

Abstract
Post-transcriptional 3′ end addition of nucleotides is important in a variety of RNA decay pathways. We have examined the 3′ end addition of nucleotides during the decay of the Hsp70 mRNA and a corresponding reporter RNA in Drosophila S2 cells by conventional sequencing of cDNAs obtained after mRNA circularization and by deep sequencing of dedicated libraries enriched for 3′ decay intermediates along the length of the mRNA. Approximately 5%–10% of 3′ decay intermediates carried nonencoded oligo(A) tails with a mean length of 2–3 nucleotides. RNAi experiments showed that the oligoadenylated RNA fragments were intermediates of exosomal decay and the noncanonical poly(A) polymerase Trf4-1 was mainly responsible for A addition. A hot spot of A addition corresponded to an intermediate of 3′ decay that accumulated upon inhibition of decapping, and knockdown of Trf4-1 increased the abundance of this intermediate, suggesting that oligoadenylation facilitates 3′ decay. Oligoadenylated 3′ decay intermediates were found in the cytoplasmic fraction in association with ribosomes, and fluorescence microscopy revealed a cytoplasmic localization of Trf4-1. Thus, oligoadenylation enhances exosomal mRNA degradation in the cytoplasm.
Keywords: mRNA decay, exosome, oligoadenylation, noncanonical poly(A) polymerase
INTRODUCTION
Eukaryotic cells use numerous pathways for RNA degradation: Regulated turnover of mRNA in the cytoplasm contributes to the control of gene expression, “surveillance pathways” serve in the accelerated disposal of all kinds of nonfunctional RNAs both in the nucleus and in the cytoplasm, and intergenic transcripts as well as leftovers of RNA processing are rapidly degraded in the nucleus (Houseley and Tollervey 2009; Stoecklin and Mühlemann 2013). In the majority of these pathways, modifications of the 3′ ends of the RNAs play important roles.
For example, both of the two main pathways of cytoplasmic mRNA decay are initiated by exonucleolytic shortening of the 3′ poly(A) tail, catalyzed mostly by the CCR4–NOT complex. Deadenylation beyond some threshold is required for the second step of mRNA degradation: In the 5′ pathway, 7-methyl GDP is cleaved off the 5′ cap structure by Nudix family enzymes like Dcp2, so that a 5′ monophosphate is exposed, which is then attacked by the processive 5′ exonuclease Xrn1. In the alternative pathway, RNA is degraded from its 3′ end by the exosome. These two pathways were initially characterized in budding yeast, but are conserved among eukaryotes as shown by the conservation of the relevant proteins as well as experimental analyses (Meyer et al. 2004; Parker and Song 2004; Stoecklin et al. 2006; Bönisch et al. 2007; Murray and Schoenberg 2007; Houseley and Tollervey 2009).
RNA degradation can be promoted not only by the removal but also by the addition of 3′-terminal nonencoded nucleotides. The best understood example is provided by the TRAMP complex of S. cerevisiae, which contains the noncanonical poly(A) polymerases Trf4p or Trf5p. With the assistance of the RNA-binding proteins Air1p or Air2p and the RNA helicase Mtr4, these enzymes add oligo(A) tails to a large number of substrates, including misfolded tRNAs, unstable transcripts of intergenic regions, intermediates of rRNA processing, and mRNAs that failed to reach export competence. Oligoadenylation favors complete degradation or 3′ trimming of these RNAs by the exosome (Kadaba et al. 2004; LaCava et al. 2005; Vanácová et al. 2005; Wyers et al. 2005; Rouegemaille et al. 2007; Saguez et al. 2008; San Paolo et al. 2009; Wlotzka et al. 2011), similar to the role of oligoadenylation in prokaryotes (Maes et al. 2012). The oligo(A) tails added by the TRAMP complex may provide an unstructured entry site for the exosome. However, as a catalytically inactive Trf4p mutant can also assist degradation (Wyers et al. 2005; Rouegemaille et al. 2007; San Paolo et al. 2009), protein–protein interactions are likely to contribute to Trf4p-dependent decay (Tudek et al. 2014). Identified substrates of TRAMP-dependent degradation or processing in yeast are so far restricted to nuclear RNAs. Similar mechanisms seem to operate in higher eukaryotes (West et al. 2006; Shcherbik et al. 2010; Lubas et al. 2011; Berndt et al. 2012). In mammalian cells, oligoadenylation of cytoplasmic mRNAs and rRNA fragments has been described. Oligoadenylated rRNA fragments were shown to be decay intermediates, but mRNA fragments were not examined in this respect, and it was not determined whether oligoadenylation promotes decay (Slomovic et al. 2010). In Chlamydomonas, oligoadenylation facilitates degradation of the upstream fragments generated by siRNA-dependent mRNA cleavage (Ibrahim et al. 2006).
The addition of U residues at the 3′ end can affect cytoplasmic mRNA decay (Norbury 2013; Scott and Norbury 2013; Lee et al. 2014a). Oligo(U) sequences have been found at the ends of mRNA poly(A) tails in S. pombe (Rissland and Norbury 2009; Malecki et al. 2013), mammalian cells (Chang et al. 2014; Lim et al. 2014), and Arabidopsis (Sement et al. 2013); short C/U tags have been found in Aspergillus (Morozov et al. 2010, 2012). In mammalian cells, mRNAs with short poly(A) tails are preferred as substrates of uridylation over mRNAs with long tails, and the modification destabilizes the RNAs (Lim et al. 2014). Evidence has been presented that oligouridylation favors cap hydrolysis (Shen and Goodman 2004; Rissland and Norbury 2009; Sement et al. 2013), presumably via binding of the Lsm1–7 complex (Song and Kiledjian 2007). In S. pombe and in mammals, oligouridylation also seems to promote 3′ decay (Hoefig et al. 2013; Malecki et al. 2013; Slevin et al. 2014), whereas the opposite may be true in Arabidopsis (Sement et al. 2013). Consistent with a role in 3′ decay, U residues are added not only to poly(A) tails but also to the upstream fragments generated by siRNA-directed mRNA cleavage in Arabidopsis and mouse cells (Shen and Goodman 2004) and to 3′ decay intermediates of mammalian histone mRNAs (Mullen and Marzluff 2008; Hoefig et al. 2013; Slevin et al. 2014). Like Trf4p and Trf5p, the enzymes responsible for the addition of U residues are members of the enzyme family of poly(U) polymerases or terminal uridilyl transferases (TUTases) and noncanonical poly(A) polymerases (Kwak and Wickens 2007; Martin and Keller 2007; Rissland et al. 2007; Rissland and Norbury 2009; Morozov et al. 2010, 2012; Schmidt and Norbury 2010; Schmidt et al. 2011; Su et al. 2013; Lim et al. 2014).
Decay of the Hsp70 mRNA in Drosophila S2 cells can be analyzed by a natural pulse-chase protocol (Bönisch et al. 2007): Under heat shock, transcription of the Hsp70 genes is induced, and mRNA decay is blocked. When the cells are returned to the normal growth temperature, transcription ceases, and decay of the RNA commences. Rapid degradation directed by the 3′ UTR proceeds via CCR4-NOT-dependent deadenylation followed by Dcp2-catalyzed cap hydrolysis and Xrn1-mediated 5′ decay. The Hsp70 mRNA transiently accumulates as a deadenylated, capped species; thus, cap hydrolysis is a slow step. The same decay pathway is used by reporter RNAs carrying the Hsp70 3′ UTR. 3′ degradation of the Hsp70 mRNA by the exosome is a minor pathway that becomes apparent only when 5′ decay is inhibited. 5′ decay is also the predominant pathway for the degradation of other unstable mRNAs, including miRNA targets, in S2 cells (Behm-Ansmant et al. 2006; Bönisch et al. 2007; Vindry et al. 2012).
Here, we have used a circularization-RT-PCR (cRT-PCR) approach (Couttet et al. 1997) as well as specific amplification of 3′ decay intermediates followed by deep sequencing to analyze the ends of Hsp70 mRNA 3′ decay intermediates. The results, which are consistent with our previous description of the decay pathway, show that intermediates of 3′ decay carry nonencoded 3′ terminal A residues, which are added primarily by the noncanonical poly(A) polymerase Trf4-1. Oligoadenylation is a cytoplasmic process and promotes degradation of the RNA by the exosome.
RESULTS
Intermediates of mRNA decay carry nonencoded A residues
Intermediates of Hsp70 mRNA decay were initially analyzed for 3′ end addition of nucleotides by cRT-PCR (Couttet et al. 1997): Purified cellular RNA is circularized by ligation at low concentration. After reverse transcription, a PCR product spanning the expected junction between 5′ and 3′ end is obtained, and sequencing of plasmid clones then reveals the structure of both ends of the RNA. If circularization is carried out without pretreatment of the RNA, only intermediates of 5′ decay are obtained, as these have lost their 5′ cap and have a ligatable 5′ monophosphate. Pretreatment of the RNA with tobacco acid pyrophosphatase (TAP) removes the cap and allows ligation, so that RNAs with intact 5′ ends can be analyzed together with those that were already decapped in the cell.
S2 cells were heat-shocked for 60 min, then total cellular RNA was isolated. At this time point, the Hsp70 mRNA consists of a mixture of fully polyadenylated molecules and decay intermediates (Bönisch et al. 2007). Transcripts of both Hsp70 genes in the A cluster were amplified. Before cRT-PCR analysis, their transcription start sites were mapped to a position 253 nucleotides (nt) upstream of the translation start codon (Supplemental Table I), which differs from the start site listed at http://flybase.org. The known major polyadenylation site (Bönisch et al. 2007), 200 nt downstream from the stop codon, was confirmed (Supplemental Table I). Hsp70 RNA, including decay intermediates, was then analyzed in the same RNA preparation by cRT-PCR. Of 19 independent clones prepared without TAP treatment, none contained nonencoded nucleotides other than the regular poly(A) tail (Table 1; Supplemental Table II). After TAP treatment, three out of 19 independent clones contained one or two nonencoded A residues (Table 1; Supplemental Table II). All were found upstream of the major polyadenylation site, i.e., they appeared to have been added to 3′ shortened mRNA molecules.
TABLE 1.
Summary of RNA species found in cRT-PCR analysis
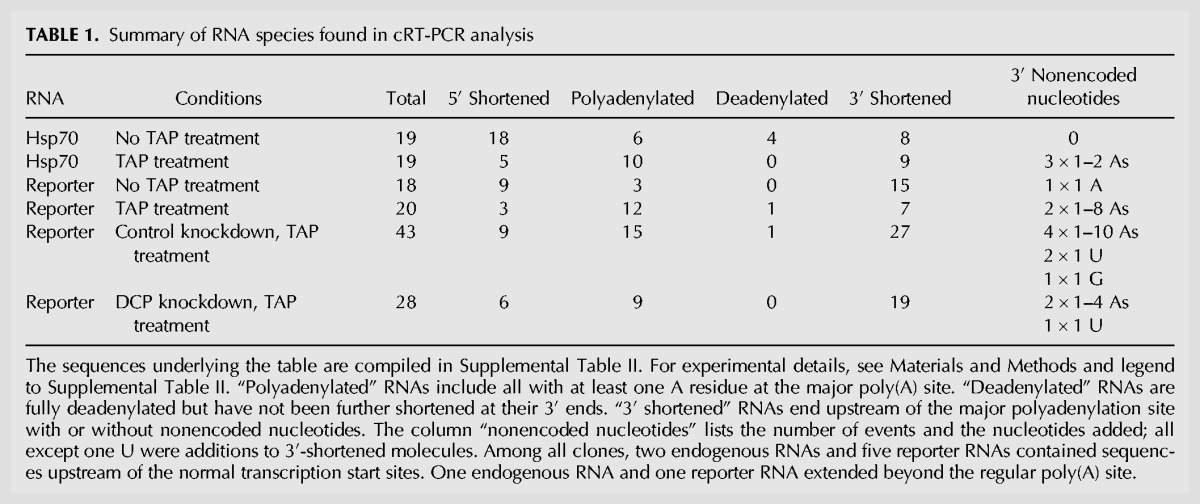
The RNA used for this analysis had been obtained from heat-shocked cells. Since heat shock inhibits mRNA deadenylation and 5′ decay (Bönisch et al. 2007), it seemed possible that 3′ end addition of nucleotides would also be affected under these conditions. Therefore, the analysis was repeated without a heat shock with a Cu2+-inducible reporter mRNA that is destabilized by the Hsp70 3′ UTR (Bönisch et al. 2007). Of 18 independent clones obtained without TAP treatment, the majority had shortened 3′ ends, with one containing a nonencoded A residue (Table 1; Supplemental Table II). Twenty cRT-PCR products were derived from TAP-treated RNA; two contained one, respectively eight nonencoded A residues.
To check whether the 3′ end addition of nucleotides is related to decapping, a combined knockdown of Dcp1 and Dcp2 was carried out. The efficiency of the knockdown was confirmed by Northern blots showing the accumulation of the deadenylated decay intermediate of the reporter RNA (data not shown) (Bönisch et al. 2007). Twenty-eight independent sequences were obtained by cRT-PCR after TAP treatment. One clone displayed a single U residue following two A residues at the poly(A) site. Nineteen clones had shortened 3′ ends, with two carrying nonencoded A residues (Table 1; Supplemental Table II). Forty-three independent sequences were obtained from cells treated with a control RNA, 27 of which had shortened 3′ ends. Two of these contained a single nonencoded U, one a G, and four had nonencoded A residues (Table 1; Supplemental Table II). Thus, inhibition of decapping did not induce a detectable increase in nucleotide addition events.
In summary, 12 out of 147 (8.2%) independent cRT-PCR-derived clones carried nonencoded A residues, with a mean number of 2.6 residues per event. The frequency of end addition of other nucleotides was very low. The nonencoded A residues were found at different sites in the 3′ UTR, but all were located at the ligation junction between 5′ and 3′ end. The chromosomal sequence excluded their origin from transcription initiation upstream of the major site; thus, they must have been derived from an end-addition activity, most likely during 3′ degradation of the mRNA.
Attempts to force the detection of oligouridylated Hsp70 mRNA species by two different PCR methods failed, suggesting that oligouridylated RNAs are barely present in the steady-state population.
Nonencoded A residues are added by the noncanonical poly(A) polymerase Trf4-1 and removed by the exosome
3′ decay intermediates of the Hsp70 reporter RNA were further analyzed by deep sequencing combined with knockdown of proteins involved in mRNA decay and, potentially, nucleotide addition. For this purpose, dedicated sequencing libraries were developed: RNA was isolated 1 h after induction of reporter gene transcription, and DNA adaptors were ligated to the 3′ ends. A primer complementary to the adaptor was used for cDNA synthesis, and Hsp70 reporter sequences were enriched by PCR. Different pairs of nested upstream primers distributed along the mRNA resulted in predominant PCR products reflecting the distance to the 3′ end of full-length deadenylated RNA. However, by selection of shorter PCR products, 3′ decay intermediates could be examined (Figs. 1, 3; see Materials and Methods). After Illumina sequencing, reads were aligned on the Hsp70 reporter sequence and then examined for nonencoded nucleotides at the 3′ end (see Materials and Methods). The numbers of reads matching the reference sequence are shown in Figure 2A,B. As two sets of experiments carried out some time apart and with different sequencing techniques showed different overall levels of oligoadenylated RNAs, the results are presented separately in Figure 2C,D, and the following discussion will refer to the two data sets separately. In spite of different levels of oligoadenylated sequences, the results were overall consistent between the two data sets. In control samples treated with luciferase dsRNA, 9.6% (4.7% in the second experiment) of all reads contained nonencoded A residues at 3′ ends upstream of the normal poly(A) site, whereas only 1.1% (1.0%) had nucleotides other than A. The predominance of A residues among the 3′ extensions is evident in a sequence logo (Fig. 2F). Similar results were obtained in additional controls not treated with any dsRNA (10.7% and 6.6% A reads, respectively). The average length of the A tracts was ∼2.4 nt (Figs. 2C–E). In two additional control samples, the ligation reaction was carried out in the absence of ATP with a pre-adenylated adaptor. The levels of oligoadenylated RNAs (Fig. 2C,D; 6.5% and 4.2%) were within the range of the samples obtained with the normal ligation procedure, and the average lengths of the A tracts were indistinguishable from the other samples. Thus, the additional nucleotides are not artifacts of A addition during the ligation procedure. Nucleotide addition events were distributed over the entire sequence examined. As the analysis of each sample was based on eight independently prepared libraries covering overlapping parts of the sequence, a quantitative comparison of A addition events is possible only locally, not over the whole sequence. Still, several hot spots, for example at nucleotide positions 338, 479, 535, 556, 560 and, most of all, 392/393, were consistent between the many different samples analyzed (Fig. 3A–C). At the 392/393 hot spot, a large fraction of reads ended with nonencoded A residues, for example 64% (Position 392) and 19% (Position 393) in Figure 3A. Overall, the data confirmed the results of the cRT-PCR analyses: Oligo(A) addition was a relatively frequent event, but U addition was below 0.7%. The frequency with which U residues were detected at the end of the “regular” poly(A) tail was below 0.1% in almost all libraries, lower than the frequency of G or C in the same position.
FIGURE 1.
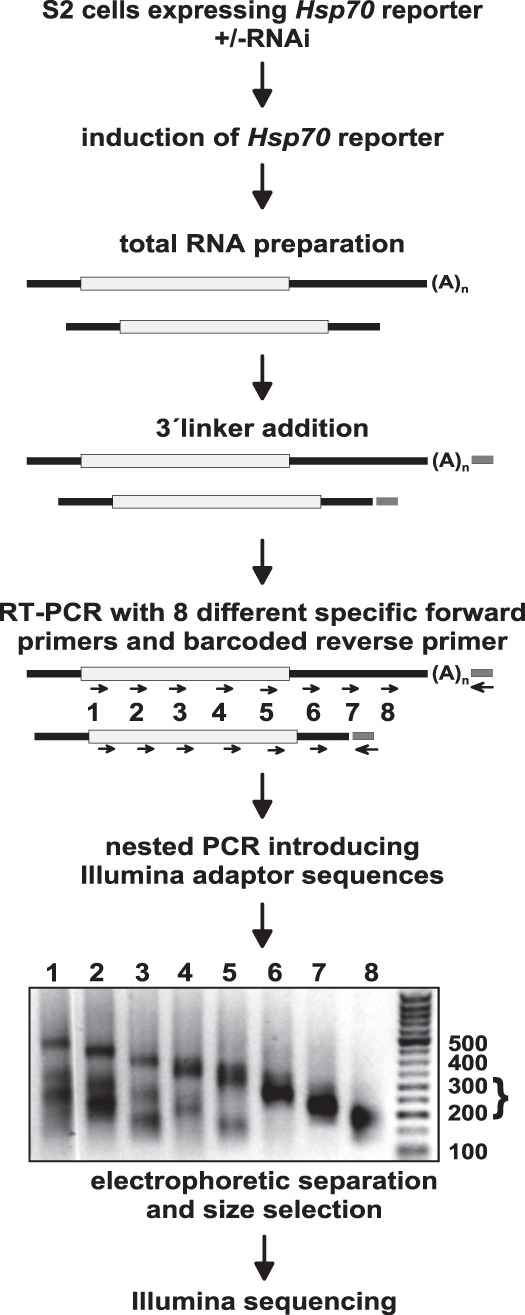
Library preparation for Illumina sequencing. S2 cells expressing the inducible Hsp70 reporter were treated with dsRNA as indicated in the text. Reporter gene transcription was induced with CuSO4 for 1 h, and total RNA was isolated. After barcoded linker addition, PCR was performed as indicated. A size selection (bracket) was performed for products of 200–300 nt, which were subsequently analyzed by Illumina sequencing.
FIGURE 3.

Distribution of oligoadenylated 3′ ends along the reporter mRNA. (A) Distribution of 3′ ends of total sequence reads (red) and of oligoadenylated sequence reads (green) over the reporter mRNA sequence in the luciferase control. Green bars represent the last encoded nucleotide in the sequence read, i.e., the position to which the A tail is added. A hot spot consistently observed at nucleotides 392/393 is indicated with an arrow. Sequence representation ends at the “regular” poly(A) addition site at position 577. Numbering of the sequence is based on the transcription start site reported by the supplier of the pMT/V5 vector; note that the transcription start site identified by the cRT-PCR analysis is 3 nt downstream (Supplemental Table II). (B) The same type of data is displayed for the Mtr3 knockdown. (C) The same type of data is displayed for the Dcp2 knockdown. All data are taken from the experiment shown in Figure 2A,C. (D) Approximate extent of the mRNA sequence covered by the deep sequencing libraries. The numbers on the left indicate the first nucleotide of the second (nested) upstream primer, defining the upper end of the sequence. The lower end depended on the gel purification procedure and is thus less precisely defined, as indicated by the dotted lines.
FIGURE 2.

Illumina sequencing analysis of nonencoded 3′ nucleotides. Data were obtained by the procedure outlined in Figure 1. Labels at the bottom of each figure refer to the dsRNA treatment. Luc, luciferase RNA control; not treated, no dsRNA added; luc-ATP, cells treated with luciferase control RNA and adaptor ligation carried out in the absence of ATP (see Materials and Methods); triple, combined knockdown of CG1091, mkg-p, and Trf4-1. (A) Read numbers of individual sequencing libraries in the first and (B) in the second experiment. In B, samples “Luc r” and “Mtr3 + Trf4-1 r” are technical replicates of the respective preceding sample. (C) Summary of nonencoded 3′ nucleotides in the first and (D) in the second experiment. In D, two samples are averages of two technical replicates as explained for B. (E) Length distributions of oligo(A) tails summarized over the three control libraries of Figure 2C at all internal positions [i.e., excluding the regular poly(A) tail] and at the 392/393 hot spot. The number of tails consisting of a single A was set to 1. This plot represents the length distribution up to 20 nt. Note that in all other analyses, only extensions up to 6 nt were included as explained in Materials and Methods. (F) Sequence composition of the nonencoded 3′ extensions in the luciferase control knockdown experiment represented as sequence logo. For this analysis, tails of between one and six nonencoded nucleotides were used independently of their sequence. Only tails from internal positions were used, i.e., the regular poly(A) tail was excluded from the analysis.
Noncanonical poly(A)/poly(U) polymerases (ncPAPs) are likely to be responsible for the nucleotide addition observed. The D. melanogaster genome contains seven genes encoding members of this enzyme family, either previously characterized experimentally or inferred from the sequence: CG1091, CG11265 (Trf4-1) (Nakamura et al. 2008), CG7163 (monkey king protein; mkg-p), CG5732 (Gld2) (Kwak et al. 2008), CG11418, CG17462 (Trf4-2) (Nakamura et al. 2008) and CG15737 (wispy; wisp) (Benoit et al. 2008; Cui et al. 2008; Lee et al. 2014b). Coding sequences for all except wispy could be amplified from S2 cell RNA. Knockdown experiments were carried out for all seven family members. In addition, a triple knockdown for what appeared to be the most highly expressed candidates, CG1091, mkg-p, and Trf4-1 was carried out. As the RNAs receiving nonencoded nucleotides are likely to be intermediates of 3′ decay catalyzed by the exosome, each ncPAP was knocked down not only individually but also in combination with a knockdown of Mtr3, a core subunit of the exosome (Lorentzen et al. 2008). A knockdown of Dcp2 was also included. Knockdown efficiencies were determined by qRT-PCR (Supplemental Table III).
After depletion of six individual ncPAPs, the percentage of reads with nonencoded 3′-terminal A residues was similar to the controls (6.5%–11.5%, all in the first data set). In contrast, knockdown of Trf4-1 caused a reduction in two independent experiments (3.6% in both data sets). Similarly, the triple knockdown, which included Trf4-1, led to a decrease of oligoadenylated reads (2.7%; first data set) (Fig. 2C). In two independent depletions of Mtr3, the number of reads displaying nonencoded oligo(A) tails increased to 10.6% and 14.8% in the first and second data sets, respectively, and their average length was slightly increased to 2.8 and 2.7 nt. Increased oligoadenylation was also seen when the Mtr3 knockdown was combined with the individual knockdown of six out of seven polymerases (10.9%–15.0% oligoadenylated reads, all in the first data set; Fig. 2C). In agreement with the effect of the single knockdown, codepletion of Trf4-1 partially reverted the increase of oligoadenylated reads caused by the Mtr3 knockdown (8.3% and 10.0% A reads in the first and second experiment, respectively). We conclude that the exosome removes the oligo(A) tails, very likely together with the RNA fragments carrying them. Trf4-1 appears to be mainly responsible for the addition of nonencoded A residues, but a contribution from additional enzymes appears possible based on the slightly stronger effect of the triple knockdown. Surprisingly, the knockdown of Dcp2 resulted in a decrease of oligoadenylated 3′ decay intermediates in three out of three experiments (5.5% and 4.7% A reads in the first and 3.1% in the second data set). The opposite would have been expected if nucleotide addition favored decapping.
Trf4-1 promotes 3′ decay of mRNA
When 5′ decay is blocked by knockdown of the decapping enzyme, additional fragments of the Hsp70 reporter RNA become visible. These were previously identified as intermediates of 3′ decay because they disappear upon a knockdown of the exosome-associated protein Ski3 (Bönisch et al. 2007). This interpretation was confirmed by a Northern blot in which the main fragment was detected by a probe placed immediately upstream of the predicted 3′ end of the fragment but not by a probe placed immediately downstream (Fig. 4A,B). This 3′ decay intermediate with a length of just below 400 nt matches the particularly consistent hot spot of oligoadenylation at nucleotides 392/393 and may be responsible for one of the shorter PCR products seen at the bottom of Figure 1. When Trf4-1 was knocked down simultaneously with the decapping enzyme, the abundance of the ∼400 nt decay intermediate was increased about twofold (Fig. 4C). This result not only supports the correspondence between this fragment and the A addition hot spot but also shows that Trf4-1 promotes mRNA decay, presumably by facilitating the activity of the exosome. While a twofold increase is modest, it was very reproducible, being observed in 11 out of 11 RNA samples obtained at different time points of four independent experiments (average 2.1-fold ± 0.64). A knockdown of Trf4-1 by itself had no effect detectable by Northern blotting (Fig. 4C). This is not unexpected, as 3′ decay makes a minor contribution to the decay of the reporter RNA under normal conditions (Bönisch et al. 2007).
FIGURE 4.

An intermediate of 3′ decay accumulates upon knockdown of Trf4-1. (A) The scheme of the reporter RNA and probes used for the Northern blot. The arrow indicates the position of the approximate 3′ end of the main 3′ decay intermediate visible in Northern blots. (B) S2 cells expressing the Hsp70 reporter were treated with Dcp2 dsRNA. Total RNA was isolated and analyzed by Northern blotting with the probes depicted in A. The ∼400 nt fragment (arrow) was detectable only with probe 1, but not with probe 2. Identification of the deadenylated RNA species is based on comparison to RNaseH/oligo(dT) digestions such as shown in C. (C) S2 cells expressing the Hsp70 reporter were treated with dsRNA against Dcp2 and/or Trf4-1 as indicated. Reporter transcription was induced and inhibited by actinomycin D after 60 min. Total RNA was isolated at the indicated time points after actinomycin D addition and analyzed by Northern blotting with a reporter-specific probe. For quantitation, the intensity of the ∼400 nt 3′ decay intermediate (arrow) was normalized to the intensity of the fully deadenylated decay intermediate (A0).
Oligoadenylation-facilitated mRNA decay can occur in the cytoplasm
A role of oligoadenylation in mRNA degradation has so far been described only as part of a nuclear surveillance pathway in yeast (Rouegemaille et al. 2007; Saguez et al. 2008). In contrast, decapping-dependent 5′ decay of mRNA is considered a cytoplasmic process (van Dijk et al. 2002; Sheth and Parker 2003; Franks and Lykke-Andersen 2008). As the appearance of the 3′ decay intermediates of the Hsp70 reporter mRNA is provoked by the inhibition of decapping, 5′ and 3′ decay pathways compete; this suggests the possibility that the 3′ decay intermediate reflects a cytoplasmic process. Decreased abundance of 3′ decay intermediates upon knockdown of Ski3 (Bönisch et al. 2007) also supports a cytoplasmic localization of the decay intermediates because Ski3 is a cytoplasmic protein. This was initially reported in yeast (Brown et al. 2000) and, judged from the protein's role in the degradation of RNA fragments generated by RNAi, is also true in Drosophila S2 cells (Orban and Izaurralde 2005). Therefore, increased abundance of these intermediates upon Trf4-1 knockdown argues that this enzyme is involved in cytoplasmic mRNA decay.
In order to further examine this point, we separated, in two independent experiments, nuclear and cytoplasmic fractions of S2 cells expressing the Hsp70 reporter. Based on a comparison to the levels of 7SL RNA and U4 snRNA in total RNA preparations from the same cell samples, the cytoplasmic fractions were enriched ∼4.5-fold and the nuclear fractions approximately threefold (Fig. 5A). Hsp70 reporter RNA was present in both fractions, but the nuclear fraction contained an unexpectedly high amount, which was also enriched for the deadenylated intermediate. Thus, we cannot exclude that 7SL RNA was not representative for the extent of cytoplasmic contamination in the nuclear RNA. However, the cytoplasmic fraction is more relevant in this experiment, which was designed to answer the question whether oligoadenylation can occur in the cytoplasm. Deep sequencing revealed that the cytoplasmic fraction contained a significant proportion of oligoadenylated decay intermediates (3.9% compared to 5.8% in nuclear and 6.6% in total RNA; Fig. 5B). If oligoadenylation were a purely nuclear process, the level of oligoadenylated RNA in the cytoplasmic fraction should only have been 1.5%, based on the level of nuclear contamination (U4 snRNA) and oligoadenylated RNA in the total preparation. The average number of A residues added was essentially identical in both fractions (Fig. 5B), and A reads showed similar distributions over the entire mRNA sequence (Supplemental Fig. S1). Thus, while a nuclear contribution cannot be excluded, the data suggest that oligoadenylation of 3′ decay intermediates are present in the cytoplasm.
FIGURE 5.
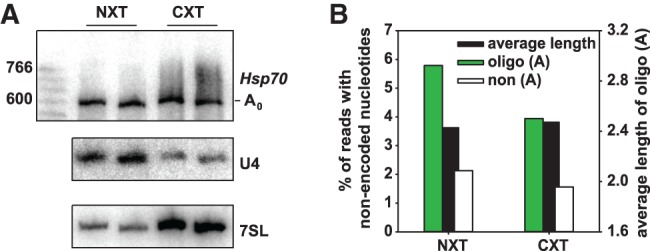
Oligoadenylated 3′ decay intermediates are present in cytoplasmic RNA. Transcription of the Hsp70 reporter was induced for 60 min before cytoplasmic and nuclear RNA was isolated as described in Materials and Methods. (A) The RNA was analyzed by Northern blotting with a reporter-specific probe and probes for U4 and 7SL RNA as markers for the nuclear and cytoplasmic fraction, respectively. The degree of enrichment was calculated from a comparison to additional lanes on the same Northern blot in which total RNA prepared from the same batch of cells was analyzed for U4 and 7SL RNA. (B) Sequencing libraries were generated from the same samples and analyzed by Illumina deep sequencing. Data represent between 1.3 and 2.9 million reads per sample and are averaged from the two independent experiments. The analysis of total RNA from the same cell sample is presented as “not treated” in Figure 2D.
Given the uncertainty of the fractionation experiment, we analyzed polysome-associated RNA after sucrose gradient centrifugation in order to probe more definitively for cytoplasmic oligoadenylation (Fig. 6A). RNA was isolated from polysomal, 80S, and RNP fractions. Northern blot analysis showed that the Hsp70 reporter RNA was mainly associated with 80S and polysomal fractions, indicating that it is translated in S2 cells. Remarkably, this was also true for the deadenylated decay intermediate (Fig. 6A,B). Sedimentation of the reporter RNA was sensitive to EDTA, in agreement with its polysome association. Polysomal RNA and, for comparison, unfractionated input RNA was ligated to a 3′ adaptor, and Hsp70 reporter sequences were amplified with a single pair of nested upstream primers (Fig. 6C). The main RT-PCR product corresponded to the size of the fully deadenylated decay intermediate (255 nt), but a second product corresponding to the expected size (∼70 nt) of the 3′ decay intermediate ending at nucleotide 392/393 was visible in both total and polysomal RNA (Fig. 6D). Sequencing of PCR products of 65–85 nt in length revealed that 5.6% of total RNA reads and 3.4% of polysomal RNA reads contained nonencoded A's. The average tail length was lower than in other samples, but almost identical in total and polysomal RNA (Fig. 6E). Within the limited window examined, a hot spot of A addition at nucleotides 392/393 was again observed in both samples (Supplemental Fig. S2). The experiment indicates that oligoadenylated mRNA 3′ decay intermediates can be associated with ribosomes.
FIGURE 6.

Oligoadenylated 3′ decay intermediates are present on polysomes. (A) Cytoplasmic extract was prepared from S2 cells expressing the Hsp70 reporter and fractioned by sucrose gradient centrifugation (see Materials and Methods). The panel on the left shows the UV trace of a sample from cycloheximide-treated cells analyzed in the presence of Mg2+; the panel on the right shows the UV trace of a comparable sample from cells not treated with cycloheximide and mixed with EDTA to disrupt ribosomes. (B) RNA was isolated from fractions 3, 7, and 14 of the gradients shown in A and analyzed by Northern blotting. 7SL RNA and 5S RNA were used as markers for the RNP fraction and ribosomes, respectively. (C) An RT-PCR procedure as outlined in Figure 1 was used to generate sequencing libraries. A single nested pair of upstream primers was used. The scheme shows how the two main products were generated by adaptors ligated to the completely deadenylated full-length RNA and to the 3′ decay intermediate at nt 392/393. (D) For analytical purposes, the same PCR was carried out with [α-32P]-dCTP, and the products were separated on a denaturing polyacrylamide gel and analyzed by phosphorimaging. Marker sizes are indicated on the left. The main product corresponds to the size expected for amplification of full-length RNA. The second most abundant product (arrowhead) corresponds to the size expected for amplification of the fragment terminating at nt 392/393. (E) Total RNA and polysomal RNA was analyzed by Illumina sequencing. Read numbers obtained were between 303,000 and 518,000 per sequencing library. The data were averaged from two independent experiments.
The hot spot of A addition at position 392/393 is 15 nt downstream from the last nucleotide of the translation stop codon. As this corresponds to the portion of mRNA that would be covered by a ribosome (Sachs et al. 2002; Ingolia et al. 2011) and oligoadenylated 3′ decay intermediates are found in association with ribosomes, slow translation termination might block 3′ decay and cause the build-up of intermediates at position 392/393. However, two experiments suggested that this is not the case. First, ribosomes that are stuck on the message can be removed in a process dependent on the protein Dom34 (Graille and Séraphin 2012). Knockdown of its Drosophila ortholog Pelota on the background of a Dcp2 depletion did not affect the abundance of the major decay intermediate, as analyzed by Northern blots (data not shown). Thus, termination of translation on the reporter RNA appears to be normal. More importantly, when the translation stop codon was moved upstream by 39 or 66 nt, the size of the decay intermediate was not changed (Fig. 7). This result strongly suggests that a terminating ribosome is not responsible for the existence of the intermediate, neither by acting as a road block to the exosome nor by eliciting endonucleolytic cleavage in the context of nonsense-mediated decay. In contrast, insertion of 22 nt immediately after the stop codon did cause a corresponding size increase of the decay intermediate (Fig. 7). Thus, a local RNA sequence or structure or perhaps an RNA–protein complex may be responsible for the transient retardation of 3′ decay.
FIGURE 7.
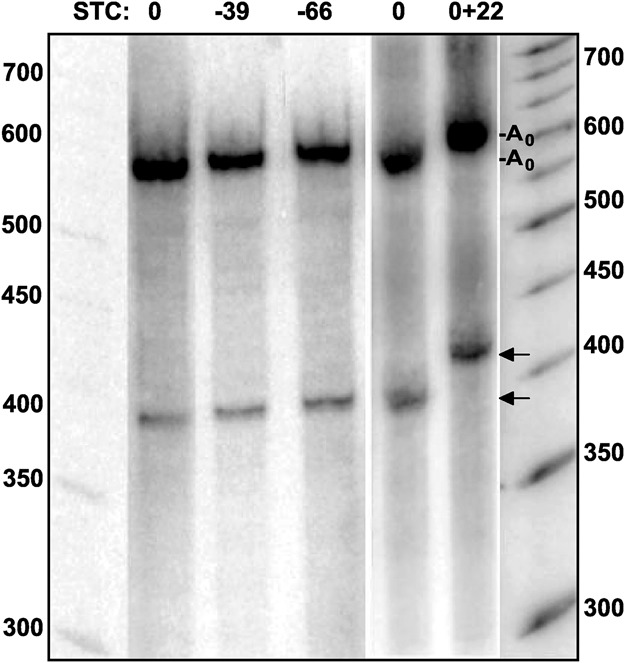
The size of the 3′ decay intermediate is independent of the stop codon. Three variants of the Hsp70 reporter construct carrying the translation stop codon at different locations were used. The position of the stop codon is indicated on top: wild-type position (0), 39 nt and 66 nt upstream. A reporter variant with a 22 nt insertion immediately downstream from the stop codon was also used. S2 cells stably transformed with the constructs were treated with the Dcp2 dsRNA. Transcription of the reporter was induced by addition of 0.5 mM CuSO4. After 1 h, total RNA was isolated and analyzed by Northern blot. DNA probes were directed against reporter regions 1–51 (0+22) and 350–400 (0, −39 and −66). Because of the unexpected result, reporter mRNA was amplified from the experimental samples and the positions of the stop codons were confirmed by sequencing.
Trf4-1 is localized in the cytoplasm
On the basis of overexpression experiments, Trf4-1 has previously been reported to polyadenylate snRNAs (Nakamura et al. 2008). The protein was found to be localized in nucleoli, which would be inconsistent with the cytoplasmic function proposed here. Therefore, the localization of C-terminally YFP-tagged Trf4-1 was re-examined by fluorescence microscopy after transfection of S2 cells. In all cells expressing the fusion protein, it was found in the cytoplasm with little fluorescence in the nucleus or nucleolus (Fig. 8A). In some Trf4-1-expressing cells, perhaps those with relatively low expression levels, the protein was widely distributed in the cytoplasm, but in many cells it was also enriched in about three to six cytoplasmic foci. Costaining with an antibody against Me31B, a P body marker (Eulalio et al. 2007) showed that the Trf4-1-containing foci were not P bodies (Fig. 8B). Although the discrepancy with the earlier localization data remains unexplained, we tentatively conclude that the localization of Trf4-1 is consistent with a function in cytoplasmic mRNA decay. The fact that the protein is not found in P bodies is consistent with its role in 3′ decay, as proteins participating in this pathway are excluded from P bodies (Brengues et al. 2005).
FIGURE 8.

The Trf4-1 protein is localized in the cytoplasm. (A) Confocal images of S2 cells transiently expressing the C-terminally YFP-tagged Trf4-1 (green). A wide cytoplasmic distribution of the protein is depicted in the upper panel. The two lower panels show the enrichment of the protein in small cytoplasmic foci. DAPI was included in the mounting medium to label the nuclei (blue). (B) Trf4-1-YFP foci are not P bodies. S2 cells transiently expressing Trf4-1-YFP (green) were stained with an antibody against Me31B. The secondary antibody was Cy3 labeled (red). Cells containing a wide distribution (upper panel) and cytoplasmic foci (middle panel) of Trf4-1-YFP are shown. An untransfected cell was included as a negative control (lower panel). DAPI staining is shown in blue. Note that plasmid yields of the original Trf4-1 clone and the Trf4-1-YFP clone were always poor, and cells containing the latter either failed to grow in liquid culture or did not grow to normal densities. As this suggests selection pressure on the plasmids, the correct sequence of the Trf4-1-YFP clone was verified in the preparation used for the transfection experiment shown here.
DISCUSSION
3′ end addition of nontemplated A residues promotes the 3′ exonucleolytic processing or decay of many different types of RNA molecules in the cell nucleus. While oligoadenylated cytoplasmic mRNA fragments have been observed (Slomovic et al. 2010), a role of such modifications in cytoplasmic mRNA decay has not been demonstrated so far. Here, small numbers of nontemplated A residues were found attached to intermediates of 3′ mRNA decay. The noncanonical poly(A) polymerase Trf4-1 was mainly responsible for A addition, and a knockdown of the enzyme inhibited exosomal 3′ decay of a reporter RNA. Importantly, oligoadenylated 3′ decay intermediates were present in polysomal RNA, and Trf4-1 was found to be present in the cytoplasm. Thus, we propose that oligoadenylation by Trf4-1 facilitates exosomal mRNA decay in the cytoplasm.
The initial cRT-PCR analysis yielded a relatively small data set, but both ends of the RNA were analyzed simultaneously. Of all independent clones analyzed after TAP treatment from cells with wild-type decapping activity, 55% were completely deadenylated, and most of the others had short tails. Still, only 17 (21%) of the same population had undergone 5′ shortening. This is consistent with the transient accumulation of deadenylated, capped species previously described for the decay of Hsp70 mRNA (Bönisch et al. 2007). As expected, the majority of clones obtained from the same RNA preparations without TAP treatment had undergone 5′ shortening (73%). Seventy-four percent of these were completely deadenylated RNAs, consistent with deadenylation preceding cap hydrolysis.
Both in the cRT-PCR analysis and in deep sequencing, ∼5%–10% of the reads displayed nonencoded 3′-terminal A residues that had been added to 3′ shortened molecules during mRNA decay by one or more noncanonical poly(A) polymerases, as supported by the following evidence: First, the A residues were present upstream of the dominant polyadenylation site of the RNA. While hot spots of A addition were identified, the sites were spread over much of the mRNA. Second, the number of A residues found at the unexpected sites was much lower than in poly(A) tails added during nuclear pre-mRNA processing: The average number was 2.6 in cRT-PCR analysis and ∼2.4 in deep sequencing. Third, knockdown of the noncanoncial poly(A) polymerase Trf4-1 led to reduced oligoadenylation.
If oligoadenylation served to promote cap hydrolysis, a knockdown of the decapping enzyme should have led to the accumulation of oligoadenylated RNAs. However, by cRT-PCR analysis, such an increase was not detectable. Surprisingly, deep sequencing even showed a reduction of oligoadenylated RNAs upon inhibition of decapping. This might simply be due to the increased abundance of deadenylated mRNA decay intermediates saturating the enzymes adding the A residues.
Oligoadenylation is associated with exosome activity. For example, a hot spot of A addition corresponds to the most highly populated intermediate of 3′ decay; reduced amounts of this intermediate upon knockdown of Ski3 imply that it is generated by the exosome (Bönisch et al. 2007). Moreover, the abundance and length of oligo(A) tails in the decay intermediates were increased upon exosome knockdown, so the additional A's appear to be degraded by this enzyme complex. These observations suggest that the exosome pauses at preferred sites during mRNA degradation, the decay intermediates are oligoadenylated, and oligoadenylation supports further digestion by the exosome. This interpretation of the data is directly supported by the increased abundance of the main 3′ decay intermediate upon knockdown of Trf4-1. While this decay intermediate was not sufficiently abundant to be visible in Northern blots of control samples, it was evident as a hot spot of A addition; thus, the events generating this intermediate are part of normal, undisturbed mRNA degradation.
In Northern blots, the oligoadenylated intermediate of 3′ decay becomes visible only when the decapping enzyme Dcp2 is depleted (Bönisch et al. 2007). Thus, the intermediate must be on a pathway competing with decapping-dependent 5′ decay. The latter is normally considered a cytoplasmic pathway, so competition with deadenylation-dependent 3′ decay would suggest a cytoplasmic localization of the latter. However, the argument is not entirely compelling, since the decapping enzyme can also act in the nucleus (Brannan et al. 2012; Geisler et al. 2012). A cytoplasmic localization of oligo(A)-stimulated 3′ decay is more convincingly supported by other observations: First, even at relatively late time points in actinomycin D chase experiments, the 3′ decay intermediate parallels the levels of full-length RNA, which is likely to be dominated by cytoplasmic molecules (Fig. 4; Bönisch et al. 2007). Second, the abundance of the intermediate decreases when Ski3 is knocked down in addition to Dcp2; presumably, this shifts the balance of degradation pathways back to 5′ decay (Bönisch et al. 2007). Ski3 is believed to be a cytoplasmic protein (Orban and Izaurralde 2005). Third, enrichment of nuclear and cytoplasmic RNAs by cell fractionation showed that similar proportions of 3′ decay intermediates were adenylated in both fractions. Fourth, and most importantly, oligoadenylated 3′ decay intermediates were found in RNA isolated from polysomal fractions. While the data do not rule out a contribution of nuclear processes, they strongly suggest that cytoplasmic, ribosome-associated mRNAs are substrates for 3′ decay and intermittent oligoadenylation. Based on cell fractionation, Schuster and co-workers also reported the presence of oligoadenylated mRNA fragments in the cytoplasm of HeLa cells. However, their relationship to mRNA decay was not examined (Slomovic et al. 2010).
As the cap and the poly(A) tail synergistically promote translation initiation (Gallie 1991; Tarun and Sachs 1996), removal of the poly(A) tail would be expected to reduce the rate of translation of mRNAs. That the deadenylated decay intermediates were associated with ribosomes to the same extent as the polyadenylated species was therefore somewhat unexpected. However, the result is consistent with the polysome association of intermediates of deadenylation-dependent 5′ mRNA decay (Hu et al. 2009; Antic et al. 2015; Pelechano et al. 2015). Deadenylation of the Hsp70 mRNA takes on the order of 20–30 min (Bönisch et al. 2007), so the deadenylated intermediate can only accumulate to significant levels if its half-life is also on the order of at least several minutes. In contrast, translation proceeds on a much shorter time-scale: With a translation rate of 6 amino acids per second (Ingolia et al. 2011), a ribosome should traverse the short ORF of the reporter construct within <20 sec, and termination is thought to be relatively fast (Arava et al. 2005). Thus, continued ribosome association of the deadenylated decay intermediate suggests ongoing initiation.
Trf4-1 appears to be mainly responsible for A addition to 3′ decay intermediates, as the number of A addition events was consistently reduced by Trf4-1 knockdown in four out of four independent experiments. In contrast, depletion of none of the other noncanonical poly(A) polymerases had a consistent effect with or without simultaneous knockdown of Mtr3 (Fig. 2). When the main 3′ decay intermediate became visible upon inhibition of decapping, its abundance was also reproducibly increased by depletion of Trf4-1 (Fig. 4). The proposed function of Trf4-1 is consistent with its ATP-specific polymerase activity observed in vitro (Nakamura et al. 2008). An involvement of the enyzme in a cytoplasmic process was unexpected, as it had been reported to be localized in nucleoli (Nakamura et al. 2008). In our experiments, however, a cytoplasmic localization of Trf4-1-YFP was apparent, in agreement with a function in mRNA decay. The reason for the discrepancy is unknown. Unfortunately, we could not repeat the localization experiment with other Trf4-1 variants, as numerous attempts to generate additional tagged versions of Trf4-1 using multiple vectors, host strains and growth media were unsuccessful. We also could not clone an active site mutant of Trf4-1. (See also legend to Fig. 8.) The Trf4 family of noncanonical poly(A) polymerases is represented by Trf4-1 and Trf4-2 in Drosophila, by a single member, GLD-4, in C. elegans and by PAPD5 and PAPD7 in mammals. A phylogenetic comparison did not suggest simple one-to-one homology relationships between family members in different organisms (Minasaki and Eckmann 2012). Nevertheless, cytoplasmic functions have been reported for several Trf4-like enzymes: C. elegans GLD-4 is partially localized in cytoplasmic RNA granules (Schmid et al. 2009), sediments with polysomes (Nousch et al. 2014) and has cytoplasmic functions (Schmid et al. 2009; Millonigg et al. 2014). Mammalian PAPD5 is localized in the cell nucleus with an enrichment in nucleoli (Lubas et al. 2011; Rammelt et al. 2011; Ogami et al. 2013) in agreement with roles in oligoadenylation of RNA polymerase I transcripts (Shcherbik et al. 2010) and snoRNA processing intermediates (Berndt et al. 2012). However, functional data are also consistent with a cytoplasmic role (Burns et al. 2011). PAPD7 is also mostly nuclear, but a cytoplasmic fraction is detectable (Ogami et al. 2013).
Only two out of 147 independent cRT-PCR clones analyzed contained a nonencoded 3′ terminal U residue, and only one of those was present at the 3′ end of an A2 sequence at the major poly(A) site. In the deep sequencing libraries, the fraction of “regular” poly(A) tails that had other nucleotides at their 3′ ends was small, typically on the order of 0.5%. Among these, U was the least abundant nucleotide. With the caveat that our analysis only allowed the analysis of short tails, this differs from the results obtained in S. pombe and in human cells, where U addition to poly(A) tails is a relatively frequent event (Rissland and Norbury 2009; Chang et al. 2014; Lim et al. 2014; Slevin et al. 2014). Also, knockdown of the decapping enzyme did not lead to the appearance of nonencoded U residues. However, we cannot exclude a technical bias against U detection at the 3′ ends. A deletion of the dis3l2+ gene of S. pombe leads to a strong increase in the abundance of 3′ mRNA decay intermediates carrying oligo(U) tails. This, together with in vitro experiments, suggests that Dis3L2 degrades such RNAs (Malecki et al. 2013). The mammalian ortholog participates in the degradation of oligouridylated histone mRNAs (Hoefig et al. 2013). The D. melanogaster genome contains a Dis3L2 ortholog; its role in mRNA decay remains to be studied before any conclusion regarding U addition can be drawn.
The data reported here and previously (Bönisch et al. 2007) lead to the following plausible model for the decay of the Hsp70 mRNA: The RNA loses its poly(A) tail while it is being translated. The deadenylated decay intermediate accumulates transiently in a capped form (Bönisch et al. 2007); slow cap hydrolysis is paralleled by continued translation of the deadenylated RNA, and it seems likely that the two phenomena are causally related. Once an RNA molecule has lost its cap, it is rapidly degraded by XRN1 without accumulation of intermediates. Alternatively, while being translated, the deadenylated, capped RNA can be digested from the 3′ end. This is a minor pathway (Bönisch et al. 2007) and proceeds through detectable paused intermediates, which are substrates for A addition by Trf4-1 and possibly additional noncanonical poly(A) polymerases. Trf4-1-dependent oligoadenylation facilitates further digestion by the exosome.
Our data are quite similar to a recent analysis of mammalian histone mRNA decay (Slevin et al. 2014). Histone mRNAs were found to be degraded by the exosome in 3′–5′ direction while being translated. A large fraction of 3′ decay intermediates had undergone 3′ end addition of nonencoded nucleotides, but these were U rather than A residues. U addition presumably facilitates exosome-catalyzed degradation of the histone mRNAs. As nonencoded U residues have also been found at the 3′ ends of other mammalian RNA decay intermediates (Wilusz et al. 2012), it appears that U addition versus A addition may be a feature specific for mammals versus Drosophila rather than for histone mRNAs versus polyadenylated mRNAs. Thus, flies and mammals use the 3′ addition of different nucleotides for the same purpose, facilitated degradation of cytoplasmic mRNAs by the exosome.
MATERIALS AND METHODS
Plasmids, oligonucleotides, and proteins
The Cu2+-inducible Hsp70 reporter construct has been described (Bönisch et al. 2007). The stop codon was moved upstream by 39 (RNPhsp70stc-39) or 66 (RNPhsp70stc-66) nt by site-directed mutagenesis. The variant with 22 nt inserted downstream from the stop codon (RNPhsp70STC+22) was generated by PCR using the Hsp70 reporter plasmid as template. For the Trf4-1-YFP expression plasmid, replacement, using SacII and AgeI, of the V5 epitope of pMT/V5 HisB (Life Technologies) by a PCR product encoding YFP resulted in pMT_YFP. The Trf4-1 ORF was amplified from the clone D. melanogaster/RE04457 (GenomeCube) and cloned into pMT_YFP between the ApaI and EcoRI sites upstream of YFP.
Me31B was cloned into pET SUMO (Invitrogen) and fused, at its C terminus, with a prescission protease site followed by the maltose-binding protein. The protein was expressed in E. coli and purified by chromatography on Ni2+-NTA and amylose resins, followed by cleavage of the tags with SUMO and prescission proteases and a final anion exchange chromatography. A rabbit antibody, raised against the purified protein by Eurogentec, was affinity-purified by incubation with the antigen blotted to a membrane, elution with glycine buffer (pH 2.3) and neutralization with Tris.
All oligonucleotides used in this study are listed in Supplemental Table IV.
Cell culture and RNA interference
Conditions for S2 cell growth, heat shock, generation of stably transformed lines and induction of the Hsp70 reporter construct have been described previously (Bönisch et al. 2007). Double-stranded RNA was synthesized by in vitro transcription (Fermentas, TranscriptAid T7 High Yield Transcription Kit) of templates obtained by PCR. In addition to Dcp2 and luciferase (Bönisch et al. 2007), templates were generated from oligo(dT)-primed cDNA derived from S2 cell RNA or genomic DNA (wispy). Logarithmically growing S2 cells (< 2 × 106 cells/mL) were pelleted and resuspended in serum-free medium to 106 cells/mL. One milliliter was transferred to a 30-mm cell culture dish containing 15 µg dsRNA, incubated for 30 min at 25°C and then mixed with 2 mL medium containing 10% serum. After 4 d, transcription was induced and blocked (Bönisch et al. 2007). The efficiency of the Dcp2 knockdown was analyzed either by the accumulation of deadenylated Hsp70 reporter RNA (Bönisch et al. 2007) or by qRT-PCR. Efficiencies of other knockdowns were analyzed by qRT-PCR.
Cell transfection and fluorescence microscopy
S2 cells were transfected with the Trf4-1-YFP expression construct in 12-well plates with Effectene (Qiagen) and 300 ng DNA per well. The plasmid expressing YFP and the empty vector served as controls. Expression of Trf4-1-YFP or YFP was induced by 0.7 mM CuSO4. For fluorescence microscopy, cells were allowed to adhere to poly-D-lysine-coated coverslips for 30 min, washed once with PBS, fixed for 15 min with 4% paraformaldehyde (Sigma), washed three times, permeabilized for 5 min with 0.3% Triton-X 100 in PBS and washed again three times. Cells were then either directly mounted in Prolong Gold antifade reagent with DAPI (Invitrogen) or first immunostained with an affinity-purified Me31B antibody as described previously (Temme et al. 2009). Anti-rabbit Cy3-labeled secondary antibody (Dianova) was diluted 1:1000. Cells were analyzed in a confocal laser scanning microscope (Leica, SP5) using a 100× TIRF lens and the Leica 3D deconvolution software.
RNA isolation and cRT-PCR analysis
Total RNA was isolated from S2 cells according to the TRIzol protocol (Invitrogen) or with the help of a total RNA isolation kit (Roche). For decapping, 25 µg RNA were treated with 5 U tobacco acid pyrophosphatase (TAP; Epicentre) in a 40 µL reaction volume for 1 h at 37°C according to the supplier's protocol in the presence of 40 U RNase inhibitor (Ribolock, Fermentas). For circularization, RNA from 8 µL of the TAP reaction or 5 µg untreated RNA were ligated overnight at 16°C with 80 U RNA ligase (NEB) in the buffer supplied supplemented with 10 mM DTT, 5% DMSO and 40 U RNase inhibitor (800 µL reaction volume). RNA was purified by phenol–chloroform extraction and ethanol precipitation. Two micrograms of the circularized RNA was reverse-transcribed with 20 pmol sequence-specific primer and 150 U M-MLV reverse transcriptase (Promega) in a 25-µL reaction volume as recommended by the supplier. For PCR, 2 µL of this reaction was used as template in a total volume of 50 µL 1× Pfu buffer (Stratagene) containing 0.2 mM dNTPs, 1 µM primer (each), and 2.5 U Pfu DNA polymerase (Stratagene). The PCR product was purified with the High Pure PCR Product Purification Kit (Roche), phosphorylated with T4 polynucleotide kinase and ligated into the PBS SK+ vector, which had been digested with EcoRV and dephosphorylated. After transformation of E. coli XL1, plasmid DNA was purified from single colonies, and the insert was sequenced. Sequences matching the Hsp70 RNA were selected. Identical sequences originating from the same PCR were counted only once.
Cell fractionation, polysome analysis, and Northern blotting
For separation of nuclear and cytoplasmic RNA, S2 cells stably transfected with the Hsp70 reporter construct were induced with CuSO4 for 1 h and lysed in 20 mM HEPES, pH 7.6, 10 mM NaCl, 20% glycerol, 0.1% Triton X-100. Nuclei were pelleted at 300g for 10 min, and RNA was isolated from the pellet and the supernatant by TRIzol extraction. For polysome analysis, cells were incubated with 0.5 mM CuSO4 for 45 min and for an additional 15 min with or without 100 µg/mL cycloheximid and lysed by dounce homogenization in 10 mM HEPES, pH 7.5, 1.5 mM magnesium acetate, 10 mM KCl, 200 µg/mL fragmented yeast RNA, 0.4 U/µL Ribolock (Fermentas), 1 mM DTT, 1 µg/mL aprotinin, 1 mM PMSF, 2 µg/mL leupeptin and 2 µg/mL pepstatin. Ribosomes in samples without cycloheximide were dissociated by the addition of 10 mM EDTA to the lysis buffer. The extracts were centrifuged at 20,000g for 5 min and the supernatants applied to 10%–45% sucrose gradients in 20 mM HEPES, pH 7.5, 150 mM KCl, 5 mM magnesium acetate ± 10 mM EDTA. After 3 h centrifugation at 40,000 rpm in a SW40Ti rotor (Beckmann), the gradient was separated into 15 fractions and RNA was isolated from fractions 3, 7, and 14 by TRIzol extraction. RNA was separated by denaturing 5% PAGE, transferred to a membrane and hybridized with synthetic oligonucleotide probes labeled with polynucleotide kinase and γ-[32P]-ATP.
Illumina sequencing
T4 RNA ligase 1 (NEB) was used according to the manufacturer's protocol to ligate a DNA oligonucleotide with a 6 nt barcode at its 5′ end to the 3′ ends of total RNA preparations. In the first data set, a control ligation in the absence of ATP was carried out with a preadenylated adaptor and T4 RNA ligase 1. In the second data set, truncated T4 RNA Ligase 2 K227Q (NEB) was used for the same type of control. After G50 gel filtration, phenol–chloroform extraction and ethanol precipitation, ligation products were reverse-transcribed (M-MLV reverse transcriptase, Promega) with an adaptor reverse oligonucleotide. The Hsp70 reporter RNA-specific library was generated by two rounds of nested PCR. Eight different forward primer combinations spanning the reporter RNA were used together with a reverse primer complementary to the 3′ ligation adaptor. The second PCR introduced Illumina-specific adaptor sequences such that sequencing was from the 3′ end of the mRNA; the first 6 nt read were part of the primer (barcode), the seventh was the 3′ most nucleotide of the mRNA. For the first data set (Fig. 2A,C), the TruSeq SR Cluster Generation Kit v5 (GA) and SBS Sequencing Kit v5 (GA) were used for sequencing 22 plus two unrelated samples mixed with 50% of a genomic library prepared from ΦX174 DNA (Illumina PhiX Control v3) in two lanes on the Illumina GA IIx platform (36 sequencing cycles). Image analysis and base calling was performed with the analysis pipeline version 1.7.0 (within SCS 2.8). For the second data set (Fig. 2B,D), 24 samples mixed with the ΦX174 control were sequenced on the Illumina HiSeq platform in two lanes (50 cycles) with the TrueSeq SR Cluster Kit v3 (HiSeq) und TrueSeq SBS Kit v3 (HS). For image analysis and base calling, the analysis pipeline version 1.8.2 (within HCS 1.4.8) was used. Sequencing was performed in the Genomics Unit of the Centre for Genomic Regulation (CRG) Barcelona, Spain.
The library for the analysis of polysomal RNA was generated using the 3′ adaptor oligonucleotide (barcode 2) for ligation and reverse transcription and only one set of nested PCR forward primers specific for the reporter RNA (RNPhsp70 336_355, RNPhsp70 355_374). Fifty nanograms of purified amplicon DNA was used in the low sample DNA protocol with the TruSeq DNA Sample Preparation Kit (Illumina) according to the manufacturer's instructions. The libraries were purified using the Ampure XP protocol (Beckman Coulter) and quantified using the Library Quantification Kit - Illumina/Universal (KAPA Biosystems) according to the manufacturer's instructions. A pool of five libraries was used for cluster generation at a concentration of 10 nM using an Illumina cBot. Sequencing of 100 bp was performed with an Illumina HighScan-SQ sequencer at the sequencing core facility of the IZKF Leipzig (Faculty of Medicine, University Leipzig) using version 3 chemistry and flow-cell according to the manufacturer's instructions. Demultiplexing of raw reads, adapter trimming and quality filtering was done as described (Stokowy et al. 2014). Subsequent analysis was limited to those reads in which the 3′ end of the Hsp70 mRNA was flanked by the adaptor sequence.
Sequencing data analysis
Illumina GA IIx and HiSeq sequencing reads were filtered for low quality, the barcode was removed, and the reverse complement of the resulting 30 nt sequences (first data set) and 44 nt sequences (second data set) was built. Illumina HiScanSQ data (polysomal samples) were trimmed according to the primer sequences used, and the 5′ ends were extended by the corresponding part of the reference sequence to obtain a total length of 44 nt for each read to allow comparison. The resulting reads of all samples were mapped against the Hsp70 reporter sequence (Bönisch et al. 2007) with Bowtie v0.12.6 (Langmead et al. 2009). Mapping parameters allowed for up to 20 nonencoded nucleotides at the mRNA 3′ end of each read with no mismatches in the anchoring 5′ part of reads (Bowtie parameters -S -l 10 -n 0 -e 800 --nomaqround --norc for 30 nt reads, and -l 24 for 44 nt reads, respectively). To detect nonencoded 3′ ends, mismatches were counted, and at most 50% matching bases (nonconsecutive) were allowed in the 3′ part after the first mismatch base. In some libraries, large numbers of reads were found with 3′ ends at a position with 4 nt sequence complementarity to the 3′ end (bar code) of the RT primer. As these reads were obviously generated by internal priming, they were excluded from the analysis. The average length of nonencoded extensions was 4.65 nt. Tracts of nonencoded 3′-terminal nucleotides longer than 6 nt tended to have mixed sequences and occurred at rates below 0.8% in the analysis of polysomal RNA (Fig. 6) and at rates of at most 5% in the first data set (Fig. 2A,C). However, in five libraries in the second data set, such tracts were found at higher levels. These were the two NXT samples and one CXT sample in Figure 5 (9.2%, 6.4%, and 6.4%); and the Mtr3/Trf4-1 double knockdown and the -ATP control in Figure 2B,D (18.7% and 10.7%). Several features suggested that they were unrelated to the phenomenon examined here and very likely artifactual. Thus, reads containing nonencoded 3′ ends longer than 6 nt were discarded in further analyses. Nonencoded extensions were classified as oligo(A) if they contained A in extensions of length 1 nt, at most one G, C, or T in extensions of length 2 nt or 3 nt, at most two non-As in extensions of 4, 5, or 6 nt, and no consecutive non-As. To generate sequence logos, 10,000 extensions were randomly selected per sample and prepared as input for WebLogo v3.3 (2012-07-02) which was run with parameters -A dna -U probability --composition none -s large --errorbars NO -c classic --scale-width NO -E YES (Schneider and Stephens 1990; Crooks et al. 2004). Custom analyses were performed with scripts written in Perl v5.8.9 and using Unix shell commands.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nadine Bley and the Imaging Facility of the Medical Faculty of Martin Luther University Halle-Wittenberg for help with microscopy; Knut Krohn and the sequencing core facility of the Medical Faculty of the University of Leipzig for sequencing the polysomal RNA libraries; Elisa Izaurralde and Christian Eckmann for helpful comments; and Christiane Rammelt for helpful discussions and comments on the manuscript. A large part of the sequencing data described in this work was generated at the Genomics Unit of the Centre for Genomic Regulation, Barcelona, Spain. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG; WA 548/11-1) to E.W.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.053942.115.
REFERENCES
- Antic S, Wolfinger MT, Skucha A, Hosiner S, Dorner S. 2015. General and microRNA-mediated mRNA degradation occurs on ribosome complexes in Drosophila cells. Mol Cell Biol 35: 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arava Y, Boas FE, Brown PO, Herschlag D. 2005. Dissecting eukaryotic translation and its control by ribosome density mapping. Nucleic Acids Res 33: 2421–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. 2006. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 20: 1885–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit P, Papin C, Kwak JE, Wickens M, Simonelig M. 2008. PAP- and GLD-2-type poly(A) polymerases are required sequentially in cytoplasmic polyadenylation and oogenesis in Drosophila. Development 135: 1969–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt H, Harnisch C, Rammelt C, Stöhr N, Zirkel A, Dohm JC, Himmelbauer H, Tavanez J-P, Hüttelmaier S, Wahle E. 2012. Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA 18: 958–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönisch C, Temme C, Moritz B, Wahle E. 2007. Degradation of hsp70 and other mRNAs in Drosophila via the 5′ 3′ pathway and its regulation by heat shock. J Biol Chem 282: 21818–21828. [DOI] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. 2012. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell 46: 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brengues M, Teixeira D, Parker R. 2005. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 310: 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JT, Bai X, Johnson AW. 2000. The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA 6: 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DM, D'Ambrogio A, Nottrott S, Richter JD. 2011. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 473: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Lim J, Kim VN, Ha M. 2014. TAIL-seq: genome-wide determination of poly(A) tail length and 3′ end modifications. Mol Cell 53: 1044–1052. [DOI] [PubMed] [Google Scholar]
- Couttet P, Fromont-Racine M, Steel D, Pictet R, Grange T. 1997. Messenger RNA deadenylylation precedes decapping in mammalian cells. Proc Natl Acad Sci 94: 5628–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Sackton KL, Horner VL, Kumar KE, Wolfner MF. 2008. Wispy, the Drosophila homolog of GLD-2, is required during oogenesis and egg activation. Genetics 178: 2017–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8: 9–22. [DOI] [PubMed] [Google Scholar]
- Franks TM, Lykke-Andersen J. 2008. The control of mRNA decapping and P-body formation. Mol Cell 32: 605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallie DR. 1991. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev 5: 2108–2116. [DOI] [PubMed] [Google Scholar]
- Geisler S, Lojek L, Khalil AM, Baker KE, Coller J. 2012. Decapping of long noncoding RNAs regulates inducible genes. Mol Cell 45: 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graille M, Séraphin B. 2012. Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nat Rev Mol Cell Biol 13: 727–735. [DOI] [PubMed] [Google Scholar]
- Hoefig KP, Rath N, Heinz GA, Wolf C, Dameris J, Schepers A, Kremmer E, Ansel KM, Heissmeyer V. 2013. Eri1 degrades the stem-loop of oligouridylated histone mRNAs to induce replication-dependent decay. Nat Struct Mol Biol 20: 73–81. [DOI] [PubMed] [Google Scholar]
- Houseley J, Tollervey D. 2009. The many pathways of RNA degradation. Cell 136: 763–776. [DOI] [PubMed] [Google Scholar]
- Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J. 2009. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 461: 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim F, Rohr J, Jeong W-J, Hesson J, Cerutti H. 2006. Untemplated oligoadenylation promotes degradation of RISC-cleaved transcripts. Science 314: 1893. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. 2004. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev 18: 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JE, Wickens M. 2007. A family of poly(U) polymerases. RNA 13: 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JE, Drier E, Barbee SA, Ramaswami M, Yin JCP, Wickens M. 2008. GLD2 poly(A) polymerase is required for long-term memory. Proc Natl Acad Sci 105: 14644–14649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. 2005. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121: 713–724. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-H, Kim B, Kim VN. 2014a. Emerging roles of RNA modification: m6A and U-tail. Cell 158: 980–987. [DOI] [PubMed] [Google Scholar]
- Lee M, Choi Y, Kim K, Jin H, Lim J, Nguyen TA, Yang J, Jeong M, Giraldez AJ, Yang H, et al. 2014b. Adenylation of maternally inherited microRNAs by Wispy. Mol Cell 56: 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Ha M, Chang H, Kwon SC, Simanshu DK, Patel DJ, Kim VN. 2014. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159: 1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentzen E, Basquin J, Conti E. 2008. Structural organization of the RNA-degrading exosome. Curr Opin Struct Biol 18: 709–713. [DOI] [PubMed] [Google Scholar]
- Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. 2011. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell 43: 624–637. [DOI] [PubMed] [Google Scholar]
- Maes A, Gracia C, Hajnsdorf E, Régnier P. 2012. Search for poly(A) polymerase targets in E. coli reveals its implication in surveillance of Glu tRNA processing and degradation of stable RNAs. Mol Microbiol 83: 436–451. [DOI] [PubMed] [Google Scholar]
- Malecki M, Viegas SC, Carneiro T, Golik P, Dressaire C, Ferreira MG, Arraiano CM. 2013. The exoribonuclease Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J 32: 1842–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Keller W. 2007. RNA-specific ribonucleotidyl transferases. RNA 13: 1834–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S, Temme C, Wahle E. 2004. Messenger RNA turnover in eukaryotes: pathways and enzymes. Crit Rev Biochem Mol Biol 39: 197–216. [DOI] [PubMed] [Google Scholar]
- Millonigg S, Minasaki R, Nousch M, Eckmann CR. 2014. GLD-4-mediated translational activation regulates the size of the proliferative germ cell pool in the adult C. elegans germ line. PLoS Genet 10: e1004647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minasaki R, Eckmann CR. 2012. Subcellular specialization of multifaceted 3′ end modifying nucleotidyltransferases. Curr Opin Cell Biol 24: 314–322. [DOI] [PubMed] [Google Scholar]
- Morozov IY, Jones MG, Razak AA, Rigden DJ, Caddick MX. 2010. CUCU modification of mRNA promotes decapping and transcript degradation in Aspergillus nidulans. Mol Cell Biol 30: 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov IY, Jones MG, Gould PD, Crome V, Wilson JB, Hall AJW, Rigden DJ, Caddick MX. 2012. mRNA 3′ tagging is induced by nonsense-mediated decay and promotes ribosome dissociation. Mol Cell Biol 32: 2585–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen TE, Marzluff WF. 2008. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev 22: 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray EL, Schoenberg DR. 2007. A+U-rich instability elements differentially activate 5′-3′ and 3′-5′ mRNA decay. Mol Cell Biol 27: 2791–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R, Takeuchi R, Takata K, Shimanouchi K, Abe Y, Kanai Y, Ruike T, Ihara A, Sakaguchi K. 2008. TRF4 is involved in polyadenylation of snRNAs in Drosophila melanogaster. Mol Cell Biol 28: 6620–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CC. 2013. Cytoplasmic RNA: a case of the tail wagging the dog. Nat Rev Mol Cell Biol 14: 643–653. [DOI] [PubMed] [Google Scholar]
- Nousch M, Yeroslaviz A, Habermann B, Eckmann CR. 2014. The cytoplasmic poly(A) polymerases GLD-2 and GLD-4 promote general gene expression via distinct mechanisms. Nucleic Acids Res 42: 11622–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogami K, Cho R, Hoshino S. 2013. Molecular cloning and characterization of a novel isoform of the non-canonical poly(A) polymerase PAPD7. Biochem Biophys Res Commun 432: 135–140. [DOI] [PubMed] [Google Scholar]
- Orban TI, Izaurralde E. 2005. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 11: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Song H. 2004. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol 11: 121–127. [DOI] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM. 2015. Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161: 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammelt C, Bilen B, Zavolan M, Keller W. 2011. PAPD5, a noncanonical poly(A) polymerase with an unusual RNA-binding motif. RNA 17: 1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ. 2009. Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol 16: 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ, Mikulasova A. 2007. Efficient RNA polyuridylation by noncanonical poly(A) polymerases. Mol Cell Biol 27: 3612–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouegemaille M, Gudipati RK, Olesen JR, Thomsen R, Seraphin B, Libri D, Jensen TH. 2007. Dissecting mechanisms of nuclear mRNA surveillance in THO/sub2 complex mutants. EMBO J 26: 2317–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs MS, Wang Z, Gaba A, Fang P, Belk J, Ganesan R, Amrani N, Jacobson A. 2002. Toeprint analysis of the positioning of translation apparatus components at initiation and termination codons of fungal mRNAs. Methods 26: 105–114. [DOI] [PubMed] [Google Scholar]
- Saguez C, Schmid M, Olesen JR, Ghazy MA, Qu X, Poulsen MB, Nasser T, Moore C, Jensen TH. 2008. Nuclear mRNA surveillance in THO/sub2 mutants is triggered by inefficient polyadenylation. Mol Cell 31: 91–103. [DOI] [PubMed] [Google Scholar]
- San Paolo S, Vanacova S, Schenk L, Scherrer T, Blank D, Keller W, Gerber AP. 2009. Distinct roles of non-canonical poly(A) polymerases in RNA metabolism. PLoS Genet 5: e1000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Kuchler B, Eckmann CR. 2009. Two conserved regulatory cytoplasmic poly(A) polymerases, GLD-4 and GLD-2, regulate meiotic progression in C. elegans. Genes Dev 23: 824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M-J, Norbury CJ. 2010. Polyadenylation and beyond: emerging roles for noncanonical poly(A) polymerases. Wiley Interdiscip Rev RNA 1: 142–151. [DOI] [PubMed] [Google Scholar]
- Schmidt M-J, West S, Norbury CJ. 2011. The human cytoplasmic RNA terminal U-transferase ZCCHC11 targets histone mRNAs for degradation. RNA 17: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DD, Norbury CJ. 2013. RNA decay via 3′ uridylation. Biochim Biophys Acta 1829: 654–665. [DOI] [PubMed] [Google Scholar]
- Sement FM, Ferrier E, Zuber H, Merret R, Alioua M, Deragon J-M, Bousquet-Antonelli C, Lange H, Gagliardi D. 2013. Uridylation prevents 3′ trimming of oligoadenylated mRNAs. Nucleic Acids Res 41: 7115–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbik N, Wang M, Lapik YR, Srivastava L, Pestov DG. 2010. Polyadenylation and degradation of incomplete RNA polymerase I transcripts in mammalian cells. EMBO Rep 11: 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Goodman HM. 2004. Uridine addition after microRNA-directed cleavage. Science 306: 997. [DOI] [PubMed] [Google Scholar]
- Sheth U, Parker R. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300: 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slevin MK, Meaux S, Welch JD, Bigler R, Miliani de Marval PL, Su W, Rhoads RE, Prins JF, Marzluff WF. 2014. Deep sequencing shows multiple oligouridylations are required for 3′ to 5′ degradation of histone mRNAs on polyribosomes. Mol Cell 53: 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomovic S, Fremder E, Staals RHG, Pruijn G, Schuster G. 2010. Addition of poly(A) and poly(A)-rich tails during RNA degradation in the cytoplasm of human cells. Proc Natl Acad Sci 107: 7407–7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M-G, Kiledjian M. 2007. 3′ Terminal oligo U-tract-mediated stimulation of decapping. RNA 13: 2356–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Mühlemann O. 2013. RNA decay mechanisms: specificity through diversity. Biochim Biophys Acta 1829: 487–490. [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Mayo T, Anderson P. 2006. ARE-mRNA degradation requires the 5′-3′ decay pathway. EMBO Rep 7: 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokowy T, Eszlinger M, Świerniak M, Fujarewicz K, Jarząb B, Paschke R, Krohn K. 2014. Analysis options for high-throughput sequencing in miRNA expression profiling. BMC Res Notes 7: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Slepenkov SV, Slevin MK, Lyons SM, Ziemniak M, Kowalska J, Darzynkiewicz E, Jemielity J, Marzluff WF, Rhoads RE. 2013. mRNAs containing the histone 3′ stem-loop are degraded primarily by decapping mediated by oligouridylation of the 3′ end. RNA 19: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarun SZ Jr, Sachs AB. 1996. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J 15: 7168–7177. [PMC free article] [PubMed] [Google Scholar]
- Temme C, Weissbach R, Lilie H, Wilson C, Meinhart A, Meyer S, Golbik R, Schierhorn A, Wahle E. 2009. The Drosophila melanogaster gene cg4930 encodes a high affinity inhibitor for endonuclease G. J Biol Chem 284: 8337–8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudek A, Porrua O, Kabzinski T, Lidschreiber M, Kubicek K, Fortova A, Lacroute F, Vanacova S, Cramer P, Stefl R, et al. 2014. Molecular basis for coordinating transcription termination with noncoding RNA degradation. Mol Cell 55: 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanácová S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. 2005. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol 3: e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Séraphin B. 2002. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J 21: 6915–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindry C, Lauwers A, Hutin D, Soin R, Wauquier C, Kruys V, Gueydan C. 2012. dTIS11 protein-dependent polysomal deadenylation is the key step in AU-rich element-mediated mRNA decay in Drosophila cells. J Biol Chem 287: 35527–35538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S, Gromak N, Norbury CJ, Proudfoot NJ. 2006. Adenylation and exosome-mediated degradation of cotranscriptionally cleaved pre-messenger RNA in human cells. Mol Cell 21: 437–443. [DOI] [PubMed] [Google Scholar]
- Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, Sharp PA. 2012. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26: 2392–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlotzka W, Kudla G, Granneman S, Tollervey D. 2011. The nuclear RNA polymerase II surveillance system targets polymerase III transcripts. EMBO J 30: 1790–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyers F, Rougemaille M, Badis G, Rousselle J-C, Dufour ME, Boulay J, Régnault B, Devaux F, Namane A, Séraphin B, et al. 2005. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.