

Abstract
Cryo-electron tomography (cryo-ET) has emerged as perhaps the only practical technique for revealing nanometer-level three-dimensional structural details of subcellular macromolecular complexes in their native context, inside the cell. As currently practiced, the specimen should be 0.1-0.2 microns in thickness to achieve optimal resolution. Thus, application of cryo-ET to intact frozen (vitreous) tissues, such as skeletal muscle, requires that they be sectioned. Cryo-ultramicrotomy is notoriously difficult and artifact-prone when applied to frozen cells and tissue, but a new technique, focused ion beam milling (cryo-FIB), shows great promise for “thinning” frozen biological specimens. Here we describe our initial results in applying cryo-FIB and cryo-ET to triad junctions of skeletal muscle.
Key Words: Cryo-electron microscopy, Cryo-electron tomography, Excitation-Contraction Coupling, Focused-Ion-Beam milling, Skeletal muscle
Current understanding of the molecular structure of the triad junction, the site of excitation-contraction in skeletal muscle, is severely limited. Most knowledge of overall triad junction architecture has come from classical transmission electron microscopy (TEM) of sectioned or freeze-fractured muscle1. To date it has not been possible to purify triad junctions, or natively preserved fragments thereof, for more detailed study, although microsomal fractions that retain some limited similarities to native triad junctions have been isolated and characterized by TEM cryo-tomography (cryo-ET).2 The two major membrane-associated components of the triad, the ryanodine receptor/ calcium release channel and the dihydropyridine receptor/voltage sensor, have been isolated and studied by single-particle cryo-EM techniques (reviewed by Hamilton et al.3 and by Wong et al.4) but little, if any, success in reconstituting these and other triad components has been reported to date.
Recent applications of cryo-ET to intact bacterial and eukaryotic cells have shown that large macromolecular complexes (e.g. ribosomes, nuclear pores), including membrane-associated ones, can be resolved and, if multiple copies are present, the individual subvolumes containing them can be computationally selected and averaged to improve the resolution (in favorable cases) to 2 nm (recently reviewed).5,6 Significant improvements in resolution are likely forthcoming (see “Outlook,” below). Cryo-ET allows subcellular structures to be imaged in situ in their native state because no fixatives and no heavy-metal stains are required prior to freezing the specimen, which is done at a sufficiently high rate (or pressure) that crystalline ice does not form. The resulting “vitreous” ice is known to allow macromolecular preservation to atomic levels. The contrast of the tomograms computed from the micrographs in the tilt series is very low, such that only membrane-bounded organelles and large macromolecular complexes are readily visualized, but cryo-imaging technology is constantly improving, and additional image processing, such as subtomogram averaging, where this is possible, can significantly improve the attainable structural information.
A major obstacle to applying cryo-ET to large eukaryotic cells and tissues, such as muscle, has been that for these specimens it is necessary to use high-pressure freezing to achieve the vitreous form of ice. It is then necessary to “thin” the specimen to less than 500 nm (100-200 nm is preferable for achieving highest resolution) and then transfer the sample to a specimen holder for insertion into the TEM, all the while maintaining low temperature and avoiding frost contamination. Until recently, the only way to achieve these steps was to transfer the block of frozen tissue or cell suspension from the high-pressure freezer to a cryo-ultramicrotome, cut vitreous cryo-sections (typically 100-200 nm thickness), transfer the sections to EM grids, and then transfer the grids to a TEM cryo-holder. Technology for cryo-ultramicrotomy has been available for several decades, but despite improvements7-12 vitreous cryo-sections suffer from artifacts such as compression in the direction of cutting (up to 50%), crevasses and knife marks, all of which adversely affect the images.10 Even more vexing for cryo-ET applications, the cryo-sections are almost invariably non-planar (wrinkled) and difficult to attach to EM grids with sufficient stability to allow collection of a suitable tilt series of micrographs (typically from - 60 to + 60 in 1- or 2-degree intervals). Consequently, few cryo-ET studies have been published using vitreous cryo-sections.
Recently, we proposed and have developed a new method for preparing thin sections of vitreously frozen cells - and also tissue - which employs focused-ion-beam (FIB) milling.13-16 Here we give an overview of the potential for cryo-ET applied to FIB-milled muscle, to resolve structural details of the triad junction. Although much progress remains to be made, conceivably the macromolecular organization of the triad at the nanometer level of detail will be achievable in the future.
Overview of the FIB milling technique
Thinning of biological specimens by FIB milling involves irradiation under vacuum with a focused beam of (usually gallium) ions, which scans the specimen in a raster fashion, removing mass from the surface by a sputtering mechanism. FIB instruments are often combined with a scanning electron microscope (SEM) so that the location and progress of the milling can be monitored. Currently, FIB-SEM tomography is a popular technique,17 often applied to a block of resin-embedded, stained biological specimen. The block face is imaged by SEM after FIB-milling to remove thin layers of the specimen, and then tomograms are formed from the stack of images obtained after each successive layer of the specimen is removed. FIB-SEM tomography is useful for visualizing rather large volumes of specimens at modest resolution but is not generally useful for visualizing macromolecular assemblies and is therefore not treated further here. Rather, we focus on the application of FIB milling to create thin sections of vitreously frozen specimens for subsequent transfer to a TEM for cryo-ET.
The utility of FIB milling for preparing thin sections of vitreously frozen cells for cryo-ET was first demonstrated by Marko and colleagues.14-16
Remarkably, when suitable precautions are taken, FIB-prepared cryo-sections do not suffer from the artefacts mentioned above that plague cryo-ultramicrotomy, and FIB-specific damage is minimal: damage and implantation from Ga+ ions, or devitrification due to warming, are restricted a depth of about 10 nm.15, 18 These demonstrated advantages of FIB-milling over cryo-ultramicrotomy for creating thin sections of vitreously frozen biological specimens for cryo-ET have stimulated several research groups to develop and implement the methodology. Our laboratory’s experience, which is not unique, indicates that the field is still in its early stages and, at present, the techniques that have been described remain cumbersome, far from routine, and require custom-modified instruments and tools.
To date, most success has been achieved with strategies that involved depositing or culturing cells on TEM grids, and freezing them by plunging the grids into a cryogen (“plunge freezing”). Then the grids are transferred into the FIB-SEM instrument for thinning a few limited regions (typically hundreds of µm2 in area). Finally, the grids are transferred into the cryo-TEM. All these steps must be done while maintaining the temperature below the devitrification temperature (< -140°C) and minimizing frost or warming during cryo-transfer steps. Several groups have implemented this strategy15,19-23 and have applied it to bacteria and unicellular eukaryotes. The cryo-ET analysis of Dictyostelium discoideum cells described by Rigort et al.21 is particularly noteworthy. The FIB-milled region was formed by ablating regions from the upper and lower portions of the cell to create a 300-nm-thick lamella in the middle of the cell that was supported on the sides by non-milled regions; this differs from the more straightforward procedure followed for smaller cells in which the cell is milled from just one side, the side facing away from the grid surface. A similar two-sided milling strategy is also necessary for tissue, as we discuss below. Another notable achievement of this study is that ten nuclear pore complexes (which span two membranes, as do triad junctions) were identified visually in one tomogram, and the authors successfully applied sub-tomogram averaging to obtain a reconstruction of the nuclear pore having a resolution of 7.9 nm.
FIB milling of tissue for CTEM (Albany implementation) 13
Specimen preparation techniques that involve deposition directly onto EM grids are not easily adapted to bulk tissue specimens, which are too large for plunge-freezing, and therefore must be high-pressure frozen (HPF) and then “pre-trimmed” in a cryo-ultramicrotome before cryo-FIB milling is used to create the final thinned lamellae. The methodology that we have developed allows specimens to be frozen in commercially available HPF specimen carriers.13. Customized accessories have been fabricated for holding, manipulating, and storing the specimen through all of the steps of the procedure. An outline of the main steps of the procedure (all carried out below - 140°C) is here presented and illustrated in Figure 1. We have implemented the procedures using a FIB– SEM instrument from Carl Zeiss (Oberkochen Germany) equipped with a cryo-stage and cryo-transfer accessories that are available from Leica Microsystems (Vienna, Austria). For a more detailed description of method and hardware see Hsieh et al. (2014).13
Fig 1.

Preparation of tissue for cryo-FIB milling. A. High-pressure freezing. Bal-Tec HPM 010 high-pressure freezer with close-up of a 3-mm diameter slot-type specimen carrier (sc) (i) and the freezing head (ii). B. The front part of an Intermediate specimen holder (ish), shown holding a specimen carrier (sc); arrow shows slot for specimen. C. Trimming a specimen carrier in the cryo-ultramicrotome. The carrier is trimmed to expose the specimen at the front (i, arrowhead) and the top and bottom (ii); the final trimmed specimen is shaped into a 1-2 mm long X 20-µm-thick edge (arrowheads), supported on the edges by remaining aluminum. D. Custom specimen block, shown with shutter open (left) and closed (right). The block can hold two intermediate specimen holders or grids (inset, arrows). E. Leica VCT-100 shuttle ready to accept the specimen block (at right). F. SEM of FIB-milled specimen. Edge-on view (i, similar to view in Cii). (ii); view (perpendicular to i), showing the four thinned regions (100-330 nm thick X 10 µm X 20 µm) appropriate for imaging in TEM. Asterisks indicate the four thinned lamellae as indicated by arrowheads in (i). (iii), SEM-FIB instrument (Zeiss Neon 40 EsB). G. Tip of modified Gatan 626 cryo-transfer holder, shown with door (arrowhead) and shutter (asterisk) open to reveal the specimen in the intermediate specimen holder (arrow). Parts of this figure were adapted from reference.13
Usually step 1 (high-pressure freezing), steps 2-3 (cryo-ultramicrotomy), steps 4-5 (FIB milling), and step 7 (cryo-ET) are carried out on separate days, with the FIB milling being the rate-limiting step.
(1) High Pressure Freezing (Fig. 1A). Deposit fresh tissue in the slot (e.g. 0.3 mm wide X 2 mm long) of a standard HPF specimen carrier (Fig. 1Ai) and freeze. Typically, several are prepared in a session, and these can be stored under liquid nitrogen indefinitely.
(2) Load the specimen carrier into the intermediate specimen holder (ISH). The custom-made intermediate specimen holder (Fig. 1B) allows the HPF specimen carrier to be conveniently manipulated throughout the following steps. This step is typically performed in the working chamber of the cryo-ultramicrotome immediately before carrying out the next step.
(3) Pre-trim the intermediate specimen holder and frozen tissue in a cryo-ultramicrotome (Fig. 1C). Most of the aluminum specimen holder is trimmed away, and the specimen is reduced to 20-30 µm in thickness (Fig. 1Cii). The aluminum backing is removed from the bottom surface of the specimen-bearing slot such that the thinned specimen area is surrounded by metal only on its sides. Milling will be initiated (step 6) at the exposed edge of specimen.
(4) Transfer the ISH containing the trimmed specimen to the cryo-SEM specimen block. The block (Fig. 1D) holds and orients the specimen in the FIB instrument. This block is a custom component of the Leica VCT-100 cryo-transfer system, which includes a cryostage for the FIB-SEM instrument and a cryo-transfer shuttle system. A shutter on the specimen block protects the specimen from contamination during cryo-transfer.
(5) Transfer the specimen block to the FIB instrument. The specimen block is inserted into the cryo-transfer shuttle (Fig. 1E). The shuttle docks onto the FIB-SEM instrument (Fig. 1Fiii) and allows the specimen block to be moved to the FIB-milling cryo-stage. The shuttle also interfaces with a vacuum evaporator that allows it to be evacuated before use to reduce exposure of the shuttle to water vapor. Loading (and unloading) of the specimen block into the shuttle is done under liquid nitrogen in a loading box.
(6) FIB milling. The thin (20 µm) platform of frozen specimen (300 µm W x 500 µm L) that was created in the cryo-ultramicrotome (Fig. 1C) is milled in selected rectangular areas (Fig. 1F; 10 µm W X 20 µm L) to the desired thickness, typically 100-300 nm. Each rectangular milled area is separated from its neighbor(s) by regions of thicker frozen specimen to provide mechanical stability, and the edges are beveled to allow tilting of the specimen to at least 60 degrees in the TEM. The milling process is done in several steps using a higher beam current (e.g. 5 nA) for the initial steps and a lower beam current (e.g. 100 pA) in the final step. Typically we create four groups of the final rectangular areas that are suitable for cryo-TEM imaging. The duration of the actual milling is about one hour for two groups of four. Thus, up to 16 regions suitable for TEM can be created from a single specimen carrier in one several-hour session. Upon completion of milling, the specimen block is removed from the FIB-SEM instrument via the shuttle, transferred from the shuttle to the loading box, and then may be stored under liquid nitrogen until needed for cryo-TEM.
(7) Transfer the ISH containing the milled specimen to the TEM cryo-holder for cryo-TEM. The specimen block containing the FIB-milled specimen(s) is transferred to the workstation that is used to load the TEM cryo-holder, and the specimen-loaded ISH is transferred to the cryo-holder (Fig. 1G). We have modified the tip of a Gatan Model 626 cryo-holder (Gatan, Inc., Pleasanton, CA) to accept the ISH instead of the usual EM grid. Cryo-EM and collection of the tilt series for tomography is conducted according to standard procedures.
Cryo-TEM and cryo-ET of FIB-milled skeletal muscle.
The skeletal muscle triad junction would appear to be well-suited for application of our method of applying FIB milling to specimens for cryo-ET. Firstly, from a practical viewpoint, muscle is constructed of repeating units (sarcomeres), thus essentially any electron micrograph of a longitudinal cryosection will contain several triad junctional regions. This is important because one of the current limitations of cryo-ET as applied to vitreous cryosections of eukaryotic cells is that a substructure of interest is usually only detectable after the tomographic data is collected. Consequently, small-copy-number structures are likely not to be present in the reconstructed volume, which typically represents a tiny fraction of the total cell volume. Secondly, from the viewpoint of structural biology, cryo-ET has the potential to reveal details of the triad junction at an unprecedented level. Although some of the triad’s major structural features, such as the spatial distribution of RyRs in the terminal cisternae region of the sarcoplasmic reticulum and that of the DHPRs in the apposed transverse tubule, have been gleaned from classical electron microscopy, the molecular details of the interaction between the RyR and the DHPRs remain unknown, as are the locations and intermolecular interactions among the numerous other proteins that contribute to the triad junction.
Figure 2 shows some of our initial micrographs from FIB-milled fast-twitch toadfish swim bladder, which has previously been extensively characterized by TEM of conventional thin-section and freeze-fracture specimens by Dr. C. Franzini-Armstrong, who assisted us with the preparation of the swimbladder muscle.24 For convenience, for our initial work to demonstrate feasibility of FIB-milling for cryo-ET, the muscle was fixed with 2.5% glutaraldehyde, but the specimens remained fully hydrated and no heavy metal stains were used. In longitudinal sections, as expected, triad junctions are prevalent, and the major structural features agree remarkably well with those described previously, albeit at reduced contrast because we are imaging native muscle. In certain orientations, the ryanodine receptors, whose cytoplasmic regions (also known as foot structures) bridge the gap between the junctional SR membrane and the T-tubule, are very well resolved as darkly contrasted dark bands that sometimes are bifurcated. This appearance arises because the RyRs are arranged as two side-by-side rows along the T-tubule. In some cases, the RyRs appear to be connected by bridges of density to the T-tubule. However, further study will be required to discern whether these connections are related to the dihydropyridine receptors, which are thought to interact and communicate directly with the RyRs to carry out the fundamental signal-transducing event of excitation-contraction. Another recurring feature is that the lumens of the T-tubules usually appear to contain a substantial amount of mass of unknown composition, but no membrane associated densities that might correspond to the dihydropyridine receptors stand out from the noise. Direct interpretation of the micrographs is difficult, not only due to the low contrast, but also because the thickness of the section (100-200 nm) is somewhat greater than is usual for conventional plastic sections, resulting in overlap of structural features.
Fig 2.
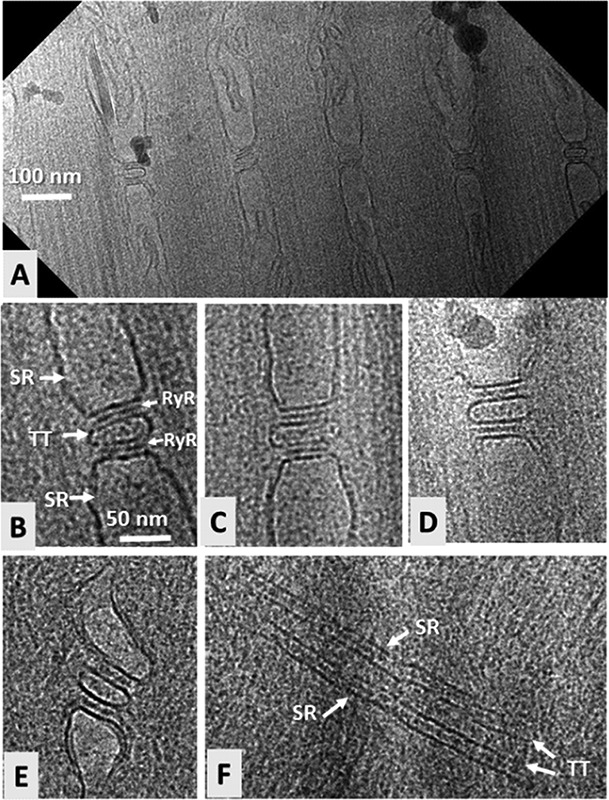
Triad junctions imaged by cryo-electron microscopy of FIB-milled muscle. A. Overview of a 300-nm-thick longitudinal section showing five triad junctions from toadfish swimbladder. B-D. Higher- magnification images of three triad junctions. Abbreviations: SR, sarcoplasmic reticulum; TT, T-tubule; RyR, ryanodine receptor. E-F. Triad junctions from zebrafish skeletal muscle. E shows a longitudinal view with the T-tubule in cross-section (as in A-D), while F shows a longitudinal view in which the T-tubule is viewed from the side (arrows adjacent to “TT” indicate the two opposing surfaces of the membrane of the T-tubule); rows of RyRs are visible in the two spaces between the TT and SR membranes. A and B adapted from reference.13
Results from our first attempt at cryo-ET of FIB-milled swimbladder muscle is shown in Figure 3.13 Shown in Figure are a series of X-Y slices through the tomogram and a three-dimensional representation that shows colored surfaces for the major recognizable structural features (the SR and T-tubule boundaries, RyRs, and the luminal density in the T-tubule) obtained by manually tracing of the visible features in each XY-plane slice of the tomogram (Fig. 3A,B, respectively).
Fig 3.

Cryo-ET of FIB-milled toadfish swimbladder muscle.13 A. X-Y slices through the tomogram determined for the triad shown in Fig. 2B (section thickness ~100 nm). Slices are 1 nm thick, spaced at 10 nm intervals along Z. B. Manually segmented model in the region of the triad junction. Green, T-tubule; blue, density containing ryanodine receptors; yellow, SR membrane; purple, unidentified mass density in lumen of T-tubule. C. View onto RyR-bearing surface of SR (from upper SR in B) showing 4-6 RyRs (numbered), which are not clearly resolved from one another. Parts of A adapted from reference.13
Figure 3C depicts an orientation of a portion of the surface model such that the arrangement of RyRs on one of the SR junctional faces is revealed. Although the individual RyRs are not resolved from one another, there is clear evidence for two rows of RyRs in accordance with previous findings. The FIB-milled section for this experiment was about 100 nanometers thick, sufficient to contain about three RyRs from each of the two rows, and the contoured model appears to contain six RyRs (numbered 1-6 in Fig. 3A), although those at the top of the panel appear to be truncated. The square-shaped outline of the RyRs, whose overall structure is known from previous work, is barely detectable due the high noise level. Poor contrast is an inherent problem in cryo-ET of native cells, and is further compounded, in this direction of view, by the missing wedge of structural information that arises from the limited tilt range in the microscope, in this case ±60 degrees. An additional unknown is the extent to which additional proteins, for instance subunits of DHPR, are packed in between the RyRs which at low resolution could confuse interpretation of the images. As more, and better, tomograms are obtained, and sub-tomogram averaging is applied to subvolumes containing RyRs, the structural organization of the triad’s components should become much better resolved.
Outlook
Presently, the main drawback of cryo-FIB preparation is low throughput, arising at several steps in the process. Even when all of the steps described above are successful, only about eight useful 10 µm X 20 µm lamellae are produced in each specimen, yielding a total area of about 1600 µm2 suitable for tomographic data collection. This area corresponds to only about 20% of the area of a single grid square of a 200-mesh grid which contains about 50 grid squares suitable for full-tilt-range imaging. Clearly, it is essential that most milled areas contain the structure(s) of interest, which is indeed the case for muscle tissue. However, for some specimens this is not achievable because structures at the surface of a vitreously frozen sample are very poorly imaged by the electron or ion beam, and structures in the depth of a sample cannot be directly located at all. In our case, it is feasible to obtain 10 or more tomograms per successful FIB-milling session, with each tomogram containing 5-6 triad junctions. In contrast to this situation, cryo-ET of bacteria and small cells that can be deposited over the entire surface of a grid, and which do not require FIB milling, can yield tens to hundreds of tomograms from a single grid, by means of automated microscopy.25,26 Much effort will be required to improve the efficiency of cryo-FIB milling to the point where it is practiced routinely. The problem of identifying regions of the frozen specimen that contain the regions of interest is being addressed by correlative light microscopy. The rationale is to label the structure of interest with a fluorescent tag, image it in 3-D by confocal fluorescence microscopy in the frozen state, and then translate the coordinates to the FIB-SEM for milling.21,27 It may also be possible to immediately check finished lamellae for presence of a desired structure by use of a scanning-transmission detector in the FIB-SEM instrument.29 With regard to improving the efficiency of the FIB milling itself, the specimen configuration and pre-trimming geometry that we use lends itself to automated production of the lamellae, once the sample is aligned in the FIB-SEM instrument. Despite the limitations of FIB-milling applications to tissues and cells, current technology should allow progress to made on favorable specimens, such as the triad junction discussed here. Already, it should be feasible to acquire, over a period of months, 100 or more triad-junction sub-tomograms from well-preserved, thin (100-200 nm) lamellae. This data should be sufficient to begin realizing the full and evolving potential of cryo-electron tomography, as exemplified by recent studies of thin (i.e. not requiring FIB), plunge-frozen single cells.5,6,28 Currently, a limitation of cryo-ET is that generally applicable strategies for labeling proteins of interest have not been perfected6. However, by means of sub-tomogram averaging, large macromolecular complexes, such as ryanodine receptors that are identifiable from their known structures, can be extracted from tomograms and combined to produce 3D reconstructions having resolutions of 2-4 nm, thereby revealing their structural state in an in situ environment. When applied to RyRs in triads, structural information on proteins that interact with RyRs, such as calmodulin, the dihydropyridine receptor, and triadin/calsequestrin should be resolved. This would certainly advance current understanding of structure-function relationships for the triad junction.
Acknowledgement
The authors thank Dr. Hongli Hu for help with preparing Figure 3. This work was supported by NIH grants R01AR40615 (to T. Wagenknecht) and R01GM097010 (to M. Marko). Microscopy infrastructure was provided by the Wadsworth Center’s 3D-EM Facility.
References
- 1.Franzini-Armstrong C. The sarcoplasmic reticulum and the control of muscle contraction. FASEB Journal 1999;13:266-70. [DOI] [PubMed] [Google Scholar]
- 2.Wagenknecht T, Hsieh CE, Rath BK, et al. Electron Tomography of Frozen-Hydrated Isolated Triad Junctions. Biophys J 2002;83:2491-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamilton SL, Serysheva II. Ryanodine Receptor Structure: Progress and Challenges. J. Biol. Chem 2009, 284, 4047-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang MC, Dolphin A, Kitmitto A. L-type voltage-gated calcium channels: understanding function through structure. FEBS Lett 2004;564:245-50. [DOI] [PubMed] [Google Scholar]
- 5.Briggs JAG. Structural biology in situ - the potential of subtomogram averaging. Curr Opinion Struct Biol 2013;23:261-7. [DOI] [PubMed] [Google Scholar]
- 6.Hoenger A. High-resolution cryo-electron microscopy on macromolecular complexes and cell organelles. Protoplasma 2014;251:417-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDowall AW Chang JJ, Freeman R.et al. Electron microscopy of frozen hydrated sections of vitreous ice and vitrified biological samples. J Microsc 1983;131:1-9. [DOI] [PubMed] [Google Scholar]
- 8.Al-Amoudi A, Dubochet J, Gnaegi H, et al. An oscillating cryo-knife reduces cutting-induced deformation of vitreous ultrathin sections. J Microsc 2003;212:26-33. [DOI] [PubMed] [Google Scholar]
- 9.Al-Amoudi A, Norlean LPO, Dubochet J. Cryo-electron microscopy of vitreous sections of native biological cells and tissues. J Struct Biol 2004;148:131-5. [DOI] [PubMed] [Google Scholar]
- 10.Al-Amoudi A, Studer D, Dubochet J. Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. J Struct Biol 2005;150:109-21. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh CE, Leith A, Mannella CA, et al. Towards high-resolution three-dimensional imaging of native mammalian tissue: Electron tomography of frozen-hydrated rat liver sections. J Struct Biol 2006;153: 1-13. [DOI] [PubMed] [Google Scholar]
- 12.Pierson J, Fernandez JJ, Bos E, et al. Improving the technique of vitreous cryo-sectioning for cryo-electron tomography: electrostatic charging for section attachment and implementation of an anti-contamination glove box. J Struct Biol 2010;169:219-25. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh C, Schmelzer T, Kishchenko G, et al. Practical workflow for cryo focused-ion-beam milling of tissues and cells for cryo-TEM tomography. J Struct Biol 2014;185:32-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marko M, Hsieh C, Moberlychan WJ, et al. Feasibility of focused ion beam miling for preparation of TEM specimens of biological material embedded in vitreous ice. Microsc Microanal 2005; 11 (suppl 2): 802C-802D. [Google Scholar]
- 15.Marko M, Hsieh C, Schalek R, et al. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nature Methods 2007;4:215-17. [DOI] [PubMed] [Google Scholar]
- 16.Marko M, Hsieh C, Moberlychan W, et al. Focused ion beam milling of vitreous water: prospects for an alternative to cryo-ultramicrotomy of frozen-hydrated biological samples. J Microsc 2006;222: 42-47. [DOI] [PubMed] [Google Scholar]
- 17.Kizilyaprak C, Daraspe J, Humbel BM. Focused ion beam scanning electron microscopy in biology. J Microsc 2014;254:109-14. [DOI] [PubMed] [Google Scholar]
- 18.Villa E, Schaffer M, Plitzko JM, Baumeister W. Opening windows into the cell: focused-ion-beam milling for cryo-electron tomography. Curr Opin Struct Biol 2013;23:771-7. [DOI] [PubMed] [Google Scholar]
- 19.Rigort A, Bauerlein FJB, Leis A, et al. Micromachining tools and correlative approaches for cellular cryo-electron tomography. J Struct Biol 2010;172:169-79. [DOI] [PubMed] [Google Scholar]
- 20.Rigort A, Gunther D, Hegerl R, et al. Automated segmentation of electron tomograms for a quantitative description of actin filament networks. J Struct Biol 2012;177:135-44. [DOI] [PubMed] [Google Scholar]
- 21.Rigort A, Villa E, Bauerlein FJB, et al. Integrative approaches for cellular Cryo-electron Tomography: Correlative imaging and focused Ion Beam Micromachining. Methods in Cell Biol 2012;111:259-81. [DOI] [PubMed] [Google Scholar]
- 22.Strunk KM, Wang K, Ke D, Gray JL, Zhang P. Thinning of large mammalian cells for cryo-TEM characterization by cryo-FIB milling. J Microsc 2012;247:220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Strunk K, Zhao GP, et al. 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. J Struct Biol 2012;180:318-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franzini-Armstrong C, Nunzi G. Junctional feet and particles in the triads of a fast-twitch muscle fibre. J Muscle Res Cell Motil 1983;4:233-52. [DOI] [PubMed] [Google Scholar]
- 25.Gan L, Jensen GJ. Electron tomography of cells. Q Rev of Biophys 2012;45:27-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu B, Margolin W, Molineux IJ, Liu J. The Bacteriophage T7 virion undergoes extensive structural remodeling during infection. Science 2013;339:576-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukuda Y, Schrod N, Schaffer M, et al. Coordinate transformation based cryo-correlative methods for electron tomography and focused ion beam milling. Ultramicroscopy 2014;143:15-23. [DOI] [PubMed] [Google Scholar]
- 28.Lucic V, Rigort A, Baumeister W. Cryo-electron tomography: The challenge of doing structural biology in situ. J Cell Biol 2013;202:407-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Winter DAM Mesman RJ Hayles MF.et al. In-situ integrity control of frozen-hydrated, vitreous lamellas prepared by the cryo-focused ion beam-scanning electron microscope. J Struct Biol 2013;183:11-8 [DOI] [PubMed] [Google Scholar]