

Abstract
Myotonic Dystrophy type 1 (DM1) is a dominantly inherited disease comprehending multiple features. Fatigue and exhaustion during exercise often represent significant factors able to negatively influence their compliance to rehabilitation programs. Mitochondrial abnormalities and a significant increase in oxidative markers, previously reported, suggest the hypothesis of a mitochondrial functional impairment. The study aims at evaluating oxidative metabolism efficiency in 18 DM1 patients and in 15 healthy subjects, through analysis of lactate levels at rest and after an incremental exercise test. The exercise protocol consisted of a submaximal incremental exercise performed on an electronically calibrated treadmill, maintained in predominantly aerobic condition. Lactate levels were assessed at rest and at 5, 10 and 30 minutes after the end of the exercise. The results showed early exercise-related fatigue in DM1 patients, as they performed a mean number of 9 steps, while controls completed the whole exercise. Moreover, while resting values of lactate were comparable between the patients and the control group (p=0.69), after the exercise protocol, dystrophic subjects reached higher values of lactate, at any recovery time (p<0,05). These observations suggest an early activation of anaerobic metabolism, thus evidencing an alteration in oxidative metabolism of such dystrophic patients. As far as intense aerobic training could be performed in DM1 patients, in order to improve maximal muscle oxidative capacity and blood lactate removal ability, then, this safe and validate method could be used to evaluate muscle oxidative metabolism and provide an efficient help on rehabilitation programs to be prescribed in such patients.
Key Words: Myotonic Dystrophy type 1, Oxidative Metabolism, Aerobic Training, Incremental exercise test, Lactate acid
Myotonic Dystrophy type 1 (DM1) is a dominantly inherited multisystemic disorder, that is now recognized as one of the most common forms of muscular dystrophy in adults. In addition to muscle atrophy and myotonia (involuntary persistence of muscle contraction), DM1 causes a consistent constellation of clinical features, including: cardiac conduction defects, posterior subcapsular iridescent cataracts, and specific set of endocrine changes, such as hyperinsulinemia, hyperglycemia and insulin insensitivity, together with testicular atrophy, premature frontal balding in men and cognitive impairment.1,2 The genetic cause of DM1 was identified in 1992 as a (CTG)n repeat in the 3’ untraslated region of a protein kinase gene, called myotonin protein kinase (DMPK), located on chromosome 19q13.3.3,4 The abnormal repeat triplet is situated in a non-coding region of the gene, so it was suggested a loss of function of DMPK protein, caused by either a transcriptional repression or a gain of function mediated by the mutant RNA transcripts. Actually, the best theory explaining the pathogenesis focuses on RNA transcripts containing pathogenic CUG repeats, which could produce defects in alternative splicing of multiple RNAs, thus providing the basis for the multisystemic features of DM1.5,6,7
One of the most common symptoms in DM1 patients is represented by fatigue and early exhaustion during exercise. These factors are characterized by a loss of voluntary force-producing capacity of the muscles during exercise, due both to central mechanisms of motor input and to peripheral involvement of the lower motor unit or sensory nerves. In patients with myopathies, muscle fatigability is enhanced because of the presence of structural changes or for altered muscle metabolism, which may affect muscle contraction.8 Some authors hypothesized an oxidative impairment in DM1 patients. Toscano et al. (2005) revealed a significant increase of several markers of oxidative stress, probably acting as a cofactor in disease progression.9 Angelini et al. (2012) assessed the neuronal nitric oxide synthase activity, a molecule implied in protecting the cellular susceptibility to oxidative stress, to state the muscle involvement, probably at the basis of precocious fatigability.10 Ono et al. found mitochondrial abnormalities in muscle biopsy of DM1 patients, such as ragged red fibers, supporting the hypothesis that an impaired respiratory chain function could be relevant for the disease.11 Moreover, the abnormal CTG repetition, responsible for an altered DMPK function, leads to a compromised myofiber metabolic homeostasis, thus increasing the susceptibility of cells to oxidative stress, through the activation of different pathways of signal transduction, as postulated by Usuki et al.12,13,14
Histological studies of skeletal muscle sections from patients with DM1 show typical findings: variability of myofiber size, ring fibers, an increase in central nuclei, together with a prevalent atrophy in type 1 (slow twitch) fibers.15 Recent studies evidenced the higher presence of reactive oxygen species (ROS) that suggests a failure in the removal system of toxic proteins that interfere with mitochondria function, inducing the release of pro-apoptotic factors.16 Mitochondria have the major role in energetic homeostasis, thus determining ATP availability in the cells. Indeed, their dysfunction will be unable to meet cellular ATP demands, thus compromising the cellular ability to adapt to physiological stress. In fact the ATP production following the oxidation of metabolic fuels depends on oxidative capacity and energetic efficiency of the mitochondrial compartment.17
It is well known that skeletal muscle acidosis could impair oxidative metabolism and a reduced respiratory capacity could contribute to the onset of fatigue during exercise, even if quantitative assessment of this parameter is hard to be explored.18,19,20 Siciliano et al. reported an early activation of anaerobic metabolism, and a lower lactate threshold during a maximal exercise in DM1 patients.21 In this study we assess the oxidative metabolism efficiency in Myotonic Dystrophy patients, by means of analysis of haematic lactate levels at rest and after an incremental submaximal exercise test on treadmill.
Materials and Methods
We enrolled eighteen patients (mean age 43,67 ± 11,28 years, 7 females and 11 males) affected by Myotonic Dystrophy type 1, followed by the Unit of Neurorehabilitation at the University Hospital of Pisa. Patients were entered into the study if they met the following criteria:
Established clinical and genetic diagnosis of DM1.22
Ability to perform physical exercise (lower limbs less impairment).
Exclusion criteria were: clinically manifest pulmonary or cardiac disorders, fever, neoplasms, diabetes and insulin insensitivity. Informed consent was given by each patient. Ethical Committee of the University Hospital of Pisa approved the study. For clinical details see Table 1. The control group was formed by fifteen healthy subjects: 10 females and 5 males, mean age 43.2 years.
Table 1.
Clinical features of patients, trend of lactate values, number of steps performed during aerobic exercise.
| Lactate values mmol/L | ||||||
|---|---|---|---|---|---|---|
| Pz | Sex/Age | Rest | 5 min | 10 min | 30 min | Steps |
| 1 | F 31 | 1.33 | 1.51 | 1.42 | 1.29 | 5 |
| 2 | M 33 | 1.6 | 1.83 | 1.93 | 1.44 | 7 |
| 3 | M 59 | 1.7 | 2.78 | 2.66 | 1.86 | 2 |
| 4 | M 39 | 1.29 | 4.51 | 3.73 | 2.01 | 11 |
| 5 | M 34 | 1.25 | 6.07 | 5.15 | 3.59 | 5 |
| 6 | F 49 | 2.6 | 4.44 | 3.52 | 3.05 | 11 |
| 7 | F 48 | 0.49 | 0.8 | 0.77 | 0.45 | 4 |
| 8 | F 57 | 1.22 | 1.53 | 1.78 | 1.48 | 11 |
| 9 | M 42 | 1.57 | 1.17 | 1.15 | 1.24 | 11 |
| 10 | F 35 | 0.74 | 1.89 | 1.4 | 0.9 | 11 |
| 11 | M 20 | 1.41 | 1.73 | 0.71 | 11 | |
| 12 | M 52 | 1.91 | 3.15 | 1.65 | 11 | |
| 13 | F 55 | 1.22 | 2 | 1.71 | 0.95 | 11 |
| 14 | M 39 | 0.73 | 1.95 | 0.95 | 9 | |
| 15 | M 42 | 1.64 | 3.75 | 3.05 | 1.74 | 7 |
| 16 | F 39 | 2.64 | 4.11 | 4 | 2.45 | 11 |
| 17 | M 64 | 2.49 | 2.54 | 2.53 | 2.22 | 11 |
| 18 | M 48 | 2.21 | 2.39 | 2.16 | 1.93 | 11 |
To study oxidative metabolism we assessed blood lactate levels before and after a submaximal incremental exercise on a treadmill.23 The exercise protocol consisted of 11 steps, of 2-min duration each step, at a constant speed of 3 km/h. The slope was 0% at the start level and increased of 2.5% at each step. Capillary hemoglobin O2 saturation and heart rate were monitored during the exercise: if the patient reached 75% of the maximum theoretical heart rate (220-patient’s age), the test was stopped, so keeping the exercise predominantly aerobic. Venous blood samples were executed at rest and at 5, 10 and 30 min after the end of exercise.24,25.
Blood samples were collected in EDTA tubes from the antecubital vein. Lactate was rapidly analyzed through an enzimatic assay, based on the conversion of lactate to piruvate and H2O2 by lactate oxidase. In the presence of the H2O2 formed, peroxidase catalyzes the oxidative condensation of chromogen precursor to produce a coloured dye with adsorption maximum at 520 nm. The increase in absorbance at 520 nm is directly proportional to lactate concentration in the sample, as described by Chisari et al.26 Analysis of the differences in mean values was performed using the Z test, performed on average values.
Results
All subjects of the control group completed the exercise (11 steps) while the patients performed 9 steps on average (Tab. 1). The lactate resting values were comparable between DM1 patients and the control group: 1.56 mmol/L vs 1.63 mmol/L respectively. Lactate values were significantly higher in DM1 patients compared to controls at any given time during the recovery period (Fig. 1).
Fig 1.
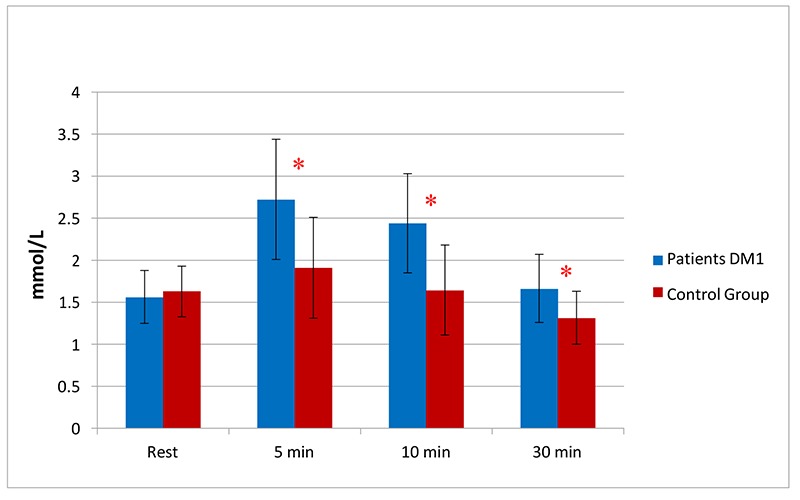
Mean lactate values in Myotonic Dystrophy group and control, * p < 0.05 at any time during recovery period from control subjects
Discussion
The first datum emerging in this study is the early exercise-related fatigue arising in DM1 patients compared to controls, as they performed a mean number of 9 steps, while the latter completed the exercise (11 steps) (Tab. 1). In the second hand, patients revealed normal lactate values at rest, which reached significantly higher levels during recovery after exercise (Fig. 1). These findings suggest an early activation of anaerobic metabolism, not proportional to workload executed, thus evidencing an alteration in oxidative metabolism of such dystrophic patients. During an increasing exercise, the working muscles and various tissues require more ATP production due to glycolytic or glycogenolytic pathway, and this mechanism leads to the final increase in lactate levels. During steady state sub-maximal exercise, the lactate concentration in the lactate pool remains constant and the rate of oxygen consumption is the measure of the whole body energy expenditure; when exercise intensities arise above steady state, a major concentration is attributed to an increase in the rate of lactate production or result from a decrease in the rate of lactate removal.19,27 The lactate anion together with decreased muscle pH seem to be involved in muscle function disturbances, such as impairing excitation-contraction coupling, modifying protein conformation, altering channel properties and depressing the activity of key enzymes in glycolysis.19 Consequently, it may be crucial for exercising muscle cells to delay pH decline and lactate accumulation in the cytosol, to prevent muscle performance impairment.20
The present study confirms what emerged in a previous one proposed by Barnes et al.28 in which it was investigated the glycogenolytic pathway by using P31 magnetic resonance spectroscopy: indeed they revealed an abnormal glycogenolytic ATP-production, during the first minute of aerobic exercise. In addition, the proportion of the total requirement provided by glycogenolysis resulted greater than in controls, revealing values similar to that in the control subject performing heavier works. During aerobic exercise in both calf muscle and flexor digitorum superficialis, phosphocreatine was depleted more rapidly in patients than in controls, evidencing how consistent is the metabolic demand in wasted muscle cells. Thomas et al. evidenced that the removal of lactate and protons from skeletal muscle is important to maintain force and prevent fatigue during continuous and intermittent supramaximal exercises in well-trained subjects.18,27 Our data confirm the findings observed in an another study conducted by Siciliano et al. They reported an early activation of anaerobic metabolism in DM1 patients, after performing an incremental bicycle exercise, as far as lactate threshold was achieved at values ranging from 30 to 50% of the predicted normal maximal power output (pnPOmax), while controls reached it at 60-70%. Hence, they concluded that the early achievement of lactate threshold represent a critical point, reflecting metabolic modifications in the oxidative metabolism.21
Briefly, an evidence of oxidative impairment in DM1 patients emerged in the work of Barnes et al.28, while an indirect assumption is proposed by Toscano et al.9 Anyway, Angelini pointed out that depletion in neuronal nitric oxide synthase is frequently found and may correlate with post-exercise fatigability in Duchenne Muscular Dystrophy and in Sarcoglicanopathy, while in DM1 two different types of fatigue may be found.10 In fact, probably the cortical atrophy and the presence of white matter lesions could explain the mechanism of central fatigue, while the muscular fibers atrophy could count for peripheral fatigue. Moreover, another aspect probably implied in the presence of abnormal lactate levels in these patients is the predominant atrophy of type 1 fibers evidenced by histological studies of skeletal muscle sections. The histochemical analysis leads to the original division of muscle fibers into type I (slow-twitch) and type II (fast-twitch), evidencing how the greater myoglobin and capillary content in red muscles contributes to their greater oxidative capacity compared with white muscles.29 It could then be argued that the prevalent atrophy of slow-twitch muscle fibers in DM1 could represent one of the possible mechanism implied in the altered oxidative capacity evidenced in such patients, thus providing basis for the abnormal lactate levels assessed performing our aerobic protocol.
So, as far as lactate determination during cycle or treadmill ergometry is used for diagnostic purpose in mitochondrial myopathies,30 this submaximal exercise could be a tolerable and safety method to assess aerobic capacity in this kind of patients. The interest of our study consists of the fact that our protocol is a safe, non exhaustive and easy reproducible method to be performed in such patients, who may frequently present early structural and functional myocardial abnormalities, strongly associated with cardiac conduction alterations.31,32
Our findings confirm the alteration in oxidative capacity of DM1 skeletal muscle, which could negatively influence motor performances of these patients. The relevance to know one of the mechanisms implied in fatigue in DM1 consists of the possibility to state aerobic capacity of these patients, in order to plane specific rehabilitative protocols.
In fact, in patients affected by mitochondrial myopathies, Taivassalo et al. investigated the role of a specific aerobic training, consisting of an 8-week aerobic program performed on a treadmill for 20 up to 30 minutes, under constant supervision and heart rate monitoring, associated with stretching exercise, warm-up and cool-down periods. They demonstrated an increase of aerobic capacity with a decrease of 30 % in lactate concentration at rest and after exercise, that was correlated with a reduced fatigue and improved tolerance to daily activities.33,34 In patients affected by Polymyositis/Dermatomyositis, Bertolucci et al. evidenced a precocious fatigability and higher lactate values after performing a submaximal aerobic exercise: they revealed an improvement both in muscle performance and in a reduction of fatigue symptom after a period of specific aerobic training.25
This safe and easily reproducible protocol could be used to address aerobic training programs, which were provided to be a safety method to increase fitness, endurance and level of physical activity,35-37 in order to improve maximal muscle oxidative capacity and blood lactate removal ability, probably implied in early fatigability that arise in these patients.
In conclusion, this kind of exercise protocol could provide a valid and safe method to evaluate aerobic capacity in Myotonic Dystrophy patients, able to reveal precocious alteration in oxidative metabolism of such patients and to direct rehabilitation trainings aiming at improving aerobic performances.
Contributor Information
Caterina Tramonti, Email: caterina.tramonti@gmail.com.
Stefania Dalise, Email: stefania_dalise@yahoo.it.
Bruno Rossi, Email: b.rossi@ao-pisa.toscana.it.dk.
References
- 1.Larkin K, Fardaei M. Myotonic Dystrophy-A multigene disorder. Brain Research Bulletin 2001;56:389-95. [DOI] [PubMed] [Google Scholar]
- 2.Schara U, Schoser B G. Myotonic dystrophies type 1 and 2: a summary on current aspects. Semin Pediatr Neurol 2006;13:71-9. [DOI] [PubMed] [Google Scholar]
- 3.Ranum LPW, Day JW. Myotonic Dystrophy: Rna Pathogenesis Come into Focus. Am J Hum Genet 2004;74:793-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wansink DG, Van Herpen REMA, Coerwinkel-Driessen MM, et al. Alternative Splicing Controls Myotonic Dystrophy Protein Kinase Structure, Enzymatic Activity, and Subcellular Localization. Mol Cell Biol 2003;23:5489-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho DH, Tapscott SJ. Myotonic Dystrophy. Emerging mechanisms for DM1 and DM2. Biochem Biophys Acta 2007;1772:195-204. [DOI] [PubMed] [Google Scholar]
- 6.Day JW, Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscular Disorder 2005;15:5-16. [DOI] [PubMed] [Google Scholar]
- 7.Wang GS, Kearney DL, De Biasi M, et al. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J of Clin Invest 2007;117:2802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Vries JM, Hagemans MLC, Bussmann JBJ, et al. Fatigue in neuromuscular disorders: focus on Guillain–Barre’ syndrome and Pompe disease. Cellular and Molecular Life Sciences 2010;67:701–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toscano A, Messina S, Campo GM, et al. Oxidative stress in myotonic dystrophy type 1. Free Radical Research 2005;39:771–6. [DOI] [PubMed] [Google Scholar]
- 10.Angelini C, Tasca E. Fatigue in muscular dystrophies. Neuromuscular Disorder 2012;22:214-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ono S, Kurisaki H, Inouye K, Mannen T. Ragged-red fibers in myotonic dystrophy J Neurol Sci 1986;74:247-55. [DOI] [PubMed] [Google Scholar]
- 12.Usuki F, Ishiura S. Expanded CTG repeats in myotonin protein kinase increase susceptibility to oxidative stress. Neuroreport 1998;9:2291-6. [DOI] [PubMed] [Google Scholar]
- 13.Usuki F, Tagahashi N, Sasagawa N, Ishiura S. Differential signal pathways following oxidative stress in mutant Myotonin Protein Kinase cDNA-transfected C2C12 cell lines. Biochem Byophis Res Commun 2000;267:739-43. [DOI] [PubMed] [Google Scholar]
- 14.Pantic B, Trevisan E, Citta A, et al. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death and Disease 2013;4:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Timchenko L. Molecular mechanisms of muscle atrophy in myotonic Dystrophies. Int J Biochem Cell Biol 2013;45:2280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loro E, Rinaldi F, Malena A, et al. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death and Differentiation 2010;17:1315–24. [DOI] [PubMed] [Google Scholar]
- 17.Crescenzo R, Bianco F, Mazzoli A, et al. Alterations in proton leak, oxidative status and uncoupling protein 3 content in skeletal muscle subsarcolemmal and intermyofibrillar mitochondria in old rats. BMC Geriatrics. 2014,14:79-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas C, Sirvent P, Perrey S, et al. Relationships between maximal muscle oxidative capacity and blood lactate removal after supramaximal exercise and fatigue indexes in humans. J Appl Physiol 2004;97:2132-8. [DOI] [PubMed] [Google Scholar]
- 19.Messonnier L, Kristensen M, Juel C, Denis C. Importance of pH regulation and lactate/H+ transport capacity for work production during supramaximal exercise in humans J Appl Physiol 2007;102:1936-44. [DOI] [PubMed] [Google Scholar]
- 20.Finsterer J, Milvay E. Diagnostic yield of the lactate stress test in respiratory chain disorders under absolute and relative workload Journal of Neuroscience Methods 2001;108:65-70. [DOI] [PubMed] [Google Scholar]
- 21.Siciliano G, Tovani S, Paquali L, et al. Alteration of lactate production during incremental exercise in Myotonic Dystrophy is not dependent by catecholamine increase. Basic Appl Myol 2002;12:231-7. [Google Scholar]
- 22.Kamsteeg EJ, Kress W, Catalli C, et al. Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. European Journal of Human Genetics 2012;20:1203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodwin ML, Harris JE, Hernández A, Gladden LB. Blood Lactate Measurements and Analysis during Exercise: A Guide for Clinicians. Journal of Diabetes Science and Technology 2007;1:558-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cupisti A, Licitra R, Chisari C, et al. Skeletal muscle and nutritional assessment in chronic renal failure patients on a protein-restricted diet. J Intern Med 2004;255:115–24. [DOI] [PubMed] [Google Scholar]
- 25.Dalise S, Bertolucci F, Simonella C, Rossi B, Chisari Cet al. Intensive aerobic training improves motor performances and oxidative metabolism efficiency in chronic polymyositis: A case report. Neuromuscular Disorders 2012;22:221–5. [DOI] [PubMed] [Google Scholar]
- 26.Bertolucci F, Neri R, Dalise S, et al. Abnormal Lactate levels in patients with Polymyositis and Dermatomyositis: the benefits of a specific Rehabilitative program. European Journal of Phisical and Rehabilitation Medicine, in press. [PubMed] [Google Scholar]
- 27.Moxnes JF, Sandbakk O. The kinetics of lactate production and removal during whole-body exercise Theoretical Biology and Medical Modelling 2012;9:2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnes PRJ, Kemp GJ, Taylor DJ, Radda GK. Skeletal muscle metabolism in myotonic dystrophy. A 31P magnetic resonance spectroscopy study. Brain 1997;120:1699–711. [DOI] [PubMed] [Google Scholar]
- 29.Binder-Macleod S A, Scott W, Stevens J. Human Skeletal Muscle Fiber Type Classifications. Physical Therapy 2001;81:1810-6. [PubMed] [Google Scholar]
- 30.Finsterer J, Milvay E. Stress lactate in mitochondrial myopathy under constant, unadjusted workload. European Journal of Neurology 2004;11:811–6. [DOI] [PubMed] [Google Scholar]
- 31.Hermans MCE, Faber CG, Bekkers M, et al. Structural and functional cardiac changes in myotonic dystrophy type 1: a cardiovascular magnetic resonance study. Journal of Cardiovascular Magnetic Resonance 2012;14: 1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelargonio G, Dello Russo A, Sanna T, et al. Myotonic Dystrophy and the heart. Heart 2002;88:665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taivassalo T, De Stefano N, Argov Z, et al. Effects of aerobic training in patients with mitochondrial myopathies. Neurology 1998;50:1055-60. [DOI] [PubMed] [Google Scholar]
- 34.Taivassalo T, Gardner JL, Taylor RW, et al. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions: Brain 2006;129: 3391-401. [DOI] [PubMed] [Google Scholar]
- 35.Ørngreen MC, Olsen DB, Vissing J. Aerobic Training in Patients with Myotonic Dystrophy Type 1 Ann Neurol 2005;57:754–7. [DOI] [PubMed] [Google Scholar]
- 36.Ansved T. Muscle training in muscular dystrophies. Acta Physiol Scand 2001;171:359-66. [DOI] [PubMed] [Google Scholar]
- 37.Kierkegaard M, Harms-Ringdahl K, Edstrom L, et al. Feasibility and effects of a physical exercise programme in adults with Myotonic Dystrophy type 1: a randomized controlled pilot study. J Rehabilitation Medicine 2011;43:695-702. [DOI] [PubMed] [Google Scholar]