

Abstract
Recombinant adeno-associated virus (rAAV) is an attractive tool for gene transfer and shows potential for use in human gene therapies. The current methods for the production and purification of rAAV from the transfected cell lysate are mainly based on cesium chloride and iodixanol density ultracentrifugation, although those are not scalable. Meanwhile, chromatography-based systems are more scalable. Therefore, in this study, we developed a novel method for the production and purification of rAAV serotype 1 (rAAV1) from serum-free culture supernatant based on ion-exchange and gel-filtration chromatography to obtain highly purified products with an ultracentrifugation-free technique towards Good Manufacturing Practice (GMP) production. The purified rAAV1 displayed three clear and sharp bands (VP1, VP2, and VP3) following sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and more than 90% of rAAV1 particles contained fully packaged viral genomes according to negative-stain electron micrographic analysis. Consequently, the resultant genomic titer of the purified rAAV1 was 3.63 × 1013 v.g./ml (the total titer was 4.17 × 1013 v.g.) from the 4 × 109 HEK293 cells. This novel chromatography-based method will facilitate scale-up of manufacturing for clinical applications in gene therapy.
Introduction
Recombinant adeno-associated virus (rAAV) is an attractive tool for gene transfer and has emerged as a versatile and leading delivery vector for gene therapy.1–3 Although rAAV can safely transfer therapeutic genes into dividing and nondividing cells in vivo, the conventional methods for rAAV production are associated with many challenges. In order to use vectors for in vivo gene transduction, the final product of rAAV should contain minimum impurities and empty particles, thus requiring simple purification processes for large-scale production. However, the conventional methods for the production and purification of rAAV from the cell lysate fractions of transfected human embryonic kidney-derived 293 (HEK293) cells entail cumbersome procedures. In addition, purification methods using cesium chloride (CsCl) or iodixanol density ultracentrifugation4,5 are not suitable for large-scale production. Therefore, development of alternative methods is required in order to use rAAV in clinical practice.
Chromatography-based systems are scalable while ultracentrifugation-based methods are not. In this regard, we used the serum-free culture supernatant of triple-transfected HEK293 cells, as opposed to the cell lysate fraction, as starting material for a chromatography procedure (ion-exchange or gel-filtration chromatography). There are several reports of chromatography-based methods for purification, which have mostly used ion-exchange6–10 and affinity chromatography.11–14 However, it is difficult to separate the particles of the viral genome from the empty particles using affinity chromatography techniques. Since the isoelectric points of vector particles (capsid proteins) are different in the presence or absence of vector genomes, it is more advisable to use ion-exchange chromatography. Consequently, rAAV serotype 1 (rAAV1) can be separated from the ion-exchange crude products on the basis of molecular size, using size-exclusion chromatography (gel-filtration chromatography). We previously reported that the viral genome particles could be effectively separated with the use of dual ion-exchange column configuration (cationic/anionic ion-exchange adsorptive membranes) chromatography, after partial purification of the cell lysate fractions of transfected HEK293 cells with step density gradient centrifugation.7 This method is useful depending on the physiological characteristics of a particular capsid of a serotype vector. Since there is generally a substantial amount of cell-derived contaminating protein from the cell lysate, for clinical applications, it is advisable to use the serum-free culture supernatant. Therefore, as the next stage of our research on the development of new techniques for the large-scale production and purification of rAAV1, we referred to ion-exchange technology.
rAAV1 can transduce the skeletal muscles more efficiently than other serotypes and is thus considered to be a promising therapeutic vector.15 Furthermore, Glybera (alipogene tiparvovec), the first rAAV-based drug to receive official approval from the European Medicines Agency, was produced from rAAV1.16 In this study, we evaluated the potential for large-scale purification of rAAV1 from the serum-free culture supernatant using ion-exchange and gel-filtration chromatography-based methods with an ultracentrifugation-free technique, with the goal of Good Manufacturing Practice (GMP)-level production.
Results
Ammonium sulfate precipitation
We used ammonium sulfate (AS) precipitation treatment as rAAV1 semi-purification procedure after the culture supernatant was ultrafiltrated with tangential flow filtration (TFF) and concentrated about 10 times, using a hollow fiber. Although half-saturated (1/2) AS precipitation is a conventional procedure, Figure 1 shows that two-step precipitation with both 1/3- and 1/2-saturated AS precipitation treatment was superior to the procedure with only the 1/2-saturated treatment in terms of purity on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis (Quick Coomassie Brilliant Blue staining, Wako, Osaka, Japan). After reducing protein debris by 1/3-saturated AS precipitation (adding half the sample volume of saturated AS made the final solution 33% (NH4)2SO4), rAAV1 was finally precipitated in the 1/2-saturated AS solution (added half the original sample volume of saturated AS) (1/3→1/2 AS). The first 1/3-saturated AS precipitation served to remove the contaminants. The second 1/2-saturated AS precipitation resulted in the precipitate and concentrated rAAV1. Both viral genomic titer of 1/3→1/2 AS and 1/2 AS was nearly equal (data not shown).
Figure 1.
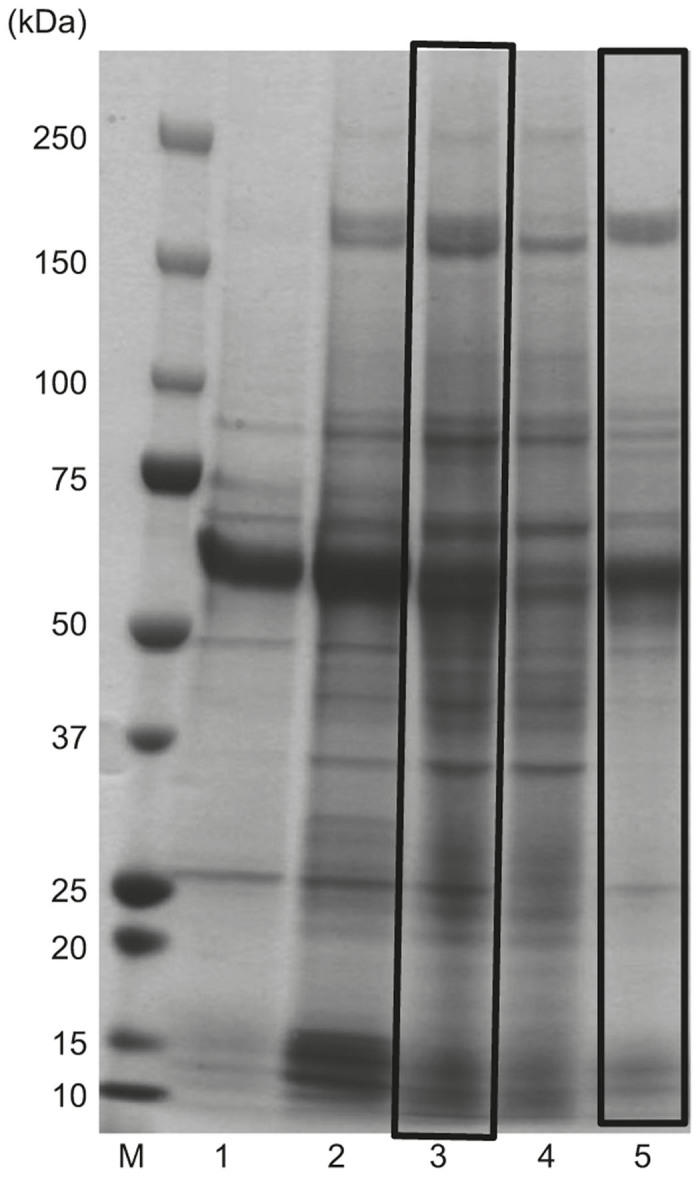
Improved purity of the culture supernatant with ammonium sulfate (AS) precipitation. Half-saturated AS (1/2 AS) precipitation was applied as the conventional procedure (Lane 3, the left black square). After reducing the amount of protein debris by 1/3-saturated AS precipitation, recombinant adeno-associated virus serotype 1 was finally precipitated in 1/2 AS solution (1/3→1/2 AS), which is our novel protocol (Lane 5, the right black square). The samples were analyzed by 5–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel (Quick Coomassie Brilliant Blue staining). Lane 1: pre-tangential flow filtration (pre-TFF); Lane 2: post-TFF; Lane 3: post-TFF preparation was directly precipitated in 1/2 AS; Lane 4: 1/3 AS precipitation; Lane 5: 1/3→1/2 AS; M: protein size marker.
Various methods attempted to remove 200 kDa protein impurities
Three plasmids (the cis AAV vector plasmid, trans-plasmid, and adenovirus helper plasmid) were transfected to HEK293 cells with polyethyleneimine (Polyethyleneimine Max, Polysciences, Warrington, PA) in serum-free medium. After harvesting culture supernatant, the supernatant was ultrafiltrated with TFF with the hollow fiber using the KrosFlo Research IIi system (Spectrum Laboratories, Rancho Dominguez, CA) and precipitated by 1/3→1/2 AS. After 1/3→1/2 AS, the sample was dissolved in 3.3 mmol/l morpholinoethanesulfonic acid, 3.3 mmol/l HEPES, and 3.3 mmol/l sodium acetate (MHN) buffer (pH 6.5) containing 50 mmol/l NaCl and 0.01% Pluronic F-68 (Gibco, Invitrogen, Grand Island, NY) and was loaded onto the dual ion-exchange column configuration (cationic: Mustang SXT; anionic: Mustang QXT, Pall Corporation, Ann Arbor, MI) chromatography. First, the tandem two Mustang SXTs were used to remove empty particles, and then, tandem two Mustang QXTs were used for absorbing fully packaged rAAV1 particles. The sample was then eluted from the Mustang QXTs by stepwise NaCl gradient elution (50–158 and 158–275 mmol/l NaCl in 3.3 mmol/l MHN buffer, pH 8.0, with 0.01% Pluronic F-68) (Supplementary Figure S1a,b). After ultrafiltration with Ultracel 30 K (nominal molecular weight cutoff 30 K, Merck Millipore, Tullagreen, Ireland), rAAV1 was finally purified by gel-filtration chromatography on Superdex 200 Increase 10/300 GL (GE Healthcare, Uppsala, Sweden) using the AKTÄ Explorer 100 HPLC system (the chromatogram results are shown in Supplementary Figure S1c,d). However, when the sample was loaded onto the ion-exchange columns in a buffer containing 50 mmol/l NaCl, the 200 kDa protein impurities could not be removed (Figure 2; Supplementary Figure S1e).
Figure 2.
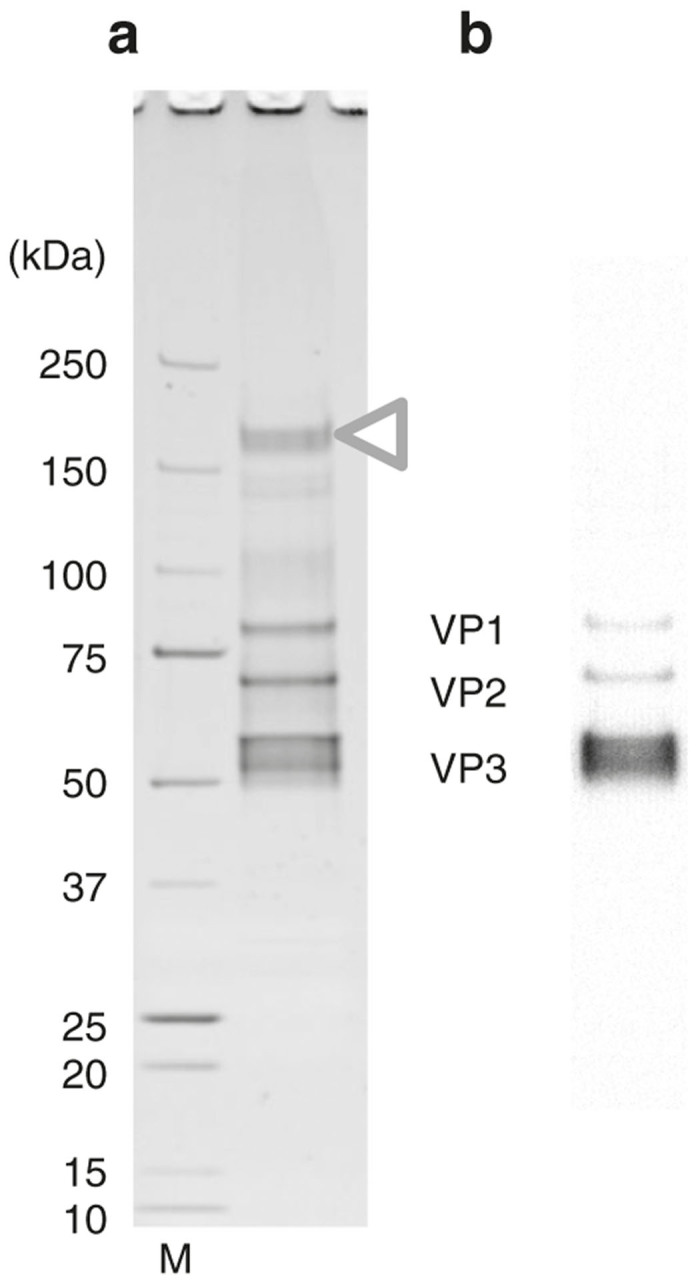
Purification with a buffer containing 50 mmol/l NaCl. (a) Recombinant adeno-associated virus serotype 1 (rAAV1) purified by ion-exchange column with a buffer containing 50 mmol/l NaCl was analyzed by 5–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel (Oriole staining) and (b) western blotting. M: protein size marker. The 200 kDa protein impurities were still present (white triangle) and the purity was low. The three rAAV1 capsid proteins, VP1 (81.4 kDa), VP2 (66.2 kDa), and VP3 (59.6 kDa), were represented.
Therefore, we tested various methods to find the most effective way to remove these impurities, which are shown in Figure 3; all methods were assayed immediately after the sample was ultrafiltrated with the TFF (post-TFF preparation). First, the sample was heated at 50 °C for 20 minutes. The low-molecular-weight molecules could be removed, but the 200 kDa protein impurities could not be removed (Figure 3a). Nevertheless, this heat treatment resulted in a higher purity with no reduction of yield (data not shown); therefore, this procedure was integrated into the overall purification protocol. Second, the sample was ultrafiltrated by Vivaspin 6 300,000 MWCO PES or 1000,000 MWCO PES (Figure 3b; Sartorius AG, Gӧttingen, Germany). Neither the impurities nor rAAV1 were ultrafiltrated, so that the impurities could not be removed with this procedure. Next, we tried to precipitate the rAAV1 with acetone (Figure 3c). After precipitating the proteins (rAAV1 and the impurities) with acetone, the precipitate was dissolved in phosphate-buffered saline (PBS). We expected that rAAV1 would be soluble in PBS while the impurities would be denatured. However, all of the proteins dissolved in PBS and therefore could not be separated with this method. Finally, we attempted Novec 7100 (methoxy-nonafluorobutane, C4F9OCH3, 3M, St. Paul, MN) treatment (Figure 3d). Similar to the heat treatment, the low-molecular-weight proteins were removed but the impurities could not be removed.
Figure 3.

Various methods were attempted to remove 200 kDa protein impurities to obtain a pure sample. All methods were assayed with post-tangential flow filtration (post-TFF) preparation using 5–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel (Quick Coomassie Brilliant Blue staining). None of these methods was effective for removing the 200 kDa protein impurities (white triangles). (a) Samples were heated at 50 °C for 20 minutes. Lane 1: pre-TFF; Lane 2: post-TFF; Lane 3: heat-treated post-TFF preparation; M: protein size marker. The star shows that the low-molecular-weight impurities were removed. (b) Samples were ultrafiltrated by Vivaspin 6 300,000 MWCO PES and 1000,000 MWCO PES (Sartorius AG, Gӧttingen, Germany). Lane 1: pre-TFF; Lane 2: post-TFF; Lane 3: fraction passed through Vivaspin 6 300,000 MWCO PES; Lane 4: concentrated with Vivaspin 6 300,000 MWCO PES; Lane 5: fraction passed through Vivaspin 6 or 1000,000 MWCO PES; Lane 6: concentrated with Vivaspin® 6 or 1000,000 MWCO PES; M: protein size marker. (c) Samples were precipitated by acetone. Lane 1: pre-TFF; Lane 2: post-TFF; Lane 3: precipitated by acetone and dissolved in phosphate-buffered saline; Lane 4: supernatant of acetone precipitation; M: protein size marker. (d) Samples were treated with Novec 7100. Lane 1: pre-TFF; Lane 2: post-TFF; Lane 3: the water layer of Novec treatment; Lane 4: the middle layer of Novec treatment; M: protein size marker. The star shows the same as (a).
Purification by ion-exchange and gel-filtration chromatography with a buffer containing 5 mmol/l NaCl
After the post-TFF preparation from 4 × 109 HEK293 cells was partially purified by 1/3→1/2 AS, we attempted to dialyze the sample against a low-salt buffer containing 5 mmol/l NaCl and 3 mmol/l MgCl2 and load it onto the tandem dual five cation-exchange columns and the five anion-exchange columns configuration with low ionic strength to remove the impurities. The resultants purified by the ion-exchange columns are shown in Figure 4a. We successfully removed the impurities using this method.
Figure 4.

Effects of chromatography-based purification of recombinant adeno-associated virus serotype 1 (rAAV1). (a) rAAV1 eluted from the five Mustang QXTs (anion-exchange column) by stepwise NaCl gradient elution was analyzed by 5–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel (Oriole staining). The black square indicates the fractions loaded on Superdex 200 10/300 GL at next gel-filtration chromatography step. The white triangle indicates the 200 kDa protein impurities. Lane 1: after dialysis; Lanes 2–4: eluted by 100 mmol/l NaCl; Lanes 5–7: eluted by 120 mmol/l NaCl; Lanes 8–10: eluted by 130 mmol/l NaCl; Lanes 11–13: eluted by 140 mmol/l NaCl; Lanes 14–16: eluted by 200 mmol/l NaCl; M: protein size marker. (b,c) The samples were subsequently gel-filtrated by the Superdex 200 10/300 GL column and elution fractions were analyzed by 5–20% gradient SDS–PAGE (Oriole staining). (b) Elution pattern of Superdex 200 10/300 GL. The y-axis: 280 nm absorbance; x-axis: fraction number. The black triangle indicates the peak fractions of rAAV1 (corresponding to the black square on (c)) and the gray arrowhead indicates ~65 kDa protein impurities. (c) Elution fractions analyzed by 5–20% gradient SDS–PAGE gel (Oriole staining). Lane 1: after dialysis; Lanes 2–11: Fr 13–22; Lane 12: Fr 27; M: protein size marker. The gray arrowhead shows the same as (b). (d–f) Peak fractions (the black square on (c)) were collected and concentrated by Ultracel 30 K to obtain the final product. (d) The final product of rAAV1 was analyzed by 5–20% gradient SDS–PAGE gel (Oriole staining), (e) western blotting, and (f) electron microscopy (negative staining). M: protein size marker. The three rAAV1 capsid proteins, VP1 (81.4 kDa), VP2 (66.2 kDa), and VP3 (59.6 kDa), were represented.
The sample was dialyzed against a low-salt concentration buffer containing 5 mmol/l NaCl and 3 mmol/l MgCl2, which was added to the buffer to stabilize the rAAV.17 The dialyzed sample was loaded onto the tandem dual five Mustang SXTs (to remove the empty particles) and the five Mustang QXTs (to absorb rAAV1). As shown in Figure 4a, the rAAV1 was mainly eluted by 120 mmol/l NaCl buffer from the Mustang QXTs, and the 200 kDa protein impurities were excluded by the 200 mmol/l NaCl buffer. The sample eluted by 120 mmol/l NaCl buffer and the first 40 ml of the sample eluted by 130 mmol/l NaCl buffer were collected and ultrafiltrated with Ultracel 30 K. We also eluted the rAAV1 with 160, 180, and 200 mmol/l NaCl-containing buffer. Although additional rAAV1 was eluted, it contains a lot of contaminated proteins and there was very few amount of rAAV1. The fractions in the black square on Figure 4a were ultrafiltrated with Ultracel 30 K and gel-filtrated by the Superdex 200 10/300 GL column (GE Healthcare, Uppsala, Sweden) (Figure 4b). The first peak represents elution of rAAV1 (black arrowhead) and the second peak represents elution of the ~65 kDa protein impurities (gray arrowhead). As shown in Figure 4c, three clear and sharp bands (VP1 (81.4 kDa), VP2 (66.2 kDa), and VP3 (59.6 kDa)) represented the product of rAAV1 obtained by gel-filtration chromatography with high purity on SDS–PAGE (Oriole staining). Peak fractions at the black square on Figure 4c were collected and concentrated by Ultracel 30 K to obtain the final product (Figure 4d). The high purity of rAAV1 was confirmed by SDS–PAGE (98% of intensity by Image J program analysis), the protein staining with Oriole (BIO-RAD, Hercules, CA), and western blotting using anti-AAV capsid-monoclonal antibody (B1, Progen, Heidelberg, Germany) (Figure 4e).
Characteristics of rAAV1
We analyzed the empty particle contamination and infectivity of rAAV1 with electron microscopy (negative staining) and fluorescence-activated cell sorting, respectively, to verify its quality. More than 90% of the rAAV1 particles contained viral vector genomes, as observed by electron micrographic analysis (Figure 4f). We titrated the rAAV1 at a point where 2.0 × 105 cells of C2C12 were transduced with rAAV1 at 0.5, 0.7, and 2.0 × 105 v.g./cell (Table 1). We compared the infectivity directly with rAAV1s before and after chromatography-based purification. We confirmed increasing infectivity of rAAV1 product depending on chromatographic purification step. Both population of EGFP-positive cells and intensity of EGFP expression were improved at all condition (Table 1). Furthermore, this observation supports the high percentage of fully packaged viral genomes by electron microscopic analysis. The resultant genomic titer of the purified rAAV1 was 3.63 × 1013 v.g./ml (the total titer was 4.17 × 1013 v.g.) from the 4 × 109 HEK293 cells, suggesting that the purified rAAV1 corresponded to about 1 × 104 v.g. per cell. These results suggest that functional rAAV1 with high purity could be obtained using our new protocol.
Table 1. Infectious titer measured by FACS.
| C2C12 | 2 × 105 cells | ||
|---|---|---|---|
| Sample | 5 × 104 | 7 × 104 | 2 × 105 |
| Final product | 11.61% | 15.13% | 39.82% |
| (6.71) | (7.85) | (18.28) | |
| Post-TFF preparation | 8.99% | 12.78% | 38.55% |
| (5.68) | (6.80) | (16.99) | |
The 2.0 × 105 of C2C12 cells were transduced with recombinant adeno-associated virus serotype 1 (rAAV1) at 0.5, 0.7, and 2.0 × 105 v.g./cell. Population of EGFP-positive cells as well as intensity was measured by FACS with propidium iodide staining solution to remove nonviable cells. The upper row is the population of the EGFP-positive cells (%) and the lower row is intensity. C2C12 cells were transduced with rAAV1s before and after chromatography-based purification. Upper row; population of EGFP-positive cells (%). Lower row: intensity of EGFP expression. FACS, fluorescence-activated cell sorting; TFF, tangential flow filtration.
A schematic representation of the processes for rAAV1 production and purification is finally shown in Figure 5.
Figure 5.
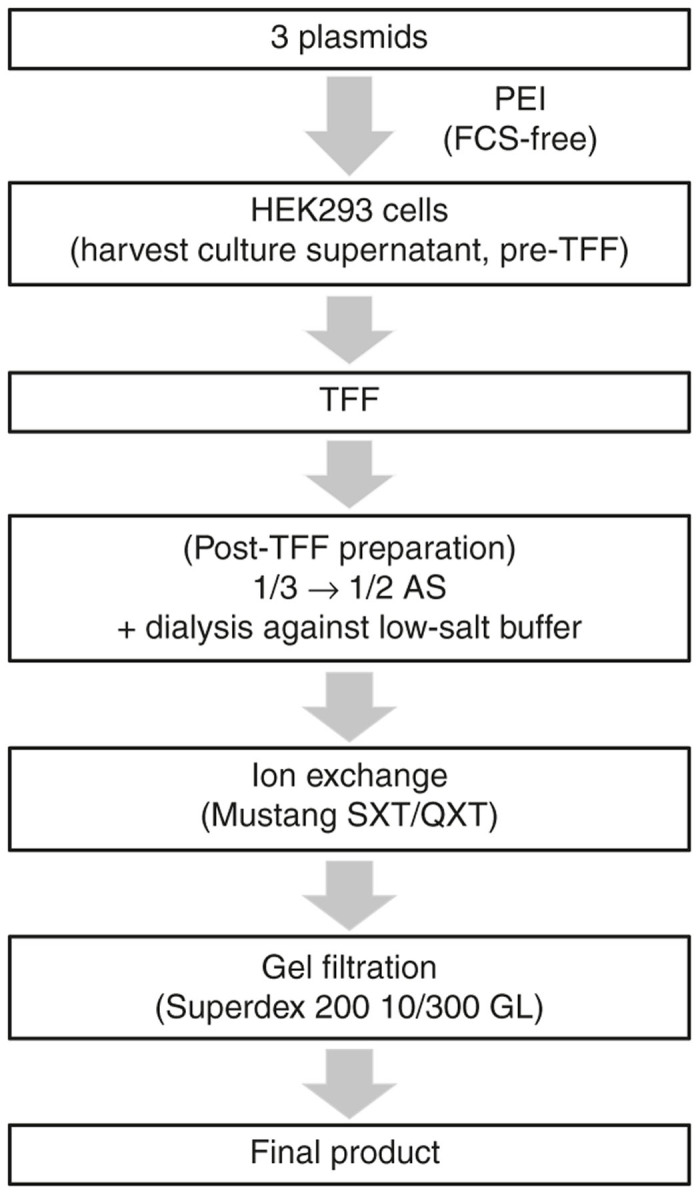
Schematic representation of the recombinant adeno-associated virus serotype 1 (rAAV1) production and purification procedure. Three plasmids (the cis AAV vector plasmid, trans-plasmid, and helper plasmid) were transfected to HEK293 cells with PEI (Polyethyleneimine Max) in serum-free medium. After harvesting the culture supernatant, it was ultrafiltrated with TFF using the hollow fiber using the KrosFlo Research IIi system. After reducing the amount of protein debris by 1/3-saturated AS precipitation, rAAV1 was finally precipitated in 1/2-saturated AS solution (1/3→1/2 AS). Then, it was dialyzed against a low-salt buffer (containing 5 mmol/l NaCl) and absorbed to ion-exchange membranes. Finally, rAAV1 eluted from the anion-exchange column was purified by gel-filtration chromatography. AS, ammonium sulfate; FCS, fetal calf serum; PEI, polyethyleneimine.
Comparison of the purity of rAAV1 obtained from ultracentrifugation and chromatography-based purification
The comparison of the purity of rAAV1 obtained from the ultracentrifugation and chromatography steps with SDS–PAGE is shown in Figure 6. The ultracentrifugation was performed using an iodixanol linear gradient operation after 1/2-saturated AS precipitation. The rAAV1 purified by ultracentrifugation was stained with Quick Coomassie Brilliant Blue, and that purified by this novel chromatography-based protocol was stained with Oriole Fluorescent Gel Stain. The sensitivity of the Oriole stain is almost equal to that of silver staining. The black square on Figure 6a shows the peak fractions of rAAV1 purified by iodixanol density ultracentrifugation. The results show that the purity of rAAV1 processed by chromatography steps was equal to or higher than that processed by ultracentrifugation (Figure 6).
Figure 6.
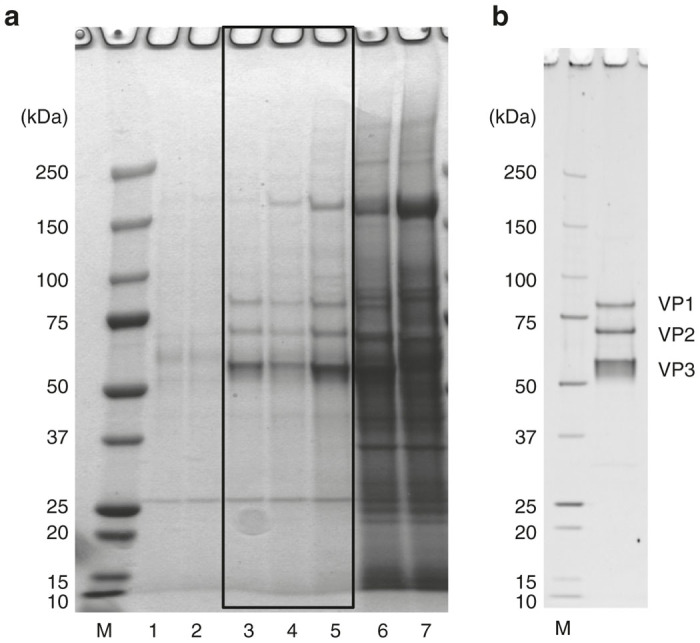
Comparison of the purity of recombinant adeno-associated virus serotype 1 (rAAV1) obtained from ultracentrifugation and chromatography-based purification. (a) The purity of rAAV1 obtained by ultracentrifugation was analyzed by 5–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel. Post-tangential flow filtration preparation was directly precipitated in 1/2-saturated ammonium sulfate (AS). Subsequently, the 1/2-saturated AS-precipitated sample was ultracentrifuged for 15 hours, 16 minutes at 125,400g with an iodixanol linear gradient. The proteins were stained by Quick Coomassie Brilliant Blue. rAAV1 was fractionated from the bottom. Lane number corresponds to fraction number. For example, lane 3 shows the fraction 3. The black square shows the peak fractions of rAAV1. Lanes 1–7: Fr 1–7; M: protein size marker. (b) The purity of rAAV1 using this novel protocol on SDS–PAGE gel (Oriole staining). The three rAAV1 capsid proteins, VP1 (81.4 kDa), VP2 (66.2 kDa), and VP3 (59.6 kDa), were represented.
Discussion
In this study, we tried to produce and purify rAAV1 from serum-free culture supernatant and chromatography (dual ion-exchange and gel-filtration chromatography) in order to avoid the disadvantages associated with purification using the ultracentrifugation technique. We took advantage of the fact that a good yield of rAAV1 can be obtained directly in the culture supernatant18 and the culture supernatant has vector-containing sample with higher purity than cell lysate from the point of view of proteins contamination, and we finally obtained highly purified and functional rAAV1 with an ultracentrifugation-free technique from the culture supernatant. Our large-scale method for the purification of rAAV1 from the culture supernatant using an ultracentrifugation-free technique for GMP-grade production is the first such protocol and thus represents a breakthrough in the field.
There are several reports related to the production and purification of rAAV1.7,10,19–21 Zolotukhin et al.10 reported the purification of rAAV1 from the cell lysate using iodixanol gradient ultracentrifugation and anion-exchange chromatography. To minimize impurities, the use of the culture supernatant has many advantages. Indeed, we previously reported7 a method for the ion-exchange (Mustang S + Mustang Q) chromatography purification of rAAV1 from the culture supernatant. We demonstrated the successful large-scale purification of rAAV1 using CsCl ultracentrifugation for the first concentration step. Vandenberghe et al.20 and Lock et al.19 reported rAAV1 purification from the serum-free culture medium. In addition to the purification of rAAV1 by CsCl or iodixanol ultracentrifugation, they did not purify it at the large scale. Furthermore, Smith et al.21 reported one-step affinity chromatography purification of rAAV1 from the baculovirus-AAV expression vector system. Although they purified high-titer rAAV1 without using the ultracentrifugation technique, we consider the baculovirus system to be problematic. Recently, a novel rhabdovirus was found in Spodoptera frugiperda (Sf9) cell lines.22 Furthermore, the glycoproteins produced in insect cells are different from those of mammalian cells, which may ultimately affect the biological function of rAAV in terms of glycosylation23 and lead to immunorejection in humans. In this context, human cells (HEK293 cells) are preferable for producing rAAV1 compared to the baculovirus system. There has been no previous report about the large-scale purification of rAAV1 from the serum-free culture supernatant to avoid the ultracentrifugation technique toward GMP production. Taken together, our present study represents an innovative method for rAAV1 purification and valuable.
Smith et al.24 reported the rAAV1 purification method using a low-salt buffer. In this study, rAAV1 was harvested from the cell lysate and purified using SOURCE15Q, a bead-based anion-exchange chromatography column. Although a good yield of rAAV1 can be obtained in the culture supernatant,18,20 there is nonetheless a substantial amount of impurities derived from the cells when purified from the cell lysate. Therefore, we purified rAAV1 from the culture medium. Compared to SOURCE15Q, Mustang columns reduce the bed volume and purification procedure for rAAV and involve simpler processes with respect to equilibration, sample loading, washing, and elution. Therefore, the use of membrane adsorbers such as Mustang columns also saves time and cost. The 200 kDa protein impurities were hard to remove by various methods but efficiently removed by using the low-salt concentration buffer. However, the specific characteristics of the 200 kDa protein impurities were not identified. Therefore, further investigations should be conducted to determine exactly what these impurities are and why the low-salt buffer can effectively remove them. A potential way to approach this question is with mass spectrometry.
Elution pattern from ion-exchange column is influenced by the temperature.25 Purified by a buffer containing 50 mmol/l NaCl using AKTA (4 °C), rAAV1 was eluted by 158 mmol/l NaCl. Furthermore, in small scale, rAAV1 was mainly eluted by 140 mmol/l NaCl (21 °C) (data not shown). In double scale (main results of this article), rAAV1 was mainly eluted by 120 mmol/l NaCl (14 °C). These indicate that temperature affects the ion-exchange column. Therefore, we eluted rAAV1 by stepwise NaCl gradient (100–200 mmol/l NaCl) where rAAV1 was eluted a certain concentration absolutely. Therefore, we would like to fix the temperature in future protocol.
This novel protocol produced a highly purified vector, which suggests further scalability with our improved culture system using chemical defined medium and membrane capsule (e.g., Mustang QXT 5, Mustang QXT 140, or Mustang QXT 5000) and its applicability to safe clinical investigations. It will need suspension cell system to produce rAAV in larger scale for clinical study. We believe that this purification method for rAAV1 secreted to serum-free medium is applicable to rAAV1 secreted to serum-free medium in suspension culture system. The median lethal dose (LD50) of CsCl in mice is 910 µg/g of body weight for i.v. injection (“Safety data for caesium chloride”; http://msds.chem.ox.ac.uk/index2.html). So the CsCl-free and the ultracentrifugation-free method has very little risk of toxicity and is thus a suitable approach for the clinical use of rAAV1. Therefore, this streamlined method with a high-performance scalable ion-exchange membrane and gel-filtration chromatography should facilitate future clinical studies.
In summary, ion-exchange chromatography was used to successfully separate rAAV according to its isoelectric point. This suggests that different serotypes and vector genomes may show differences in absorption to an ion-exchange column. Thus, the purification of other serotypes without using ultracentrifugation should be investigated in the future.
Materials and Methods
Cell culture
HEK293 cells (~4 × 109 cells in total) were plated in 22 square culture dishes (245 × 245 × 18 mm, 500 cm2; Corning, New York, NY) with a total surface area of 11,000 cm2. The cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO) with 8% fetal calf serum (Biowest, Nuaillé, France), penicillin, and streptomycin (Sigma-Aldrich, St. Louis, MO) at 37 °C in a 5% CO2 incubator for 2 days.
Production of rAAV1
After the cells were grown to more than 90% confluence, they were transfected with the cis AAV vector plasmid (with AAV-type 2 inverted terminal repeats), the trans-plasmid (with the AAV-type 2 Rep gene and the type 1 Cap gene; p5E18RXC1; a gift from James M. Wilson, University of Pennsylvania), and an adenovirus helper plasmid (with an essential region from the adenovirus genome; pHelper, Stratagene, La Jolla, CA) at a ratio of 1:1:226,27 in serum-free medium and GlutaMAX-І (1×; Gibco Life Technologies, Grand Island, NY) using polyethyleneimine (Polyethyleneimine Max, Polysciences) at a DNA:polyethyleneimine ratio of 1:219 for 5 days. The cis AAV vector plasmid expressing the EGFP gene under the control of the CBA promoter, pdsAAV-CB-EGFP, was used in this study (provided by Dr. Arun Srivastava, University of Florida). Subsequently, the cells were cotransfected with the three plasmids (90.1 µg of the helper plasmid, 44.8 µg of the trans-plasmid, and 44.8 µg of the cis plasmid per square dish) in serum-free medium and GlutaMAX-І with polyethyleneimine; the medium was not replaced for 5 days. As most rAAV1 particles were obtained directly in the culture supernatant,18,20 they were harvested from the culture supernatant at 120 hours after transfection. After filtration through a 0.45-µm filter, the culture supernatant was ultrafiltrated with TFF and concentrated about 10 times using a hollow fiber (UFP-750-E-3MA, 750,000 nominal molecular weight cutoff, GE Healthcare, Westborough, MA) with the KrosFlo Research IIi TFF system (Spectrum Laboratories, Rancho Dominguez, CA). Subsequently, 1 µl/5 ml of benzonase (250 U/µl, Merck Millipore, Billerica, MA) was added to the sample and incubated for 30 minutes at 37 °C. After stopping the benzonase reaction with 0.5 mol/l ethylenediaminetetraacetic acid, the crude rAAV1 fractions were heated for 20 minutes at 50 °C, and the denatured low-molecular-weight proteins were removed with three cycles of centrifugation at 13,000g for 10 minutes. After reducing protein debris by 1/3-saturated AS precipitation (adding half the sample volume of saturated AS made the final solution 33% (NH4)2SO4), rAAV1 was finally precipitated in the 1/2-saturated AS solution (added half the original sample volume of saturated AS) (1/3→1/2 AS). Subsequently, the precipitated rAAV1 was dissolved in 10 ml of PBS (+) (in 3 mmol/l MgCl2, Sigma-Aldrich).
Various methods attempted to remove 200 kDa protein impurities
All methods were assayed post-TFF preparation. First, the sample was heated at 50 °C for 20 minutes. Second, ultrafiltrated by Vivaspin 6 300,000 MWCO PES and 1000,000 MWCO PES (Sartorius AG) at 500g for 10 minutes. Third, precipitated by acetone. After adding four times volumes of acetone, the sample was incubated at −80 °C for 1 hour. After centrifugation at 13,000g for 15 minutes, the supernatant was removed and the pellet was dissolved in PBS. Fourth, treated with Novec 7100 (3M, St. Paul, MN). Novec treatment is similar to chloroform extraction. After the same quantity of Novec was added to the sample, the sample was voltexed and centrifuged at 18,800g for 15 minutes. Subsequently, the water layer and the middle layer were collected and dissolved in PBS.
Ion-exchange and gel-filtration chromatography
Initially, the resultant rAAV1 sample was dialyzed against 20 mmol/l Tris–HCl (pH 8.0) buffer containing 50 mmol/l NaCl and 3 mmol/l MgCl217 for 3–4 hours. In the next step, the rAAV1 dialysate was dialyzed against 20 mmol/l Tris–HCl (pH 8.0) buffer containing 5 mmol/l NaCl and 3 mmol/l MgCl2 (dialysis buffer) for 20 hours with one fresh buffer replacement. The sample was then diluted with dH2O until the conductivity decreased to around 1.0 mSV/cm. The conductivity was measured with an electroconductivity sensor (LAQUA Twin, HORIBA, Tokyo, Japan). The diluted sample was loaded at a rate of 1 ml/minute to tandem five cation-exchange membranes and five anion-exchange membranes with a column volume of 0.86 ml (Acrodisc unit with Mustang SXT and the Mustang QXT membrane, Pall Corporation, Ann Arbor, MI) equilibrated with the equilibration buffer which was diluted dialysis buffer with dH2O (conductivity = 1.2 mSV/cm) by a syringe pump (TERUMO, Tokyo, Japan). The membrane was washed with over 10 column volumes of wash buffer which was diluted dialysis buffer with dH2O (conductivity = 1.0 mSV/cm). After removing the Mustang SXT columns, rAAV1 was eluted from the Mustang QXTs using 120 ml of 100 mmol/l NaCl, 120 mmol/l NaCl, 130 mmol/l NaCl, 140 mmol/l NaCl, and 200 mmol/l NaCl in 20 mmol/l Tris–HCl (pH 8.0) buffer with 3 mmol/l MgCl2. All of the 120 mmol/l NaCl-containing buffer and the first 40 ml sample of the elution with 130 mmol/l NaCl in 20 mmol/l Tris–HCl (pH 8.0) buffer with 3 mmol/l MgCl2 were used for gel-filtration chromatography, at next purification step.
After ultrafiltration with Ultracel 30 K (Merck Millipore, Tullagreen, Ireland) and buffer exchange (PBS(+)), rAAV1 was finally purified by size-exclusion chromatography (gel-filtration chromatography) on the Superdex 200 10/300 GL system using the AKTÄ Explorer 100 HPLC (GE Healthcare, Little Chalfont, UK) system equipped with a 2 ml sample loop with 3.3 mmol/l morpholinoethanesulfonic acid, 3.3 mmol/l HEPES, and 3.3 mmol/l sodium acetate buffer (MHN, pH 6.5) containing 300 mmol/l NaCl and 0.01% nonionic surfactant Pluronic F-68 (Gibco, Invitrogen, Grand Island, NY); 0.5 ml fractions were collected.
Peak fractions based on 280 nm absorption were collected and ultrafiltrated with Ultracel 30 K. The genomic titer was determined by quantitative PCR. The biological activity (infectious titer) was determined by transduction to C2C12 cells and evaluating the cells for EGFP positivity. The purity of the final rAAV1 product was assessed by SDS–PAGE and western blotting. The contents of empty particle contamination of the final rAAV1 were determined by electron microscopy.
Quantitative PCR
Two microliters of benzonase (250 U/µl) was added to the purified rAAV1 for 30 minutes. After stopping the reaction with the addition of 0.5 mol/l ethylenediaminetetraacetic acid and 1× Tris–ethylenediaminetetraacetic acid, the lysis buffer (AL Buffer, QIAGEN, Hilden, Germany) was added to extract the viral genome and was heated at 56 °C for 10 minutes. After dilution by 100-fold with dH2O, the benzonase-resistant genome titer was measured by SYBR green dye real-time Q-PCR (7500 Fast Real-Time PCR System, Applied Biosystems, Foster City, CA) using an EGFP-targeted primer (forward primer: 5′-AGCAGCACGACTTCTTCAAGTCC-3′, reverse primer: 5′-TGTAGTTGTACTCCAGCTTGTGCC-3′). We used a linearized pdsCBA-EGFP DNA (digestion by SacІ restriction enzyme) as a standard plasmid. After 95 °C holding stage for 10 seconds, two-step PCR cycling stage was performed at 95 °C for 5 seconds, followed by 60 °C for 34 seconds for 40 cycles.
SDS–PAGE and western blotting analysis
Purified rAAV1 was separated on 5–20% gradient polyacrylamide gels (Wako, Osaka, Japan) with SDS running buffer. Oriole Fluorescent Gel Stain (BIO-RAD, Hercules, CA) or Quick Coomassie Brilliant Blue (Wako) was applied to visualize and analyze the SDS–PAGE bands.
For western blotting analysis, the samples were transferred to a polyvinylidene fluoride membrane overnight. After transfer, the membrane was blocked with 3% bovine serum albumin/Tris-buffered saline with Tween 20 and incubated with the primary antibody (monoclonal antibody B1; Progen, Heidelberg, Germany) for 1 hour. After rinsing with Tris-buffered saline with Tween 20, the membrane was probed with horseradish peroxidase-labeled anti-mouse secondary antibody (GE Healthcare, Little Chalfont, UK). The protein was detected using the ECL Plus Western Blotting Detection System (GE Healthcare, Buckinghamshire, UK).
The biological activity (infectious titer)
The 2.0 × 105 of C2C12 cells were transduced with rAAV1 at 0.5, 0.7, and 2.0 × 105 v.g./cell. Population of EGFP-positive cells as well as intensity was measured by fluorescence-activated cell sorting (FACSCalibur, Becton, Dickinson and Company, Franklin Lakes, NJ) with propidium iodide staining solution (Immunostep, Salamanca, Spain) to remove nonviable cells.
Electron microscopy
A carbon-stabilized copper grid (Nisshin EM Corporation, Tokyo, Japan) was placed onto a sample and incubated for 1 minute. The sample on the grid was then negatively stained for 1 minute with 1% uranyl acetate. Finally, the grid was viewed with the H-7650 transmission electron microscope (Hitachi, Tokyo, Japan) at an accelerating voltage of 80 kV. The ratio of empty particles to encapsidated particles was determined by counting the number of packaged and unpackaged particles in the electron micrographs. We investigated 11 fields on electron microscopy.
Acknowledgments
We thank James Wilson at the University of Pennsylvania for providing the AAV packaging plasmid (p5E18RXC1). The authors declare no conflicts of interest. We wish to thank Tomoko Chiyo for advice on experimental design.
References
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu, S, Fujimoto, K, Kato, S, Mizukami, H, Asari, S, Ikeguchi, K et al. (2010). A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Ther 18: 1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth, WW, Aleman, TS, Kaushal, S, Cideciyan, AV, Schwartz, SB, Wang, L et al. (2008). Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther 19: 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermens, WT, ter Brake, O, Dijkhuizen, PA, Sonnemans, MA, Grimm, D, Kleinschmidt, JA et al. (1999). Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum Gene Ther 10: 1885–1891. [DOI] [PubMed] [Google Scholar]
- Merten, OW, Gény-Fiamma, C and Douar, AM (2005). Current issues in adeno-associated viral vector production. Gene Ther 12 (suppl. 1): S51–S61. [DOI] [PubMed] [Google Scholar]
- Urabe, M, Xin, KQ, Obara, Y, Nakakura, T, Mizukami, H, Kume, A et al. (2006). Removal of empty capsids from type 1 adeno-associated virus vector stocks by anion-exchange chromatography potentiates transgene expression. Mol Ther 13: 823–828. [DOI] [PubMed] [Google Scholar]
- Okada, T, Nonaka-Sarukawa, M, Uchibori, R, Kinoshita, K, Hayashita-Kinoh, H, Nitahara-Kasahara, Y et al. (2009). Scalable purification of adeno-associated virus serotype 1 (AAV1) and AAV8 vectors, using dual ion-exchange adsorptive membranes. Hum Gene Ther 20: 1013–1021. [DOI] [PubMed] [Google Scholar]
- Davidoff, AM, Ng, CY, Sleep, S, Gray, J, Azam, S, Zhao, Y et al. (2004). Purification of recombinant adeno-associated virus type 8 vectors by ion exchange chromatography generates clinical grade vector stock. J Virol Methods 121: 209–215. [DOI] [PubMed] [Google Scholar]
- Brument, N, Morenweiser, R, Blouin, V, Toublanc, E, Raimbaud, I, Chérel, Y et al. (2002). A versatile and scalable two-step ion-exchange chromatography process for the purification of recombinant adeno-associated virus serotypes-2 and -5. Mol Ther 6: 678–686. [DOI] [PubMed] [Google Scholar]
- Zolotukhin, S, Potter, M, Zolotukhin, I, Sakai, Y, Loiler, S, Fraites, TJ Jr. et al. (2002). Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods 28: 158–167. [DOI] [PubMed] [Google Scholar]
- Auricchio, A, Hildinger, M, O’Connor, E, Gao, GP and Wilson, JM (2001). Isolation of highly infectious and pure adeno-associated virus type 2 vectors with a single-step gravity-flow column. Hum Gene Ther 12: 71–76. [DOI] [PubMed] [Google Scholar]
- Koerber, JT, Jang, JH, Yu, JH, Kane, RS and Schaffer, DV (2007). Engineering adeno-associated virus for one-step purification via immobilized metal affinity chromatography. Hum Gene Ther 18: 367–378. [DOI] [PubMed] [Google Scholar]
- Zolotukhin, S, Byrne, BJ, Mason, E, Zolotukhin, I, Potter, M, Chesnut, K et al. (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6: 973–985. [DOI] [PubMed] [Google Scholar]
- Tamayose, K, Hirai, Y and Shimada, T (1996). A new strategy for large-scale preparation of high-titer recombinant adeno-associated virus vectors by using packaging cell lines and sulfonated cellulose column chromatography. Hum Gene Ther 7: 507–513. [DOI] [PubMed] [Google Scholar]
- Chao, H, Liu, Y, Rabinowitz, J, Li, C, Samulski, RJ and Walsh, CE (2000). Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol Ther 2: 619–623. [DOI] [PubMed] [Google Scholar]
- Burnett, JR and Hooper, AJ (2009). Alipogene tiparvovec, an adeno-associated virus encoding the Ser(447)X variant of the human lipoprotein lipase gene for the treatment of patients with lipoprotein lipase deficiency. Curr Opin Mol Ther 11: 681–691. [PubMed] [Google Scholar]
- Turnbull, AE, Skulimowski, A, Smythe, JA and Alexander, IE (2000). Adeno-associated virus vectors show variable dependence on divalent cations for thermostability: implications for purification and handling. Hum Gene Ther 11: 629–635. [DOI] [PubMed] [Google Scholar]
- Xiao, X (2010). Adeno-associated viral vectors found free in media. Hum Gene Ther 21: 1221–1222. [DOI] [PubMed] [Google Scholar]
- Lock, M, Alvira, M, Vandenberghe, LH, Samanta, A, Toelen, J, Debyser, Z et al. (2010). Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther 21: 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe, LH, Xiao, R, Lock, M, Lin, J, Korn, M and Wilson, JM (2010). Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum Gene Ther 21: 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, RH, Levy, JR and Kotin, RM (2009). A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther 17: 1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H, Galvin, TA, Glasner, DR, Shaheduzzaman, S and Khan, AS (2014). Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J Virol 88: 6576–6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oers, MM, Pijlman, GP and Vlak, JM (2015). Thirty years of baculovirus-insect cell protein expression: from dark horse to mainstream technology. J Gen Virol 96 (Pt 1): 6–23. [DOI] [PubMed] [Google Scholar]
- Smith, RH, Yang, L and Kotin, RM (2008). Chromatography-based purification of adeno-associated virus. Methods Mol Biol 434: 37–54. [DOI] [PubMed] [Google Scholar]
- Acikara, OB (2013). Column Chromatography. In: Martin, D (ed.). Chromatography and its applications. InTech, Rijeka, Croatia <http://www.intechopen.com/books/column-chromatography/ion-exchange-chromatography-and-its-applications>. pp.31–58. [Google Scholar]
- Grimm, D, Kern, A, Rittner, K and Kleinschmidt, JA (1998). Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther 9: 2745–2760. [DOI] [PubMed] [Google Scholar]
- Salvetti, A, Orève, S, Chadeuf, G, Favre, D, Cherel, Y, Champion-Arnaud, P et al. (1998). Factors influencing recombinant adeno-associated virus production. Hum Gene Ther 9: 695–706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.