

Abstract
In the microRNA (miRNA) pathway, Dicer processes precursors to mature miRNAs. For efficient processing, double-stranded RNA-binding proteins support Dicer proteins. In flies, Loquacious (Loqs) interacts with Dicer1 (dmDcr1) to facilitate miRNA processing. Here, we have solved the structure of the third double-stranded RNA-binding domain (dsRBD) of Loqs and define specific structural elements that interact with dmDcr1. In addition, we show that the linker preceding dsRBD3 contributes significantly to dmDcr1 binding. Furthermore, our structural work demonstrates that the third dsRBD of Loqs forms homodimers. Mutations in the dimerization interface abrogate dmDcr1 interaction. Loqs, however, binds to dmDcr1 as a monomer using the identified dimerization surface, which suggests that Loqs might form dimers under conditions where dmDcr1 is absent or not accessible. Since critical sequence elements are conserved, we suggest that dimerization might be a general feature of dsRBD proteins in gene silencing.
Keywords: Loquacious, Dicer, dsRBD, microRNA, gene silencing, fluorescence spectroscopy, Drosophila
INTRODUCTION
MicroRNAs (miRNAs) are conserved small noncoding RNAs that regulate gene expression at the level of translation and mRNA stability (Bartel 2009; Dueck and Meister 2014). MiRNAs are generated as primary transcripts (pri-miRNAs), from which stem-loop-structured precursors (pre-miRNAs) are processed by the RNase III Drosha. Pre-miRNAs are exported to the cytoplasm, where Dicer, a second RNase III enzyme, generates a 20–25 nucleotide (nt) double-stranded (ds) miRNA intermediate (Carthew and Sontheimer 2009; Kim et al. 2009). One strand of the Dicer product is selected and loaded into the RNA-induced silencing complex (RISC), where it directly binds to a member of the Argonaute (Ago) protein family (Meister 2013; Dueck and Meister 2014). Both Drosha and Dicer require the help of a double-stranded RNA-binding protein (dsRBP). DGCR8/Pasha helps to recruit and correctly position Drosha on the substrate pri-miRNA (Han et al. 2004, 2006; Landthaler et al. 2004; Nguyen et al. 2015). In mammals, Dicer cooperates with TRBP and/or PACT (Chendrimada et al. 2005; Haase et al. 2005; Lee et al. 2006). Both dsRBPs have been implicated in miRNA biogenesis and contain three dsRNA binding domains (dsRBDs). It has been demonstrated that TRBP uses its first and second dsRBD to interact with the Dicer-produced miRNA intermediate while the third dsRBD is a protein–protein interaction domain and utilized for the interaction with the helicase domain of Dicer (Daniels et al. 2009; Wilson et al. 2015). In Drosophila, miRNA and short interfering RNA (siRNA) pathways are separated and two specialized Dicers exist. Dicer2 (dmDcr2) is responsible for processing of dsRNAs into siRNAs and requires the help of the dsRBP R2D2 (Liu et al. 2003; Lee et al. 2004; Tomari et al. 2004). Dicer1 (dmDcr1), in contrast, is specialized on miRNA processing and is supported by the dsRBP Loquacious (Loqs) (Forstemann et al. 2005; Jiang et al. 2005; Saito et al. 2005; Ye et al. 2007). Like other Dicer proteins, dmDcr1 is characterized by several different domains (Tsutsumi et al. 2011): two RNase III domains that cleave the two strands of the miRNA precursor (Zhang et al. 2004), a dsRBD that supports the cleavage activity (Kidwell et al. 2014), a PAZ domain, which interacts with the ds end of the pre-miRNA and positions it on human Dicer for cleavage (Park et al. 2011) and a helicase domain, which binds ATP and might be involved in substrate recognition (Sinha et al. 2015). Loqs contains three dsRBDs (Fig. 1A). DsRBD1 and dsRBD2 are classical RNA-binding domains, which interact with the dsRNA during Dicer processing and/or RISC loading. The third dsRBD, however, appears to interact with dmDcr1. The presumably unstructured linker between dsRBD2 and 3 supports this interaction (Forstemann et al. 2005). While the interface between TRBP and human Dcr has recently been structurally characterized (Wilson et al. 2015), the molecular basis of this interaction platform both on dmDcr1 and on the dsRBD3 of Loqs is not yet understood.
FIGURE 1.

Structure of Loqs-dsRBD3, a non-RNA-binding dsRBD. (A) Schematic representation of Loqs-PB. RNA-binding dsRBDs 1 and 2 are colored orange and the nonbinding dsRBD3 is shown in green. (B) Structure model of a Loqs-dsRBD3 monomer. Regions mediating RNA contacts in other dsRBDs are highlighted in yellow. The side chain of phenylalanine (F) 419 is shown. (C) Superposition of the structures of TRBP dsRBD2 bound to RNA (PDB = 3adl, yellow), RBD5 of Staufen1 (PDB = 4dkk, blue), and Loquacious dsRBD3 (green). An arrow highlights different positions of the L2 loop. (D) Structure-based alignment of Loqs-dsRBD3 with representatives of the most closely related structures identified by DALI. Secondary structure elements are indicated; positions with <3 Å distance from the equivalent in Loqs are shaded in gray. The three RNA-binding regions of dsRBDs are marked, and RNA-binding residues are shown in bold. Loqs F419 and residues at equivalent positions in other domains are highlighted in red.
To unravel the molecular and functional details underlying the dmDcr1–Loqs interaction, we have solved the structure of the dsRBD3 of Loqs at 2.65 Å. We find that a specific loop and a linker region contact dmDcr1 and point mutations in these regions abolish or weaken dmDcr1 interaction. Furthermore, the crystal packing of dsRBD3 of Loqs suggests a homodimer, which we find to be stable in solution. Critical residues in the dimerization interface are also essential for dmDcr1 binding and activity. As on the one hand the dimer interface is conserved in other dsRBPs that function in small RNA gene silencing, and on the other hand, the helix bundle of Dicer which forms the interaction module for TRBP is also conserved across species, the dsRBD3-dimer surface is probably a bifunctional interaction platform.
RESULTS
Crystal structure of Loqs dsRBD3
It has been reported that Loqs dsRBD3 interacts with Dicer (Forstemann et al. 2005). The underlying structural features that shift the dsRBD3 from an RNA-binding domain to a Dicer-interaction platform are still unclear. To clarify this issue, we solved the structure of dsRBD3 by X-ray crystallography (see Supplemental Tables 1 and 2 for statistics, parameters, and conditions). The dsRBD3 of Loqs folds into the canonical α-β-β-β-α structure described for dsRBDs (Fig. 1B). The secondary structure elements can be almost perfectly overlaid onto several dsRBD structures identified by DALI (Holm and Rosenstrom 2010) with overall rmsd values ∼2 Å. These include dsRBDs of Staufen1, Dicer, or RNase III. However, considerable diversity is observed in the loop conformations, especially of L2 connecting the β-sheets β1 and β2 (Fig. 1C).
Three highly conserved regions in dsRBDs have been shown to be crucial for dsRNA binding (for review, see Masliah et al. 2013). Particularly, a glutamate side chain in helix α1, a GPxH motif in the loop L2, and the positively charged patch KKxAK in helix α2, make important contacts with the RNA (Fig. 1B, highlighted in yellow). In Loqs dsRBD3, only region 1 is conserved (Fig. 1D). Region 2 in loop L2 is shortened and the critical GPxH motif is missing. In region 3, all positively charged residues are replaced by AADAQ, leaving the region with a negative overall electrostatic potential. In a structure-based alignment, L2 shows the highest degree of variability (Fig. 1D). The L2 of Loqs dsRBD3 is shorter than its equivalent in RNA-binding competent dsRBDs. Thus, it is not able to reach the minor groove that is specifically bound by a histidine residue in dsRBD2 of TRBP and most other RNA-binding dsRBDs (Fig. 1C; Yang et al. 2010; Masliah et al. 2013). In addition, Loqs L2 adopts a specific conformation, pointing away from the surface bound by RNA in the related structures (Fig. 1C). As this conformation is observed in all six monomers present in the asymmetric unit of our structure irrespective of the crystal environment, we are confident that it is a natural structural feature. Reconstitution of the lysine residues in region 3 and the GPxH motif in region 2 is not sufficient to restore RNA-binding activity in gelshift assays, probably due to the shorter loop L2, which prevents the histidine side chain to reach into the dsRNA minor groove (data not shown).
L2 conformations similar to Loqs-dsRBD3 have been observed in the C-terminal dsRBD of hsDicer and the RBD5 domain of human Staufen1 (Fig. 1C,D) and are indicative of a protein rather than an RNA-binding function of these domains.
The L2 loop within dsRBD3 contributes to dmDcr1 interaction
The histidine residue involved in RNA binding in TRBP-dsRBD2 is replaced by phenylalanine F419 in Loqs-dsRBD3, which is fully accessible to the solvent (highlighted in Fig. 1B). We hypothesized that this exposed hydrophobic ring might be involved in the interaction of dsRBD3 with the helicase domain of dmDcr1. To test this hypothesis, we performed pulldown experiments using wild-type (wt) dsRBD3 as well as different mutants (Fig. 2A, “dsRBD3”). Indeed, while GST-dsRBD3 efficiently precipitates overexpressed His-dmDcr1 from cell lysates, the F419A mutant is strongly impaired in dmDcr1 binding (Fig. 2B). In contrast, mutation of the partially exposed F423 in strand β2 has no effect on the Loqs–Dicer interaction, demonstrating that F419 specifically contributes to dmDcr1 binding. Mutations that reconstitute side chains participating in RNA binding in Loqs–dsRBD1 and -2 like A444K have no influence on Dicer binding (Fig. 2B), indicating that different surfaces of the dsRBDs are used for RNA and protein binding.
FIGURE 2.

Residues in Loop L2 and the interdomain linker contribute to dmDcr1 binding. (A) Schematic representation of GST-tagged proteins used for the pulldown assays. Positions of mutated amino acids are indicated with red lines. (B) Pulldown assay of His-dmDcr1 from SF21 cell lysate with GST-Loqs-dsRBD3 and point mutants as indicated. GST alone served as control. Bound dmDcr1 is detected by immunoblot using a specific antibody and bait proteins were stained with Coomassie blue. (C) Pulldown assay as in B using Loqs constructs (Loqs0-4) containing sequential truncations of the interdomain linker. (D) GST-pulldown with various mutations within the minimal linker Loqs-3 construct and quantification of the dmDcr1 signal normalized to the wt construct.
The linker between dsRBD2 and dsRBD3 strongly enhances dmDcr1 binding
Four splice isoforms of Loqs (Loqs-PA-PD) have been described, which differ in their C-terminal regions (Hartig et al. 2009). Loqs-PA and -PB contain all three dsRBDs with PB having a longer linker region between dsRBD2 and 3. Loqs-PC and -PD lack dsRBD3 and PD has an alternative C-terminal region. Of these isoforms, mainly Loqs-PB and to a lesser extent Loqs-PA are copurified with dmDcr1 in immunoprecipitation experiments, demonstrating a role of dsRBD3 and also the extended linker in the PB isoform in Dicer binding (Forstemann et al. 2005). In order to investigate the contribution of the PB-linker region, we expressed and purified GST-dsRBD3 protein containing different lengths of linker sequence. As expected, dsRBD3 containing the full linker sequence had markedly higher pulldown efficiency for dmDcr1 compared to the dsRBD3 alone (Fig. 2C, Loqs-0, Loqs-4). Sequential truncation of the linker (Fig. 2A) revealed a minimal region starting with amino acid (aa) 368, which contributes to dmDcr1 binding in addition to dsRBD3 (Fig. 2C).
Sequence analysis of the minimal linker region predicts a propensity to form an amphipathic helix with residues L371, N373, L379/380/382, and L387 making up the hydrophobic face. While mutation of L371A and N373E had no effect, mutation of L387A and K378E impairs dmDcr1 binding, suggesting that the linker makes hydrophobic interactions with a cognate surface on dmDcr1 (Fig. 2D). Combination of this mutant with the F419A mutation in dsRBD3 further diminishes the binding activity, which argues for a concordant binding of both regions to the dmDcr1 helicase domain (Fig. 2D).
Our truncation experiments suggested that not only the third dsRBD but also the preceding linker region is important for dmDcr1 binding. To analyze the individual contributions of both regions in detail, we directly compared the binding activity of dsRBD3 alone and together with the linker region (Fig. 3A). Although both constructs interact with dmDcr1, the addition of the linker region elevated binding by ∼10-fold. To analyze the individual contributions of these interactions more quantitatively, we performed fluorescence cross-correlation spectroscopy (FCCS) (Bacia and Schwille 2007). FCCS is a single-molecule-sensitive method to measure concentration and molecular mobility of fluorescently labeled molecules and provides access to equilibrium and rate constants of molecular interactions.
FIGURE 3.

Contribution of the interdomain linker to dmDcr1-binding affinity. (A) GST-pulldown assay of Loqs-3 and dsRBD3 constructs and quantification of the dmDcr1 signal normalized to the Loqs-3 construct. (B) Overview of the FCCS setup. Two laser lines illuminate an overlapping microscopic detection volume (1). Molecules entering/leaving the detection spot give rise to a fluctuation curve (2). Auto- and cross-correlation of the fluctuation curves gives specific information on various biophysical data (3). (C) Fluorescently labeled Loqs-3AF647 was bound to GFP-dmDcr1 and subsequently competed with different amounts of unlabeled Loqs-3 or dsRBD3. IC50 values were obtained and Ki values were calculated according to Cheng-Prusoff.
dmDcr1 was fused to GFP and a complex was formed with the Alexa Fluor (AF) 647-labeled Loqs-3 construct (Loqs-3AF647), which contains both dmDcr1-binding sites (Fig. 2A). Affinity determination of the interaction of labeled components yielded a Kd-value of 6.7 ± 2.2 nM. Additionally, we measured the equilibrium binding constants of fluorescent dsRDB3 (dsRDB3AF647) to GFP-dmDcr1 and observed a substantially decreased affinity (191.5 ± 27.6 nM) (data not shown). To rule out influences of the fluorescent dye, we tested both unlabeled Loqs-3 and dsRDB3 in competition experiments. For this, increasing amounts of unlabeled Loqs dsRBD3 alone or the Loqs-3 construct were added to the sample to allow competition for dmDcr1 binding. Loss of cross-correlation between the two fluorophores can be measured depending on the concentration of the added unlabeled protein (Fig. 3B). Using this approach, we determined an IC50 value of 104 nM and a Ki value of 43.6 nM for dsRBD3 alone (Ki values are the Kd values of inhibitor binding, i.e., the unlabeled Loqs-dsRBD3). Consistent with our binding studies, the IC50 and the corresponding Ki values decreased ∼10-fold to 9.3 nM and 3.6 nM, respectively, for the Loqs-3 construct (Fig. 3C).
In summary, we verified two specific dmDcr1 contact sites located in the linker region as well as the dsRBD3. While the dsRBD3 interaction might be important for specificity, the linker region strongly contributes to high affinity binding.
Loqs forms homodimers
Analysis of the protein contact interfaces in the crystal using PISA (Krissinel and Henrick 2007) revealed a symmetric dimerization interface of ∼700 Å2 between the dsRBD molecules when the effect of an observed domain swap, which most likely is a crystallization artifact (see Supplemental Fig. S1 in the extended view), was corrected. The observed interface is predicted to be highly stable in solution. We therefore modeled a dimer consisting of two native dsRBD3 molecules (Fig. 4A). The dimer interface is mainly hydrophobic and brings the strands β3 of both monomers into close proximity in a parallel orientation, enabling the first half of the strand (aa 434–438) to engage in beta sheet interactions.
FIGURE 4.

Loqs dsRBD3 dimerizes via a conserved interface. (A) Structure model of a dimer of native Loqs-dsRBD3 molecules. The side chain of residue L426 is shown as yellow sticks and I461 as red sticks. (B) Crosslinking assay of purified His-dmDcr1 and HA-Loqs-3 (aa 368–463). Concentrations of DSS-crosslinker are indicated on top. Both proteins are detected by immunoblot—Loqs by an anti-HA-antibody (green) and dmDcr1 by a specific antibody (red). Positions of monomeric and dimeric Loqs-3 are indicated on the right. (C) Size-exclusion chromatography of Loqs-dsRBD2 (green), Loqs-3 (black), Loqs-3 L426R (orange), Loqs-3 K460E (red), and Loqs-3 KI460/61EA (blue). Migration of a protein standard is shown on top. (D, top) Pulldown assay of His-dmDcr1 with GST-Loqs-3 and mutants as indicated. Bound dmDcr1 is detected by immunoblotting using a specific antibody; bait proteins are visualized by Coomassie staining. (Bottom) HA-tagged wt Loqs (lanes 2,4) or HA-Loqs L426R (lanes 1,3) were cotransfected with myc-dmDcr1 into HEK 293 cells. Myc-dmDcr1 was immunoprecipitated using anti-myc antibodies and the immunoprecipitates were analyzed by Western blotting using anti-dmDcr1 (upper panel) or -HA antibodies (lower panel). (E) Sequence alignment of Loqs-dsRBD3 with functional homologs of the TRBP and PACT protein families from different organisms.
To further solidify our structural model, we investigated homodimerization of Loqs via dsRBD3 in more detail. First, we incubated dsRBD3 together with recombinant dmDcr1 and chemically crosslinked interacting proteins (Fig. 4B). Consistent with our dimerization model, the Loqs-3 construct can be readily crosslinked to a species migrating as a dimer in SDS-PAGE. It is also crosslinked to dmDcr1 and does not form larger homomeric aggregates (Fig. 4B). Second, we analyzed the migration of dsRBD3 in size-exclusion chromatography (Fig. 4C). Loqs-3 (Fig. 2A) migrates at an apparent molecular weight (MW) of 26 kDa, which roughly equals twice the apparent MW of dsRBD2, which elutes close to the theoretical value of 14 kDa (Fig. 4C; compare black and green graphs). From our structural model, we predicted that L426, which is located in strand β2, participates in dimer interactions. To disrupt the dimer, we substituted L426 by a more bulky arginine (L426R). Strikingly, the L426R mutation resulted in shifting the peak in size-exclusion chromatography to a MW of 13 kDa representing the monomeric form (Fig. 3C, orange graph). In order to assess a potential requirement of the dimerization interface for dmDcr1 binding, we tested the L426R mutant in the GST-pulldown assay described above and found that the interaction with dmDcr1 is almost completely abrogated (Fig. 4D). Mutation of K460 or K460 and I461, which contribute to the dimer interface via a potential salt bridge and hydrophobic contacts, leads to a destabilization of the dimer reflected by an intermediate elution in gel filtration (Fig. 4C, blue and red graphs). On Dicer binding, they show only a modest reduction (Fig. 4D). Furthermore, we analyzed the interaction of full-length Loqs-PB with dmDcr1 by coimmunoprecipitation after cotransfection into HEK293 cells. Consistently, the L426R mutation reduced binding to dmDcr1 also in the full-length protein context (Fig. 4D, lower panel) indicating that either dimer formation is required to generate the dmDcr1-binding site on Loqs or that dimerization and dmDcr1 binding are alternative processes requiring the same protein surface. In agreement with the latter model, it has been shown recently that TRBP-dsRBD3 forms a highly similar interaction surface, which is used for direct Dicer binding (Wilson et al. 2015).
The residues participating in dimer formation are very poorly conserved between Loqs dsRBD3 and other dsRBDs of known structure (Fig. 1D). However, comparison with functionally homologous domains of the TRBP and PACT protein families reveals a clear conservation of these residues, thus implicating that dimerization via dsRBD3 might be a conserved feature of this class of proteins (Fig. 4E). Indeed, homomeric affinity of PACT dsRBD3 as well as a dimeric state of purified recombinant TRBP have been reported (Hitti et al. 2004; MacRae et al. 2008). In addition, it has been shown that the dsRBPs TRBP and PACT can form heterodimers (Kok et al. 2007) presumably via their dimerization surfaces.
Loqs dsRBD3 interacts with dmDcr1 as monomer
To investigate whether dsRBD3 interacts with dmDcr1 as mono- or dimer, we established and performed single-molecule immunoprecipitation analysis (Fig. 5). This method is highly sensitive and can probe the stoichiometry of biomolecular complexes (Jain et al. 2011). To this end, we preformed a complex consisting of GFP-dmDcr1 and AF647-labeled Loqs-3 (including the preceding linker region). The complex was immobilized to a quartz slide for single-molecule total internal reflection fluorescence (TIRF) microscopy using a biotinylated anti-GFP antibody (Fig. 5A). Single AF647 fluorescence spots were detectable in the dmDcr1 containing reaction, while signals were largely absent when dmDcr1 was omitted (Fig. 5B,C) providing evidence for the specificity of the immobilization strategy. Single fluorescent emitters show an abrupt decrease in fluorescence intensity, a phenomenon termed photobleaching, which can be exploited to count the number of molecules present at an individual fluorescent spot. In case one Loqs molecule is bound, only one bleaching step should be observed. However, if two molecules are interacting with dmDcr1, two bleaching steps would be expected (Fig. 5D). We recorded the fluorescence intensity of individual AF647-labeled Loqs molecules bound to dmDcr1 over time and performed a photobleaching analysis. Indeed, when we quantified the photobleaching steps, we observed that ∼90% of the Loqs molecules underwent a single bleaching step and <10% exhibited a two-step photobleaching event (Fig. 5E). Therefore, our single-molecule analysis clearly demonstrates that although dsRBD3 forms dimers in solution, dmDcr1 interacts with a single molecule of Loqs.
FIGURE 5.

Stoichiometry of Loquacious bound to Dicer using single-molecule coimmunoprecipitation (co-IP) analyses. (A) Schematic representation of the single-molecule co-IP assay. (B) Total internal reflection fluorescence (TIRF) images of Alexa647-labeled molecules (Loqs-3AF647) pulled down via Dicer. Dicer was genetically fused to GFP, enabling immobilization on the quartz slide via a biotinylated anti-GFP antibody (scale bar: 10 µm). As a control, Loquacious was flushed over the slide without prior incubation with GFP-dmDcr1. (C) Average numbers of Loqs-3AF647 molecules per imaging area (6800 μm2). (D) Representative single-molecule fluorescence transients of Loqs-3AF647 molecules that exhibit one-step and two-step photobleaching. (E) Photobleaching step distribution for Loqs-3AF647 bound to dmDcr1 indicating a 1:1 stoichiometry for the Dicer–Loquacious complex (molecule numbers accumulated from four independent experiments, a total of 1558 molecules were analyzed).
Analysis of Loqs–dmDcr1 interactions in an in vivo reconstitution system
We sought to test the functional relevance of the identified Dicer-interaction modules in Loqs for miRNA processing by dmDcr1. To this end, we performed rescue experiments, in which murine embryonic fibroblasts (MEFs) deficient in Dicer are complemented with Drosophila dmDcr1 (Bogerd et al. 2014). In contrast to mouse Dicer, Drosophila Dcr1 alone is not able to process murine miRNA precursors efficiently. Upon cotransfection of Loqs-PB, however, the processing activity is restored, thus giving a direct readout of functionality of different Loqs mutants (Fig. 6). We first tested single-point mutations located within the linker region that binds to dmDcr1 (Fig. 6A). As expected, L371A, which had only marginal effects on dmDcr1 binding (Fig. 2D), was comparable to wt Loqs in these in vivo assays. N373E had markedly reduced effects on dmDcr1 activity (Fig. 6B), which is in contrast to our binding experiments (Fig. 2D), where N373E binds dmDcr1 efficiently. This might be due to technical differences in the used assays or additional features present in the complex in vivo reconstitution assay. All other mutants tested (K378E, L387A, L379/80/82A) showed reduced dmDcr1 activation (Fig. 6B), which is consistent with reduced binding to dmDcr1 (Fig. 2D). The naturally occurring splice variant Loqs-PA, which does not contain the dmDcr1 interaction site located on the linker region between dsRBD2 and dsRBD3 (Fig. 6C, top panel) and therefore binds dmDcr1 with reduced affinity (Forstemann et al. 2005), is not able to reconstitute dmDcr1 activity in our in vivo assay, indicating that interaction via the linker region is essential for dmDcr1 function (Fig. 6C). We next analyzed mutations located in the dsRBD3. The F419A mutation, which decreases binding to dmDcr1 in in vitro binding assays, also reduces dmDcr1 activity in the Dicer−/− MEFs, indicating that the L2 loop contributes to dmDcr1 binding in the full-length context as well and the high-affinity interaction with the linker region alone is not sufficient for reconstituting dmDcr1 activity in vivo (Fig. 6B).
FIGURE 6.

An in vivo Dicer reconstitution assays for the functional analysis of dmDcr1–Loqs interactions. (A) Schematic overview of full length Loqs-PB. Point mutations are indicated as red lines. (B) HA-dmDcr1 (lanes 4–11), wt Loqs (lane 5) as well as the indicated Loqs mutants (lanes 6–11) were cotransfected into Dicer-deficient MEFs. Additionally, HA-GFP and HA-hDcr (lanes 2,3) were transfected as controls. Endogenous let-7a from Dcr+/+ MEFs (lane 1) was loaded as a marker and processing in the deficient cells was analyzed by Northern blotting (upper panel). HA-dmDcr1 (middle panel) or HA-Loqs (lower panel) expression was analyzed by Western blotting using anti-HA antibody. Loading of the Northern blot samples was controlled by EtBr staining and loading of Western blots via an actin blot. (C) Comparison of the Loqs-PB and Loqs-PA isoforms in the rescue assay. (D) Influence of the dimerization mutants L426R, K460E, and KI460/61EA on miRNA processing.
Finally, we tested the requirement for the dimerization interface for Loqs activity by using the L426R, K460E, and the KI460/461EA mutant constructs. Consistent with the complete lack of dmDcr1 binding, Loqs L426R is unable to support miRNA biogenesis (Fig. 6D). Of the other two mutants that weaken the dimer interface, K460E and K460E/I461A, only the double mutant, which also showed reduced binding to dmDcr1, is clearly impaired in miRNA biogenesis (Fig. 6D). Taken together, using an in vivo reconstitution assay, we demonstrate that both dmDcr1 contact regions of Loqs are required for dmDcr1 interaction and activity in the full-length context, too.
DISCUSSION
Most RNase III enzymes involved in small RNA processing function in concert with a dsRBP. These proteins are required for the correct binding and placement of the dsRNA substrate as well as strand selection processes (Ghildiyal and Zamore 2009). Here we have unraveled the molecular details of Loqs–dmDcr1 interactions in Drosophila. Using X-ray crystallography we solved the structure of Loqs dsRBD3 and provide evidence for the existence of an interaction surface that can either bind dmDcr1 or dimerize. Furthermore, Loqs uses not only its dsRBD3, but also the linker region preceding this domain for efficient dmDcr1 interaction. Using several different experimental approaches, we provide evidence that dsRBD3 provides a low and the linker region a high-affinity dmDcr1-binding site. Nevertheless, the interaction with dsRBD3 is essential since a point mutation within the dsRBD3 (F419) strongly affects dmDcr1 activity in the in vivo reconstitution assay (Fig. 6B). It is therefore tempting to speculate that dsRBD3 might form a specificity platform with low affinity and upon dmDcr1 contact and recognition, linker interactions establish a strong interaction between the two proteins. Such a two-step binding model might allow for proofreading during pre-miRNA positioning. Such a model, however, needs to be experimentally further verified.
The dsRBD3 crystallized as a dimer using a high confidence interaction surface. Interestingly, a highly similar interaction surface is used by TRBP to interact with human Dicer (Wilson et al. 2015). Loqs uses this interface to establish a low-affinity interaction with dmDcr1 as well. We found that Loqs dsRBD3 forms dimers in the absence of dmDcr1. Using single-molecule analyses, we demonstrate that dmDcr1 binds dsRBD3 monomers and liberates them from the preformed dimers. In a speculative model, we suggest that Loqs forms dimers when it is not bound to dmDcr1 (Fig. 7). This could either resemble a quickly available Loqs storage pool or Loqs could have dmDcr1-independent functions that require dimerization. Consistently, it has been demonstrated that dsRBD3 of TRBP and PACT can form homo- and also heterodimers as well (Hitti et al. 2004; Kok et al. 2007; MacRae et al. 2008). Moreover, DGCR8, the dsRBP acting together with Drosha, has been suggested to form dimers as well (Faller et al. 2007; Senturia et al. 2010, 2012). The physiological roles of such dimerization events, however, are still elusive. Nevertheless, as all structural features required for dmDcr1 binding are present in dsRBPs acting in small RNA processing pathways in various organisms, we expect the mode of Dcr-binding to be widely conserved.
FIGURE 7.
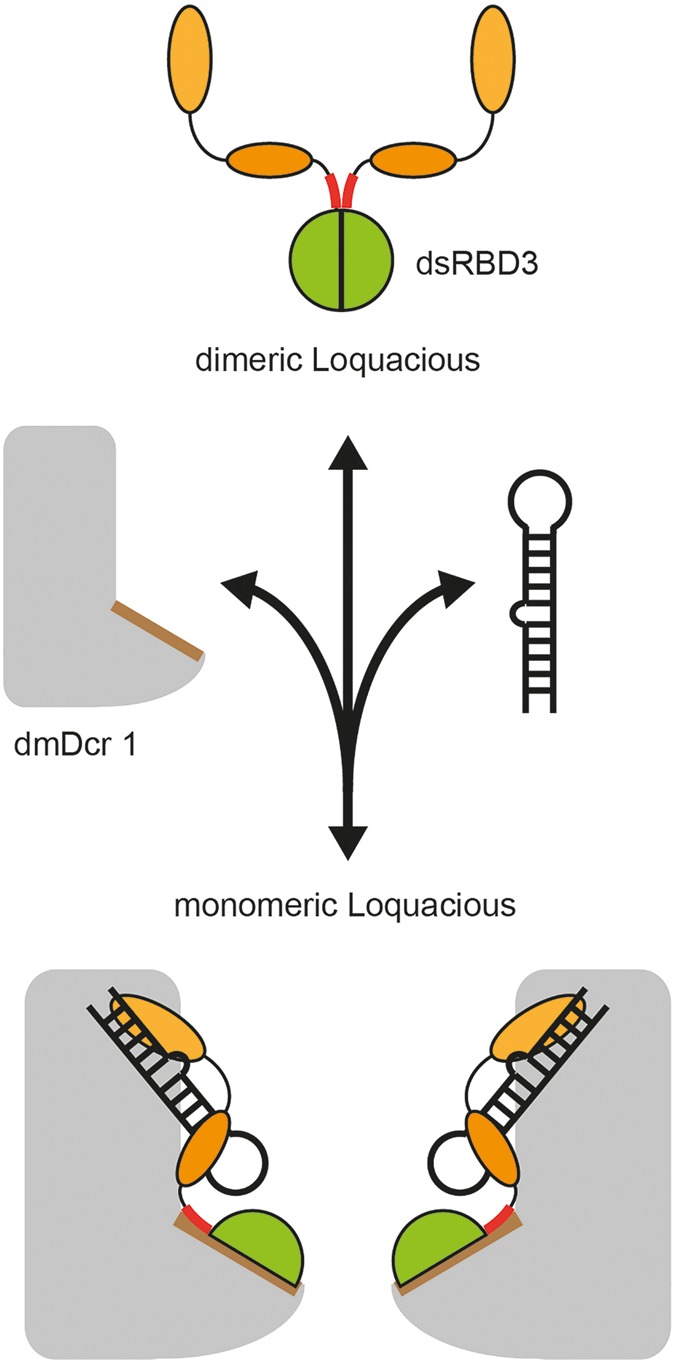
Model for Loqs dimerization and dmDcr1 binding. In the absence of dmDcr1, Loqs forms homodimers via the dimerization interface located on dsRBD3 (green). DsRBD1 and 2 are indicated as orange circles. In the presence of dmDcr1, Loqs binds to the helicase domain of dmDcr1 via the dimerization interface of dsRBD3 and a high-affinity binding site located in the linker preceding dsRBD3 (highlighted in red).
MATERIALS AND METHODS
DNA constructs
The pFastBac dmDcr1 and Loqs-PB constructs were described in Ye and Liu (2008). The sequences of the primer-pairs (P1-26) are listed Supplemental Table 3 (see extended content). Cloning into the bacterial expression vectors pET32a and pGEX-4T-1 was performed via BamHI and SalI restriction sites where a TEV protease recognition site was integrated into the forward primer. The crystallization construct of Loqs-PB (392–463, P1) also contains a spacer of two glycines for a more efficient cleavage. Linker construct Loqs-0 was introduced with P2, Loqs-1 with P3, Loqs-2 with P4, Loqs-3 with P5, Loqs-4 with P6 and Loqs-3 HA construct with P7. For eukaryotic expression, constructs were cloned into a modified pIRESneo (Clontech) and pCS2 expression vectors via FseI and AscI restriction sites. P8 was used to generate the Loqs-PB 1-463, P9 for the dmDcr1 construct from the respective pFastBac template. For point mutations the following mutagenesis primers were used: L371A with P10, P11 for L371A full-length construct, N373E with P12, K378E with P13, triple mutant L379/80/82A with P14, L387A with P15, F419A with P16, F423A with P17, A444K with P18, L426R with P19, K460E with P20, P21 for K460E full-length construct, KI460/61EA with P22 and P23 for KI460/61EA full-length construct. The Loqs-3 construct for the labeling reaction was generated with P24 for C386S, P25 for Q406C, and P26 for C425S.
Protein expression and purification
All purification steps of recombinant proteins were performed at 4°C. Protein concentration was determined spectrophotometrically at 280 nm. GST-tagged proteins were expressed from pGEX-4T-1, His6-Thioredoxin tagged proteins from pET32a.
Expression of proteins was performed in Escherichia coli BL21 (DE3). Cells were grown at 37°C to an OD600 of 0.6 and protein expression was induced by the addition of 1 mM isopropyl-β-D-thiogalactopyranosid (IPTG). Cells were grown overnight at 18°C.
The His6-thioredoxin tagged dsRBD3 was affinity purified with a 15-mL IMAC column equilibrated with His-A buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, and 10 mM imidazole). Elution was performed with His-B buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 300 mM imidazole) and protein-containing fractions were pooled and dialyzed against HisA buffer and the tag was simultaneously cleaved with a TEV-protease. After a second 15-mL IMAC, the flowthrough was concentrated by ammonium sulfate precipitation, resuspended in SEC-1 buffer (50 mM Hepes-NaOH, pH 7.5, 250 mM NaCl, 1 mM dithiothreitol [DTT]), and loaded onto a Superdex 75 26/60 column (GE Healthcare). Methylation of the protein required for crystallization was performed according to Walter et al. (2006).
For purification of GST-tagged proteins the lysate was loaded onto a 5-mL GSTrap FF column (GE Healthcare) equilibrated with GST-A buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl). Bound protein was eluted with GST-B buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 3 mg/mL glutathione). GST fusion protein containing fractions were pooled and applied to a Hiprep 26/10 desalting column and buffer was exchanged to GST-A.
For the GST-tagged Loqs-3 HA construct and the Loqs-3 construct for labeling (C386S, Q406C and C425S), the GST-tag was removed with TEV protease, which, together with the GST tag, was removed with a second 5-mL GSTrap FF column. The flowthrough was loaded onto a Superdex 75 10/300 column (GE Healthcare) equilibrated with SEC buffer and SEC buffer without DTT for the Loqs-3 labeling construct.
Constructs containing the interdomain linker and the dsRBD2 were additionally washed with GST-A containing 1 M NaCl to remove RNA contaminations.
For the generation of recombinant baculovirus encoding His-tagged dmDcr1 and His-tagged GFP-dmDcr1, the Bac-to-Bac system (Invitrogen) was used. For this, SF21 cells were transferred in Sf-900 III SFM medium (Invitrogen) at a density of 0.5–1 × 106 and infected 1:100 with V1 virus stock. Overexpressed dmDcr1 was purified according to Ye and Liu (2008).
Crystallization and structure determination
Methylated Loqs dsRBD3 was used in sparse matrix screening at 15 mg/mL and a single crystallization condition containing 10% (w/v) PEG-1000 and 10% (w/v) PEG-8000 was identified. Further refinement resulted in crystals suitable for X-ray diffraction grown in 2% (w/v) PEG-1000 and 12% (w/v) PEG-8000. Crystals were briefly soaked with cryobuffer (20% Glycerol, 2% PEG-1000, 12% PEG-8000) and frozen in liquid nitrogen. X-ray diffraction experiments were carried out at the Beamline 14.2 of the BESSY synchrotron (Berlin, Germany). Native data could be obtained up to 2.65 Å resolution. Data set statistics are given in Supplemental Table 1. For phasing, selenomethionine-substituted protein was crystallized as described above and MAD (multiple anomalous diffraction) data were measured (Supplemental Table 1). The data were processed using XDS (Kabsch 2010) and selenium sites were identified from the MAD data using AutoSHARP (Vonrhein et al. 2007). The model was manually built in COOT (Emsley et al. 2010) and refined with PHENIX.refine (Adams et al. 2010) using NCS restraints and simulated annealing. At the final stages, TLS-refinement was carried out. Model statistics are given in Supplemental Table 2.
Cell culture
HEK 293T and MEF (Dcr+/+ and Dcr−/−) cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich) supplemented with 10% FBS (Sigma-Aldrich) and 1% penicillin–streptomycin (Sigma-Aldrich) under standard conditions (37°C, 5% CO2).
Western blotting
Samples for Western blot analysis were mixed with 2× Laemmli Buffer (eluates from co-IP) or 5× Laemmli Buffer (inputs, eluates from GST-pulldown) and incubated at 95°C for 5 min. Dicer and full-length Loqs constructs were separated by SDS-PAGE with 8% polyacrylamide gels. Loqs was transferred by semidry blotting and Dicer by wet blotting to a Hybond ECL (GE Healthcare) nitrocellulose membrane. For immunodetection, the following antibodies were used: mouse-anti-HA (16B12, Covance; 1:1000), rabbit-anti-myc (Sigma), rabbit-anti-dmDcr1 antibody (ab4735, Abcam, 1:1000), mouse-anti-α-tubulin (DM 1A, Sigma, 1:10,000).
GST-pulldown assays
For each pulldown sample, 40 µL of Glutathione 4 Fast Flow Sepharose (GE Healthcare) were washed with PBS twice. Recombinant GST proteins (300 µg each) were added to the beads and incubated for 1 h at 4°C. Then the supernatant was removed and beads were washed three times with PBS and 1 mL of SF21 lysate containing overexpressed dmDcr1 was applied to the beads. For the dsRBD3-only constructs, a threefold more concentrated lysate was applied to the coupled beads. After incubation for 3 h at 4°C while rotating, the supernatant was removed and the beads were washed once with PBS supplemented with 1 mM DTT and 1 mM AEBSF and then three times with a wash buffer containing 25 mM Tris, pH 7.5, 350 mM NaCl, 1 mM MgCl2, 1 mM DTT, 1 mM AEBSF and 0.1% NP40. The elution was performed with 40 µL elution buffer (GST B) for 30 min at RT. The eluate was supplemented with Laemmli sample buffer (5×), separated on an 8% SDS–polyacrylamide gel and Western blotting was performed as described above. As a loading control, 1% of each elution sample was separated on a 10% SDS–polyacrylamide gel and Coomassie-stained.
RNA extraction
For the isolation of the RNA, cells were lysed with TRIzol reagent and extraction was performed according to manufacturer's protocol. Solubilized RNA samples were frozen and stored at −80°C.
Northern blotting
Northern blots were conducted as described earlier (Pall and Hamilton 2008). RNA samples from Dicer rescue assays were separated on 12% urea gels (UreaGel System, National Diagnostics), semidry-blotted, and EDC-crosslinked. All Northern blots were hybridized with a radiolabeled probe against let-7a (5′-AACTATACAACCTACTACCTCA-3′) overnight at 50°C. After the incubation, the blots were washed twice with 5× SSC, 1% SDS and once with 1× SSC, 1% SDS. Signals were detected by exposure to a screen and scanning with the phospho-imaging system PMI (Bio-Rad).
Dicer rescue assay
For the dicer rescue assay, 800,000 Dicer−/− MEF-cells were seeded to a 6-well plate and grown overnight. On the next day, cells were transfected by Lipofectamine LTX, using 1 µg of Loqs and 2 µg of dmDcr1 supplemented with 2.5 µL Plus Reagent and 8 µL Lipofectamine LTX. The transfection mixture was applied to the cells and incubated for 6 h. Then cells were split to a 10-cm cell culture dish and after incubation for 2 d, cells were harvested. Sixty-five percent of the cells were used for Western blot analysis and 35% for RNA extraction. Cells for Western samples were resuspended in 80 µL PBS supplemented 20 µL 5× Laemmli buffer and lysed via sonication. Western blotting was performed as described before. For Northern blotting analysis, 10 µg of extracted total RNA from the rescued Dicer−/− cells was used and 0.5 µg of the Dicer+/+ total RNA as a wt control for let-7a.
Coimmunoprecipitation of Loquacious with dmDcr1
HEK293-T cells cotransfected with myc-tagged dmDcr1 and FLAG/HA-tagged Loqs constructs were harvested and lysed for coimmunoprecipitation 48 h after transfection. For each IP transfected cells from one 15-cm plate were lysed in 1 mL IP-lysis buffer (150 mM KCl, 25 mM Tris, pH 7.4, 2 mM EDTA, 1 mM NaF, 0.5% NP-40, 1 mM AEBSF, 1 mM DTT, 100 µg/mL RNase A [Thermo Scientific]). Lysates were cleared by centrifugation after an incubation time of 10 min at 4°C and 10 min at room temperature. As input samples 1.6% of the cleared lysates were taken. The input was then incubated with 50 µL Protein-A-Sepharose beads (GE Healthcare) precoupled to 2.5 µg anti-c-myc antibody (Sigma). The IP reaction was performed for 3 h at 4°C. As control, normal rabbit IgG (Santa Cruz) was used. After incubation with the input, beads were spun down and washed five times with IP wash buffer (300 mM KCl, 50 mM Tris, pH 7.4, 1 mM MgCl2 and 0.5% NP-40) and once with PBS buffer. The elution of the precipitated protein complexes was performed by adding 50 µL SDS-PAGE loading buffer to the beads followed by boiling at 95°C for 5 min. For analysis via Western blot, 40% of each IP-eluate was taken for PAGE.
DSS crosslinks
For protein–protein crosslinks, disuccinimidyl suberate (DSS, Thermo Fisher Scientific) was dissolved in water-free dimethylformamide (DMF), resulting in a concentration of 400 mM. Dilution series were also performed in water-free DMF and the crosslinker was added 1:100 to 4 µM of each protein. The crosslinking was performed for 3 h on ice and stopped by incubation with 100 mM bicarbonate for 15 min. Crosslinked dmDcr1 containing samples were separated on 6%, Loqs-3-HA containing samples on 15% SDS–polyacrylamide gels. Western blot analysis was performed as described above.
Protein labeling
For protein labeling, mutations C386S and C425S were introduced into the Loqs-3 construct to reduce the amount of cysteine available for labeling. Additionally Q406 located in L1 and pointing away from the dimerization- and Dicer-interaction surface was mutated to cysteine, which is accessible for the fluorescent dye. Mutation of C437, which is part of the dimerization surface, led to destabilization of the dimer and could not be mutated.
The labeling reaction was performed using the Alexa Fluor 647 (A647) (Invitrogen) malemide derivate. 1 mg of protein was incubated with a fivefold molar excess of dye for 10 min at room temperature (21°C). The reaction was stopped with β-mercaptoethanol and the protein was loaded onto a Superdex 75 10/300 size-exclusion column where absorption at 280 and 651 nm was monitored. The protein eluted as a single-labeled dimer and a double-labeled monomer (see Supplemental Fig. S2). The dimer fraction was used for the fluorescence spectroscopic measurements.
Single-molecule coimmunoprecipitation experiments
Single-molecule coimmunoprecipitation assays on immobilized proteins were carried out in custom-built flow chambers based on fused silica slides passivated with polyethylene glycol (PEG). Flow chambers were prepared and assembled as described previously (Gietl et al. 2014).
For fluorescence measurements on single immobilized proteins, the flow chamber was incubated with 0.1 mg/mL NeutrAvidin (Thermo Scientific) in PBS for 5 min and washed with 500 µL PBS. Afterwards, the chamber was incubated with 15 nM biotinylated goat anti-GFP-antibody (Abcam) in PBS and washed with 500 µL PBS, 0.5 mg/mL BSA (Roche). In order to test for unspecific binding of Loqs-3AF647, the chamber was incubated with LoquaciousA647 in increasing concentrations (varying from 50 to 200 pM) for 5 min, washed with 500 µL PBS/BSA and incubated for 5 min with oxygen scavenging buffer (Rasnik et al. 2006). In order to measure the oligomerization state of Loqs-3AF647 in complex with GFP-dmDcr1, the chamber was incubated with preformed 50–200 pM Dicer-GFP/Loqs-3AF647 complexes in PBS/BSA for 5 min, washed with 500 µL PBS/BSA, and incubated for 5 min with oxygen scavenging buffer. The GFP-dmDcr1/Loqs-3AF647 complex was preformed by incubating GFP-dmDcr1 (final concentration 1.1 µM) with Loqs-3AF647 (final concentration 1.6 µM) for 2 h on ice before dilution with PBS/BSA. This procedure yielded a completely saturated GFP-dmDcr1 surface (this was needed because of the comparably low-affinity interaction between Dicer-Loqs) and immobilized Loqs-3A647 density of approximately one molecule per 4 µm2. Afterwards, the flow chamber was flushed and incubated for 5 min with 1× PBS/BSA containing the glucose oxidase/catalase oxygen scavenging system. As a control, free Alexa647 dye (200 pM) was flushed over an antibody-GFP-dmDcr1 surface and incubated for 5 min followed by oxygen removal. This did not lead to unspecific binding of the dye to the surface (average of 14 molecules per imaging area).
Single-molecule fluorescence measurements were performed on a homebuilt prism-type total internal reflection (TIRF) setup based on a Leica DMi8 inverse research microscope. Fluorophores were exited with a 488-nm solid-state laser (Coherent OBIS) with a power of 10 mW and 637 nm diode laser (Coherent OBIS, clean-up filter ZET 635/10, AHF Göttingen) with a power of 50 mW. The fluorescence was collected by a Leica HC PL Apo 63x N.A. 1.20 water-immersion objective and split by wavelength with a dichroic mirror (HC BS 560, AHF Göttingen) into two detection channels that were further filtered with a 635-nm long-pass filter (LP Edge Basic, AHF Göttingen) in the red detection channel. Both detection channels were recorded by one EMCCD camera (Andor IXon Ultra 897, EM-gain 20, frame rate 10 Hz, 200–300 frames) in a dual-view configuration (TripleSplit, Cairn Research).
The videos were analyzed employing the iSMS software (Preus et al. 2015) using the program's default settings. Molecule spots were detected using a threshold of 200 for Alexa647 spots. Single-molecule data were acquired as the average number of Loqs-3AF647 fluorescent molecules per imaging area (6800 μm2) as shown in the histograms. The error bars represent standard deviation of the mean values from 11 imaging areas. The number of fluorescence photobleaching steps of Alexa 647-labeled Loquacious was determined for detected spots with a fluorescence intensity of at least 600 (arbitrary units) for single-step bleaching and 1200 for double-step bleaching. The number of molecules showing one or two bleaching steps was accumulated from four independent experiments to obtain the stoichiometry of the complex.
Fluorescence cross-correlation spectroscopy (FCCS)
FCCS measurements were carried out on a ConfoCor2 FCS unit connected to an Axiovert 100M stand equipped with a C-Apochromat 40-fold water immersion lens, NA 1.2 (Carl Zeiss, Jena, Germany). GFP was excited by a 488-nm laser line of an argon-ion laser while AF647 was excited with a 633 helium–neon laser. The emissions were directed over an HFT 488/633 and NFT 635/IR beam splitter. The green fluorescence emitted by GFP was detected after passing a BP 505–550 filter while the red fluorescence emitted by AF647 was recorded after passing a LP 650 filter. Measurements were done in 384-well glass bottom assay plates with acquisition settings of 8 sec 8 times.
In titration experiments, GFP-dmDcr1 was diluted appropriately in PBS-0.2% BSA to a final concentration of ∼20 nM. The protein solution was dispensed as 20 µL aliquots into the 384-well glass bottom plates. Loqs-3AF647 was added to the first well in high concentration (200 nM), followed by a serial dilutions from well to well. Thereby Loqs-3AF647 concentration was titrated in the range of 200–1 nM. After incubation of 15 min at ambient temperature, the FCCS measurements were done on the ConfoCor2 instrument. Each sample was measured for 8 sec with 8 repetitions. The fluorescence fluctuations were auto- and cross-correlated and a fitting formalism employed accounting for two diffusing species for Loqs-3AF647 and one diffusing species for GFP-dmDcr1. Affinities (the dissociation constant Kd) were calculated on the basis of free and bound fractions of both labeled interactors and the concentration of doubly labeled dsRBD-Dicer complexes.
For competition experiments GFP-dmDcr1 was prepared as 20 nM in PBS–0.2% BSA and dispensed as 20 µl portions into wells of the 384-well glass bottom plate. A cascade dilution for the purified dsRBD3 and Loqs-3 was prepared therein by serial dilution steps, ranging from 0.01 nM to 4.2 µM in case of Loqs-3 and 0.02 nM to 8 µM for dsRBD3, respectively. The samples were incubated at ambient temperature for 10 min. The tracer Loqs-3AF647 in PBS-0.2 % BSA was added to each well to final 20 nM. The samples were incubated for an additional 10 min at ambient temperature and subsequently measured by FCCS. IC50 values were determined by plotting the concentration of doubly labeled particles (complex) against the competitor concentration. The resulting IC50 value was translated into a Ki value by application of the Cheng Prusoff equation.
DATA DEPOSITION
Structural coordinates have been deposited at the RCSB Protein Data Bank with the identification code 4X8W.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sigrun Ammon and Corinna Friederich for technical assistance and Qinghua Liu for plasmids. We thank Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beam time and the staff of the HZB MX group for support during data acquisition. D.G. would like to thank Andreas Gietl for his advice in constructing a PRISM TIRF microscope. Our research is supported by grants from the Deutsche Forschungsgemeinschaft (SFB 960, FOR2127), the European Research Council (ERC grant 242792 “sRNAs,” ITN RNATrain), the Bavarian Genome Research Network (BayGene), and the Bavarian Systems-Biology Network (BioSysNet). Work in the Grohmann laboratory is funded by grants from the Deutsche Forschungsgemeinschaft (SFB 960, GR 3840/2-1).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055426.115.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacia K, Schwille P. 2007. Practical guidelines for dual-color fluorescence cross-correlation spectroscopy. Nat Protoc 2: 2842–2856. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Whisnant AW, Kennedy EM, Flores O, Cullen BR. 2014. Derivation and characterization of Dicer- and microRNA-deficient human cells. RNA 20: 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136: 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. 2005. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436: 740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels SM, Melendez-Pena CE, Scarborough RJ, Daher A, Christensen HS, El Far M, Purcell DF, Laine S, Gatignol A. 2009. Characterization of the TRBP domain required for dicer interaction and function in RNA interference. BMC Mol Biol 10: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueck A, Meister G. 2014. Assembly and function of small RNA–argonaute protein complexes. Biol Chem 395: 611–629. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller M, Matsunaga M, Yin S, Loo JA, Guo F. 2007. Heme is involved in microRNA processing. Nat Struct Mol Biol 14: 23–29. [DOI] [PubMed] [Google Scholar]
- Forstemann K, Tomari Y, Du T, Vagin VV, Denli AM, Bratu DP, Klattenhoff C, Theurkauf WE, Zamore PD. 2005. Normal microRNA maturation and germ-line stem cell maintenance requires Loquacious, a double-stranded RNA-binding domain protein. PLoS Biol 3: e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. 2009. Small silencing RNAs: an expanding universe. Nat Rev Genet 10: 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietl A, Holzmeister P, Blombach F, Schulz S, von Voithenberg LV, Lamb DC, Werner F, Tinnefeld P, Grohmann D. 2014. Eukaryotic and archaeal TBP and TFB/TF(II)B follow different promoter DNA bending pathways. Nucleic Acids Res 42: 6219–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase AD, Jaskiewicz L, Zhang H, Laine S, Sack R, Gatignol A, Filipowicz W. 2005. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep 6: 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. 2004. The Drosha–DGCR8 complex in primary microRNA processing. Genes Dev 18: 3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. 2006. Molecular basis for the recognition of primary microRNAs by the Drosha–DGCR8 complex. Cell 125: 887–901. [DOI] [PubMed] [Google Scholar]
- Hartig JV, Esslinger S, Bottcher R, Saito K, Forstemann K. 2009. Endo-siRNAs depend on a new isoform of loquacious and target artificially introduced, high-copy sequences. EMBO J 28: 2932–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitti EG, Sallacz NB, Schoft VK, Jantsch MF. 2004. Oligomerization activity of a double-stranded RNA-binding domain. FEBS Lett 574: 25–30. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38: W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Liu R, Ramani B, Arauz E, Ishitsuka Y, Ragunathan K, Park J, Chen J, Xiang YK, Ha T. 2011. Probing cellular protein complexes using single-molecule pull-down. Nature 473: 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Ye X, Liu X, Fincher L, McKearin D, Liu Q. 2005. Dicer-1 and R3D1-L catalyze microRNA maturation in Drosophila. Genes Dev 19: 1674–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MA, Chan JM, Doudna JA. 2014. Evolutionarily conserved roles of the dicer helicase domain in regulating RNA interference processing. J Biol Chem 289: 28352–28362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139. [DOI] [PubMed] [Google Scholar]
- Kok KH, Ng MH, Ching YP, Jin DY. 2007. Human TRBP and PACT directly interact with each other and associate with dicer to facilitate the production of small interfering RNA. J Biol Chem 282: 17649–17657. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797. [DOI] [PubMed] [Google Scholar]
- Landthaler M, Yalcin A, Tuschl T. 2004. The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol 14: 2162–2167. [DOI] [PubMed] [Google Scholar]
- Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, Carthew RW. 2004. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 117: 69–81. [DOI] [PubMed] [Google Scholar]
- Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. 2006. The role of PACT in the RNA silencing pathway. EMBO J 25: 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Rand TA, Kalidas S, Du F, Kim HE, Smith DP, Wang X. 2003. R2D2, a bridge between the initiation and effector steps of the Drosophila RNAi pathway. Science 301: 1921–1925. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Ma E, Zhou M, Robinson CV, Doudna JA. 2008. In vitro reconstitution of the human RISC-loading complex. Proc Natl Acad Sci 105: 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah G, Barraud P, Allain FH. 2013. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci 70: 1875–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G. 2013. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14: 447–459. [DOI] [PubMed] [Google Scholar]
- Nguyen TA, Jo MH, Choi YG, Park J, Kwon SC, Hohng S, Kim VN, Woo JS. 2015. Functional anatomy of the human microprocessor. Cell 161: 1374–1387. [DOI] [PubMed] [Google Scholar]
- Pall GS, Hamilton AJ. 2008. Improved northern blot method for enhanced detection of small RNA. Nat Protoc 3: 1077–1084. [DOI] [PubMed] [Google Scholar]
- Park JE, Heo I, Tian Y, Simanshu DK, Chang H, Jee D, Patel DJ, Kim VN. 2011. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature 475: 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preus S, Noer SL, Hildebrandt LL, Gudnason D, Birkedal V. 2015. iSMS: single-molecule FRET microscopy software. Nat Methods 12: 593–594. [DOI] [PubMed] [Google Scholar]
- Rasnik I, McKinney SA, Ha T. 2006. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat Methods 3: 891–893. [DOI] [PubMed] [Google Scholar]
- Saito K, Ishizuka A, Siomi H, Siomi MC. 2005. Processing of pre-microRNAs by the Dicer-1-Loquacious complex in Drosophila cells. PLoS Biol 3: e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senturia R, Faller M, Yin S, Loo JA, Cascio D, Sawaya MR, Hwang D, Clubb RT, Guo F. 2010. Structure of the dimerization domain of DiGeorge critical region 8. Protein Sci 19: 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senturia R, Laganowsky A, Barr I, Scheidemantle BD, Guo F. 2012. Dimerization and heme binding are conserved in amphibian and starfish homologues of the microRNA processing protein DGCR8. PLoS One 7: e39688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha NK, Trettin KD, Aruscavage PJ, Bass BL. 2015. Drosophila dicer-2 cleavage is mediated by helicase- and dsRNA termini-dependent states that are modulated by Loquacious-PD. Mol Cell 58: 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomari Y, Matranga C, Haley B, Martinez N, Zamore PD. 2004. A protein sensor for siRNA asymmetry. Science 306: 1377–1380. [DOI] [PubMed] [Google Scholar]
- Tsutsumi A, Kawamata T, Izumi N, Seitz H, Tomari Y. 2011. Recognition of the pre-miRNA structure by Drosophila Dicer-1. Nat Struct Mol Biol 18: 1153–1158. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. 2007. Automated structure solution with autoSHARP. Methods Mol Biol 364: 215–230. [DOI] [PubMed] [Google Scholar]
- Walter TS, Meier C, Assenberg R, Au KF, Ren J, Verma A, Nettleship JE, Owens RJ, Stuart DI, Grimes JM. 2006. Lysine methylation as a routine rescue strategy for protein crystallization. Structure 14: 1617–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RC, Tambe A, Kidwell MA, Noland CL, Schneider CP, Doudna JA. 2015. Dicer-TRBP complex formation ensures accurate mammalian microRNA biogenesis. Mol Cell 57: 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SW, Chen HY, Yang J, Machida S, Chua NH, Yuan YA. 2010. Structure of Arabidopsis HYPONASTIC LEAVES1 and its molecular implications for miRNA processing. Structure 18: 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Liu Q. 2008. Expression, purification, and analysis of recombinant Drosophila Dicer-1 and Dicer-2 enzymes. Methods Mol Biol 442: 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Paroo Z, Liu Q. 2007. Functional anatomy of the Drosophila microRNA-generating enzyme. J Biol Chem 282: 28373–28378. [DOI] [PubMed] [Google Scholar]
- Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. 2004. Single processing center models for human Dicer and bacterial RNase III. Cell 118: 57–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.