low melting temperature agarose gel. Gels were run at 72 V for 3 h 50 min, vacuum-dried onto Whatman paper, and exposed to phosphorimaging screens, which were digitized with a Typhoon Scanner (Molecular Dynamics). For the heparin challenge experiments in Figure 3B, the 2× native gel loading buffer contained between 1 and 10 mg mL−1 heparin sulfate, and the splicing reactions were separated on 1.6% low melting temperature agarose gels run at 72 V for 3 h 30 min. Gels were dried and digitalized as described above, and quantified with the ImageQuant software (Molecular Dynamics). To determine A complex stability, A complex band intensity was compared relative to total lane intensity and normalized to samples treated with 2× native gel loading buffer containing 1 mg mL−1 heparin sulfate. Final values reported were derived from technical replicates of 2–4 independent assays.
snRNA-depletion of HeLa nuclear extracts
HeLa NEs were depleted of U2, U4, and U6 snRNAs using a combination of RNase H and DNase I digestion. First, NEs were incubated in the presence of endogenous RNase H, 2 mM magnesium acetate, 2 mM ATP, and 10 µM DNA oligonucleotides complementary to the snRNA to be depleted for 60 min at 30°C. Then, DNase I was added and incubated for 10 min at 30°C to digest the DNA oligonucleotides. The following DNA olignonucleotides were used for depletion: U2, 5′-ATCGCTTCTCGGCCT-3′ (Black et al. 1985); U4, 5′-AGCTTTGCGCAGTGG-3′, and 5′-CTAATTGAAAACTTTTCC-3′ (Black and Steitz 1986); U6, 5′-ACGCAAATCGTGAAGCG-3′ (Black and Steitz 1986). Depletion of extracts was confirmed by SYBR Gold direct staining of nucleic acids isolated from 10 µL of NEs and separated on a 7% (v/v) denaturing polyacrylamide gel.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the University of California Cancer Research Coordinating Committee, the Santa Cruz Cancer Benefit Society, and National Institutes of Health (grant R01GM72649 to M.S.J.) and by the Paul and Anne Irwin Graduate Fellowship in Cancer Research (awarded to K.A.E.).
Author contributions: M.S.J., K.A.E., V.K.U., and A.K.G. conceived and designed the experiments, analyzed the data, and wrote the paper. K.A.E., V.K.U., and B.E.P. performed the experiments.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.053108.115.
REFERENCES
- Agafonov DE, Deckert J, Wolf E, Odenwalder P, Bessonov S, Will CL, Urlaub H, Luhrmann R. 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31: 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert BJ, Mcpherson PA, O'Brien K, Czaicki NL, Destefino V, Osman S, Li M, Day BW, Grabowski PJ, Moore MJ, et al. 2009. Meayamycin inhibits pre-messenger RNA splicing and exhibits picomolar activity against multidrug-resistant cells. Mol Cancer Therapeutics 8: 2308–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K, Moore MJ. 1997. Bimolecular exon ligation by the human spliceosome. Science 276: 1712–1716. [DOI] [PubMed] [Google Scholar]
- Arai K, Buonamici S, Chan B, Corson L, Endo A, Gerard B, Hao MH, Karr C, Kira K, Lee L, et al. 2014. Total synthesis of 6-deoxypladienolide D and assessment of splicing inhibitory activity in a mutant SF3B1 cancer cell line. Org Lett 16: 5560–5563. [DOI] [PubMed] [Google Scholar]
- Black DL, Steitz JA. 1986. Pre-mRNA splicing in vitro requires intact U4/U6 small nuclear ribonucleoprotein. Cell 46: 697–704. [DOI] [PubMed] [Google Scholar]
- Black DL, Chabot B, Steitz JA. 1985. U2 as well as U1 small nuclear ribonucleoproteins are involved in premessenger RNA splicing. Cell 42: 737–750. [DOI] [PubMed] [Google Scholar]
- Blencowe BJ, Sproat BS, Ryder U, Barabino S, Lamond AI. 1989. Antisense probing of the human U4/U6 snRNP with biotinylated 2′-OMe RNA oligonucleotides. Cell 59: 531–539. [DOI] [PubMed] [Google Scholar]
- Brosi R, Hauri HP, Kramer A. 1993. Separation of splicing factor SF3 into two components and purification of SF3a activity. J Biol Chem 268: 17640–17646. [PubMed] [Google Scholar]
- Coltri P, Effenberger K, Chalkley RJ, Burlingame AL, Jurica MS. 2010. Breaking up the C complex spliceosome shows stable association of proteins with the lariat intron intermediate. PLoS One 6: e19061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertini P, Shen M, Potter PM, Palacios G, Lagisetti C, de la Grange P, Horbinski C, Fondufe-Mittendorf YN, Webb TR, Stamm S. 2014. Sudemycin E influences alternative splicing and changes chromatin modifications. Nucleic Acids Res 42: 4947–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Minana B, Valcarcel J. 2011. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RD. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Perriman RJ, Bray WM, Lokey RS, Ares M Jr, Jurica MS. 2013. A high-throughput splicing assay identifies new classes of inhibitors of human and yeast spliceosomes. J Biomol Screen 18: 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Anderson DD, Bray WM, Prichard BE, Ma N, Adams MS, Ghosh AK, Jurica MS. 2014. Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J Biol Chem 289: 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskens FA, Ramos FJ, Burger H, O'Brien JP, Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ, Baselga J, et al. 2013. Phase I, pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res 19: 6296–6304. [DOI] [PubMed] [Google Scholar]
- Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. 2011. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol 6: 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco EG, Coil KE, Reed R. 2011. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev 25: 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vogt A, Forsyth CJ, Koide K. 2013. Comparison of splicing factor 3b inhibitors in human cells. Chembiochem 14: 49–52. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Anderson DD. 2012. Enantioselective total synthesis of pladienolide B: a potent spliceosome inhibitor. Org Lett 14: 4730–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Chen ZH. 2013. Enantioselective syntheses of FR901464 and spliceostatin A: potent inhibitors of spliceosome. Org Lett 15: 5088–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Li J. 2011. A stereoselective synthesis of (+)-herboxidiene/GEX1A. Org Lett 13: 66–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Chen ZH, Effenberger KA, Jurica MS. 2014a. Enantioselective total syntheses of FR901464 and spliceostatin A and evaluation of splicing activity of key derivatives. J Org Chem 79: 5697–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Ma N, Effenberger KA, Jurica MS. 2014b. Total synthesis of GEX1Q1, assignment of C-5 stereoconfiguration and evaluation of spliceosome inhibitory activity. Org Lett 16: 3154–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Veitschegger AM, Sheri VR, Effenberger KA, Prichard BE, Jurica MS. 2014c. Enantioselective synthesis of spliceostatin E and evaluation of biological activity. Org Lett 16: 6200–6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Feld R, Reed R. 1996. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes Dev 10: 233–243. [DOI] [PubMed] [Google Scholar]
- Gozani O, Potashkin J, Reed R. 1998. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol 18: 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundluru MK, Pourpak A, Cui X, Morris SW, Webb TR. 2011. Design, synthesis and initial biological evaluation of a novel pladienolide analog scaffold. Medchemcomm 2: 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Miura T, Kuzuya K, Inoue A, Won Ki S, Horinouchi S, Yoshida T, Kunoh T, Koseki K, Mino K, et al. 2011. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol 6: 229–233. [DOI] [PubMed] [Google Scholar]
- He H, Ratnayake AS, Janso JE, He M, Yang HY, Loganzo F, Shor B, O'Donnell CJ, Koehn FE. 2014. Cytotoxic spliceostatins from Burkholderia sp. and their semisynthetic analogues. J Nat Prod 77: 1864–1870. [DOI] [PubMed] [Google Scholar]
- Hilliker AK, Mefford MA, Staley JP. 2007. U2 toggles iteratively between the stem IIa and stem IIc conformations to promote pre-mRNA splicing. Genes Dev 21: 821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan JO, Chalkley RJ, Burlingame AL, Jurica MS. 2013. Rearrangements within human spliceosomes captured after exon ligation. RNA 19: 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. 2007. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 3: 576–583. [DOI] [PubMed] [Google Scholar]
- Konforti BB, Konarska MM. 1995. A short 5′ splice site RNA oligo can participate in both steps of splicing in mammalian extracts. RNA 1: 815–827. [PMC free article] [PubMed] [Google Scholar]
- Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. 2007. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 3: 570–575. [DOI] [PubMed] [Google Scholar]
- Lagisetti C, Pourpak A, Jiang Q, Cui X, Goronga T, Morris SW, Webb TR. 2008. Antitumor compounds based on a natural product consensus pharmacophore. J Med Chem 51: 6220–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagisetti C, Pourpak A, Goronga T, Jiang Q, Cui X, Hyle J, Lahti JM, Morris SW, Webb TR. 2009. Synthetic mRNA splicing modulator compounds with in vivo antitumor activity. J Med Chem 52: 6979–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagisetti C, Palacios G, Goronga T, Freeman B, Caufield W, Webb TR. 2013. Optimization of antitumor modulators of pre-mRNA splicing. J Med Chem 56: 10033–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagisetti C, Yermolina MV, Sharma LK, Palacios G, Prigaro BJ, Webb TR. 2014. Pre-mRNA splicing-modulatory pharmacophores: the total synthesis of herboxidiene, a pladienolide-herboxidiene hybrid analog and related derivatives. ACS Chem Biol 9: 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardelli RM, Thompson JX, Yates JR III, Stevens SW. 2010. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA 16: 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Biswas S, Berg MG, Antapli CM, Xie F, Wang Q, Tang MC, Tang GL, Zhang L, Dreyfuss G, et al. 2013. Genomics-guided discovery of thailanstatins A, B, and C As pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J Nat Prod 76: 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada M. 2004. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo) 57: 188–196. [DOI] [PubMed] [Google Scholar]
- Muller S, Mayer T, Sasse F, Maier ME. 2011. Synthesis of a pladienolide B analogue with the fully functionalized core structure. Org Lett 13: 3940–3943. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. 1996a. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo) 49: 1204–1211. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. 1996b. New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 49: 1196–1203. [DOI] [PubMed] [Google Scholar]
- Perriman RJ, Ares M Jr. 2007. Rearrangement of competing U2 RNA helices within the spliceosome promotes multiple steps in splicing. Genes Dev 21: 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriman R, Ares M Jr. 2010. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol Cell 38: 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriman R, Barta I, Voeltz GK, Abelson J, Ares M Jr. 2003. ATP requirement for Prp5p function is determined by Cus2p and the structure of U2 small nuclear RNA. Proc Natl Acad Sci 100: 13857–13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal GA, Jurica MS. 2010. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res 38: 6664–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Tsujita T, Akiyama T, Yoshida T, Mizukami T, Akinaga S, Horinouchi S, Yoshida M, Yoshida T. 2002. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. II. The effects on cell cycle progression and gene expression. J Antibiot (Tokyo) 55: 863–872. [DOI] [PubMed] [Google Scholar]
- Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y. 2004. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. I. Taxonomy, fermentation, isolation and screening. J Antibiot (Tokyo) 57: 173–179. [DOI] [PubMed] [Google Scholar]
- Villa R, Mandel AL, Jones BD, La Clair JJ, Burkart MD. 2012. Structure of FD-895 revealed through total synthesis. Org Lett 14: 5396–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa R, Kashyap MK, Kumar D, Kipps TJ, Castro JE, La Clair JJ, Burkart MD. 2013. Stabilized cyclopropane analogs of the splicing inhibitor FD-895. J Med Chem 56: 6576–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman DA, Steitz JA. 1992. Interactions of small nuclear RNA's with precursor messenger RNA during in vitro splicing. Science 257: 1918–1925. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Ogawa S. 2014. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA 5: 445–459. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.