

Summary
Natural killer (NK) cells and cytotoxic T lymphocytes (CTL) use cytotoxic granules containing perforin and granzymes to lyse infected or malignant host cells and provide immunity to intracellular microbes and tumors. Perforin is essential for cytotoxic granule-mediated killing. Perforin expression is regulated transcriptionally and correlates tightly with the development of cells that can exhibit cytotoxic activity. Although a number of genes transcribed by T cells and NK cells have been studied, the cell-specificity of perforin gene expression makes it an ideal model system in which to clarify the transcriptional mechanisms that guide the development and activation of cytotoxic lymphocytes. In this review, we discuss what is known about perforin expression and its regulation, then elaborate on recent studies that utilized chromosome transfer and bacterial artificial chromosome (BAC) transgenics to define a comprehensive set of cis-regulatory regions that control transcription of the human PRF1 gene in a near-physiological context. In addition, we compare the human and murine Prf1 loci and discuss how transcription factors known to be important for driving CTL differentiation might also directly regulate the cis-acting domains that control Prf1. Our review emphasizes how studies of PRF1/Prf1 gene transcription can illuminate not only the mechanisms of cytotoxic lymphocyte differentiation but also some basic principles of transcriptional regulation.
Keywords: natural killer cells, cytotoxic T cells, transcription factors, cell differentiation, perforin, gene regulation
Introduction
Intracellular infections and tumor cells offer unique challenges to the immune system, because their pathogenicity is ‘cloaked’ from direct recognition by immune cells. To meet these challenges, the immune system must recognize and destroy aberrant host cells that harbor microbes, or that have become transformed. Although additional specialized subsets of killer cells have been described, two prototypical categories of killer lymphocytes, natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), possess mechanisms to recognize and directly lyse aberrant host cells. NK cells are cells of the innate immune system that derive from immature precursors in the bone marrow; once developed, they circulate as constitutively-armed effector cells that do not require extensive sensitization to elicit cytotoxic activity. In contrast, CTLs are specialized effector cells that differentiate from naive T cells that have been activated through their T-cell receptors (TCRs) by antigens from infected or malignant host cells, in conjunction with appropriate costimulatory and immunogenic signals. Although other cytotoxic mechanisms exist, the exocytosis of cytotoxic granules that contain the effector proteins perforin and granzymes is the main, non-redundant, mechanism used by NK cells as well as CTLs to kill target cells during an immune response (1).
Perforin and granzymes represent two distinct categories of cytotoxic proteins that collaborate to induce target cell-death under physiological conditions. Perforin is a pore-forming protein that polymerizes and forms transmembrane channels in plasma membrane lipid bilayers (2). The granzymes are a family of serine esterases with distinct proteolytic specificities that cleave numerous intracellular substrates; granzymes both initiate and directly mediate apoptosis of target cells (3). Perforin delivers granzymes into the cytosol of target cells and is the only currently known delivery molecule for the granzymes (2). Although the exact process by which delivery occurs is still unclear, analysis of perforin-deficient mice has demonstrated that perforin is absolutely required for target cell-death mediated by the granule exocytosis pathway (1). In contrast, multiple granzymes exist and exhibit some degree of functional redundancy. Mice deficient in individual granzymes have mild or as-yet undescribed phenotypes, and deficiency of multiple granzymes appears necessary for killer lymphocytes to mediate cell death and protect the animal from intracellular pathogens. Perforin is less broadly expressed than most granzymes, and some granzymes may have additional functions besides their role in cytotoxicity (3). Overall, therefore, perforin rather than granzyme expression serves as the definitive marker of cells with the capacity to kill.
Accordingly, the perforin gene is an attractive model system to elucidate the molecular mechanisms of cytotoxic lymphocyte development and activation. Perforin is a single gene with a simple genomic structure, flanked by genes that do not have an obvious specific role in the development or function of cytotoxic lymphocytes. Perforin expression is tightly restricted to cytotoxic lymphocytes, whereas its flanking genes are expressed in a range of tissues and do not appear to be co-regulated (4). The expression of perforin is induced developmentally in NK cells; in contrast in T cells, its expression is regulated dynamically both during thymocyte development and later during peripheral differentiation of effector and memory T cells during infection. Thus, studying the chromatin level and transcriptional regulation of the perforin gene is likely to yield general insights into how the transcription of developmentally regulated genes is controlled and more specifically how extrinsic signals induce or activate transcription factors that then act directly to induce the cytolytic gene program in lymphocytes. The stage has been set by recent studies that have identified a complete set of cis-acting transcriptional regulatory regions in the human perforin gene as well as transcription factors that act directly at these regulatory regions to govern perforin gene transcription in NK cells and CD8+ T cells. The regulation of cytokine genes has been examined in great detail in both CD4+ and CD8+ T cells, but these studies have the disadvantage that the secreted cytokine products of the very genes that are being studied feed back upon and influence gene expression. In contrast, perforin is an intracellular protein whose transcriptional regulation can be studied in the absence of feedback effects.
In this review, we describe what is known about the regulation of perforin gene transcription. We focus on studies describing the identification and characterization of cis-acting regions in the perforin genes of both humans and mice, highlight what remains unknown, and suggest hypotheses for how the locus might be regulated. Our hope is to inspire new avenues of investigation that will advance our understanding of how cytotoxic lymphocytes differentiate and mature and how perforin transcription is controlled.
The cell-type specific and developmental expression of perforin
Perforin is expressed in cytotoxic lymphocytes, most of which can be classified as either NK cells or CTLs. Perforin is not expressed in non-lymphoid cells, B lymphocytes, or cells of the myeloid lineage under normal conditions (3, 4). Analysis of perforin expression in human NK and CTL subsets has been facilitated by the availability of an excellent antibody that recognizes intracellular human perforin. In contrast, there is no correspondingly reliable antibody that can be used to detect mouse perforin by intracellular staining and flow cytometry, a problem that has prevented careful delineation of lymphocyte subsets that express perforin in mice (MEP, AR and MGL, unpublished data).
NK cell perforin expression
Human NK cell development has been divided into five stages based on extensive phenotypic and functional criteria, and perforin expression has been correlated with NK cell maturation (5). In both humans and mice, bone marrow-derived lymphocyte progenitor cells differentiate into mature NK cells in a step-wise fashion (5, 6). Perforin is upregulated after commitment to the NK cell lineage, and its expression correlates inversely with the level of CD56 expression. Initially, perforin is expressed in some cells of stage 4 (CD56bright), which are in the process of maturing, and most cells of stage 5 (CD56dim), which are considered ‘terminally’ differentiated; a small number of CD56dim cells are perforin-negative, but these represent an immature population that is upregulating CD56 (5). In mature peripheral blood NK cells, CD56dim cells express 10-fold higher amounts of perforin relative to CD56bright cells (7). Therefore, upregulation of perforin expression is a late event during NK cell maturation, and perforin might be expressed at different levels in different subsets of mature NK cells.
In mice, the development of NK cells has also been characterized by phenotype and function. However, these developmental stages have been analyzed for perforin mRNA expression only by RT-PCR, and expression of functional perforin protein has been assessed only by cytotoxic activity (6, 8). The earliest precursors of murine NK cells do not express perforin mRNA and are lineage-negative CD122+ cells in the bone-marrow (8). These cells then upregulate expression of NK1.1 (a marker unique to C57Bl/6 mice), and as they mature they induce expression of DX5, a late marker of NK cell development. Perforin mRNA is not expressed in CD122+NK1.1+DX5− NK cell precursors, but it is expressed later during development in cells that are CD122+NK1.1+DX5+ (8). This population contains mature NK cells that are capable of mediating strong cytotoxic activity ex vivo (8). Another study showed that Mac-1 is a definitive marker for the most mature mouse NK cells and that it correlated with enhanced cytotoxic activity, although perforin expression was not analyzed (9). Flow cytometric methods have not been used to characterize perforin protein expression during development of murine NK cells, but the earliest studies that used immunocytochemical methods indicated that all NK1.1+ splenocytes expressed perforin protein. More recent studies have now shown that mature murine NK cells express perforin mRNA but might require an activation signal to express perforin protein (discussed below). In any case, perforin expression is upregulated late during murine NK cell development and coincides with the development of NK cell cytotoxic function.
Perforin expression in thymocytes
Perforin is first expressed in T cells during thymocyte development. Perforin mRNA is undetectable by Northern analysis in Tcra−/− thymocytes arrested at the double positive (DP) stage (MEP, unpublished observations), but is detected by reverse transcriptase polymerase chain reaction (RT-PCR) in wildtype DP thymocytes that are undergoing positive selection into the CD8+ lineage (10). Subsequently, perforin transcripts continue to be expressed in single positive (SP) CD8 but not SP CD4 thymocytes (10). Perforin protein expression has not been reported in thymocytes. Basal transcriptional competence of the perforin gene might begin as early as the double negative (DN) stage, as mice transgenic for a hCD4 reporter gene driven by the mouse perforin promoter and 5 kb of its upstream flank showed hCD4+ cells in DN thymocytes, and in all stages thereafter (11). Together, these reports suggest that developmental programming of perforin gene transcription in T cells occurs in the thymus and that some degree of transcriptional competence is established in SP CD8+ thymocytes.
Perforin expression in naive CD8+ T cells
Like their immediate precursors, mature CD8+ thymocytes, naive CD8+ T cells express perforin mRNA but do not express perforin protein. Based on Northern analysis, perforin mRNA is expressed in naive CD8+ T cells purified by negative selection from C57Bl/6 and Balb/c mice, as well as naive CD8+ T cells from Tcra−/− P14 TCR transgenic mice that are uniformly negative for surface CD122 (12–15, and authors’ unpublished observations). However, western blotting indicates that perforin protein is absent or very weakly expressed in naive murine CD8+ T cells (12). The situation is not as clear in human cells. Although it is generally assumed that perforin mRNA is not expressed by naive human CD8+ T cells, this has not been demonstrated conclusively. In fact, perforin mRNA is expressed in some CD8+ T cells that exhibit phenotypic features of naive cells, although it was proposed that these cells could represent a transitional effector cell population based on their lower content of TCR gene excision circles (implying more extensive replicative history) (16). Nevertheless, there is clear evidence that naive human and murine CD8+ T cells from peripheral lymphoid organs or the blood do not express perforin protein and do not exhibit cytotoxic function without prior stimulation (4, 17). Overall, it is likely that naive CD8+ T cells express perforin mRNA but do not express perforin protein until after activation, which raises the possibility that perforin protein expression is repressed through posttranscriptional mechanisms in naive CD8+ T cells.
Perforin expression in activated CD8+ T cells
Perforin expression is dynamically regulated in naive CD8+ T cells activated through their TCRs. Based on transcriptional profiling using microarrays, perforin mRNA is downregulated in effector CD8+ T cells on day 4 after acute viral infection but then is upregulated in effector cells on day 8, the peak of the CD8+ T-cell response (14, 15). At this time, perforin is more highly expressed in KLRG-1highIL-7Rαlow effector cells which are considered more terminally differentiated as they tend to die after pathogen clearance, compared to KLRG-1lowIL-7Rαhigh effector cells tend to survive and contain more long-lived memory CTL precursors (14, 15, 18). These dynamic changes in perforin expression are recapitulated in naive CD8+ T cells activated under controlled conditions in cell-culture. Naive CD8+ T cells express perforin mRNA, but it is downregulated by day 4 after TCR stimulation and then is highly expressed on day 6 (and continues to increase until day 8) in effector phenotypic cells (12). Perforin mRNA is also inversely expressed relative to IL-7Rα mRNA in vitro (13). Another study, which analyzed purified CD8+ T cells activated in culture by single-cell RT-PCR, showed that the frequency of activated cells bearing perforin transcripts peaked around day 7 (19). Collectively, these studies suggest that after initial activation of naive CD8+ T cells, perforin mRNA is upregulated at late time points, and the highest expression of perforin is observed in effector CD8+ T cells that exhibit a more fully differentiated surface phenotype.
In humans, perforin protein is predominantly expressed in CD8+ T cells with an effector or an effector-memory phenotype (17, 20). These are a minority of all CD8+ T cells in the peripheral blood of healthy adult humans, but that are expanded dramatically during acute viral infection (21). By contrast, CD8+ T-cell subsets that demonstrate a naive or central memory phenotype and that appear to primarily home to lymphoid organs exhibit low or undetectable perforin protein expression (17, 20, 22). Because staining of intracellular perforin in murine CD8+ T cells has not been reliable, similar studies have not been performed at the protein level. However, not all effector CD8+ T cells that become activated upon viral infection express perforin and display cytolytic activity. Lysispot, single-cell RT-PCR, and limited gene arrays have all shown that only a fraction of all effector CD8+ T cells express perforin or can kill (23–25). Nevertheless, most effector or effector memory phenotypic CD8+ T cells generated by infection upregulate perforin mRNA and protein expression at some point. To clarify which activated CD8+ T cells differentiate into perforin-expressing cells and can be formally classified as CTLs in murine models of infection, it will be necessary to develop improved antibodies for intracellular staining of murine perforin, or generate mice bearing knockin perforin reporter alleles that faithfully recapitulate perforin protein expression.
Perforin expression in other cytotoxic lymphocytes
T cells that express the γδ TCR genes can also develop into CTLs. As with αβ T cells, γδ T cells can be divided into subsets that represent naive, effector, or effector memory-like cells (26). The vast majority of γδ T cells from cord blood and infant peripheral blood demonstrate a naive phenotype. By adulthood, the phenotype of nearly all γδ T cells from the peripheral blood is effector-like; they express perforin and exhibit robust cytotoxic potential directly ex vivo (26, 27). Although γδ T cells may develop both within and outside the thymus, γδ thymocytes from adults lack perforin protein expression and cytolytic potential (27, 28). Thus, as with αβ T cells, it appears that perforin protein expression and cytolytic potential of γδ CTLs is acquired by activation in the periphery and is maintained constitutively in effector-like populations.
Perforin is expressed in some murine effector CD4+ T cells that contribute to protection from lethal influenza infection (29, 30), and its expression has also been demonstrated in human CD4+CD25+ T-regulatory cells (31). The conditions in which naive CD4+ T cells are initially activated can increase their potential to express perforin (30). Perforin mRNA is undetectable by Northern blotting in CD4+ T cells cultured under Th2 conditions (4); in contrast, CD4+ T cells differentiated under Th1 conditions can express perforin mRNA, but in amounts ≥20 times lower than observed in activated CD8+ T cells differentiated in culture. Thus, although some CD4+ T cells can upregulate and then employ perforin, CD8+ T cells exhibit a much higher propensity for perforin expression and cytolytic activity. It is likely that CD4+ and CD8+ T cells use different mechanisms to control perforin expression.
Perforin is also expressed by NKT cells, a heterogeneous group of T cells that co-express both TCRs and surface receptors characteristic of NK cells (i.e. NK1.1 in mice, or CD56 in humans). They consist of classical NKT cells, a lineage that expresses a restricted αβ TCR repertoire which are selected on the non-classical MHC molecules of the CD1 family (32), and a subset of conventional αβ T cells that acquire NK receptors following acute viral infection (33–35).
Perforin is expressed in a diverse array of adaptive and innate lymphocytes from the T and NK cell lineages. A recurring theme is that perforin expression is activated at late times during development or in mature cells after immunogenic activation. This pattern of expression suggests that specific environmental signals experienced by mature cells are likely to be important for acute activation of perforin expression during development and differentiation. A future challenge is to identify the molecules and mechanisms that enable the expression of perforin in mature cytotoxic lymphocytes and those that are activated in mature cells by specific signals that acutely induce perforin expression upon stimulation.
Regulation of perforin expression in NK cells
The regulation of perforin expression in NK cells during development and upon differentiation of naive CD8+ T cells after TCR activation is not entirely understood. Collective evidence from mouse and human cells suggests that perforin is regulated by both transcriptional and posttranscriptional mechanisms in both NK cells and in CD8+ T cells.
Most if not all mature NK cells constitutively express perforin mRNA and protein. Immunocytochemistry, flow cytometry, and in situ hybridization have shown that perforin mRNA and protein are homogeneously expressed by all human NK cells in the absence of stimulation (7, 36–38). Nuclear run-on analysis showed that primary unstimulated NK cells transcribe perforin constitutively (38). Stimulation with cytokines such as IL-2, IL-6, IL-12, IL-15, IL-21, or IFN-β can increase perforin expression in human NK cells (38–42). The response to IL-2 and IL-12 was found to be due to increased transcriptional rates (38, 43). Thus, ongoing transcription underlies the continuous expression of perforin in human NK cells, and it is generally accepted that perforin mRNA expression is correlated in NK cells with constitutive expression of perforin protein.
Perforin expression in NK cells appears to be regulated by transcriptional as well as posttranscriptional mechanisms. Murine NK cells constitutively express perforin mRNA, and initial studies indicated that they also express perforin protein ex vivo (44). In addition, studies of NK cell development showed that mature NK cells are fully armed to kill (8, 9). However, a recent careful comparison of perforin mRNA and protein expression in mature splenic NK cells from wildtype mice revealed that perforin protein expression is regulated posttranscriptionally (45). Fehniger et al. (45) confirmed that perforin mRNA is constitutively expressed in mature murine NK cells but showed that stimulation with cytokines (namely IL-2 and IL-15) is required to induce perforin protein expression and cytotoxic activity and that perforin expression is upregulated during viral infection. The molecules and mechanisms that mediate this regulation have not yet been clarified.
Regulation of perforin expression in CTLs
Perforin expression is upregulated progressively after naive CD8+ T-cell activation. Northern blot analysis of perforin mRNA expression kinetics showed that purified naive murine CD8+ T cells activated with a very strong TCR stimulus (plate-bound anti-CD3 and anti-CD28 antibodies, or specific peptide presented on live antigen presenting cells) did not show a change in perforin mRNA expression in the first two days of activation but upregulated perforin strongly by day 6 after activation (12). The in vivo data are consistent: the highest frequency of antigen-specific CD8+ T cells that express perforin mRNA and protein occurs around one week following acute viral infection of mice (23, 46). These studies are consistent with the hypothesis that differentiation of effector CD8+ T cells into effector CTLs (as defined by cytotoxic activity) occurs subsequent to, rather than during, the initial TCR stimulus.
TCR signals are not sufficient to induce perforin expression and drive CTL differentiation of naive CD8+ T cells; additional signals are necessary. IL-2 receptor (IL-2R) signaling is an important regulator of perforin transcription after naive CD8+ T-cell activation. Perforin mRNA expression is reduced in activated T cells lacking IL-2Rβ (47). This implied a requirement for IL-2, IL-15, or both, since both cytokines signal through the intermediate affinity IL-2 receptor comprised of IL-2Rβ and the common γ chain (γc). High concentrations of soluble IL-15 did not induce perforin mRNA in TCR-activated naive CD8+ T cells isolated from TCR transgenic mice (MEP, unpublished observations), suggesting that perforin expression is primarily induced by IL-2. However, another study showed that high concentrations of IL-15 could induce perforin expression in human CD8+ T cells (48); the difference between the data from mouse and human T cells may reflect the difficulty in identifying genuinely naive CD8+ T cells in humans or fundamental differences between humans and mice.
The concentration of IL-2 in culture determines whether perforin expression is downregulated or re-induced after TCR activation (12, 49). After primed naive CD8+ T cells have been removed from the TCR stimulus, strong IL-2R signals induce perforin expression, whereas perforin expression is completely downregulated in cells that experience weak IL-2R signals (49). In addition, IL-2Rα-deficient CD8+ T cells in mixed bone marrow chimeric mice infected with LCMV have impaired perforin mRNA expression and cytolytic function, indicating that IL-2R signaling stimulated by high affinity IL-2 binding is necessary for normal perforin expression in vivo (49). The effect of IL-2 on perforin expression is likely to be transcriptional, because IL-2 induces increased transcription initiation of the perforin gene (50), rather than increasing mRNA stability (51). In addition, increased perforin expression is independent of IL-2-regulated proliferative or cell survival effects (52). Thus, TCR stimulation of naive CD8+ T cells initiates CTL differentiation, and IL-2/IL-2R signaling induces transcription of the perforin gene during this process. However, while perforin expression is reduced in cells with IL-2R deficiencies, it was not eliminated, indicating that additional stimuli are no doubt important for inducing perforin in activated CD8+ T cells.
In contrast to recently primed naive CD8+ T cells, previously activated CD8+ T cells rapidly induce perforin expression upon secondary stimulation. Mitogens such as PHA or ConA, pharmacological reagents such as phorbol esters or calcium ionophore, anti-CD3 agonistic antibodies, as well as the cytokines IL1, IL-2, IL-4, IL-6, IL-7, IL-12, and IL-15 have each been reported to directly or indirectly induce perforin mRNA accumulation in whole peripheral blood mononuclear cells (PBMCs), total purified T cells, sorted T-cell subsets, or cell-line CTL models (27, 32, 46, 51, 53–57). These subsets contain relatively large numbers of previously activated cells and are the most likely source of perforin that is rapidly induced upon stimulation. In fact, several days after initial activation of purified naive murine CD8+ T cells, TCR restimulation strongly induces perforin mRNA and protein within 6 h (49). Thus, a secondary encounter with antigen or a cytokine-rich environment is likely to result in strong induction of perforin in previously activated CD8+ T cells.
Transcription of perforin in NK cells and CTLs is an important mechanism for regulating overall perforin expression. However, additional regulation exists, but there has not been as much focus on these aspects. The most recent studies have shown that perforin mRNA and perforin protein are not always correlated in mature NK cells, and stimulation with cytokines affects this balance (45). Moreover, similar regulation might be important in CD8+ T cells, because perforin mRNA and protein are not always correlated, especially early after activation (49). Furthermore, it is known that human perforin protein is synthesized in an inactive pro-perforin format and must be processed prior to packaging and storage in cytotoxic granules (58). Although a similar analysis of murine perforin has not been performed, the study of human perforin processing suggests that some degree of regulation might exist at the level of compartmentalizing perforin into cytolytic granules. The molecules and mechanisms mediating these types of regulation are awaiting discovery.
The organization and transcriptional control of the PRF1/Prf1 loci
The developmentally restricted and dynamic expression of the perforin gene in cytotoxic lymphocytes makes it a unique model system in which to investigate the transcriptional mechanisms that specifically control cytotoxic lymphocyte differentiation. Cis-regulatory regions from the human and mouse perforin genes have been used to drive artificial reporter genes in a transgenic context; these studies have shown that developmental and activation-dependent expression of perforin are both controlled transcriptionally (Fig. 1). The transcriptional regulation of many genes expressed by T and NK cells (e.g. Cd2, Cd4, Cd8, IFNg, IL4, IL2, Tcra/d, Gzmb) has been studied in depth, but none of these gene products correlates as well with cytotoxic function in distinct lymphocyte lineages as the human perforin (PRF1) and murine perforin (Prf1) genes.
Fig. 1. Summary of transgenic studies of the PRF/Prf1 loci.
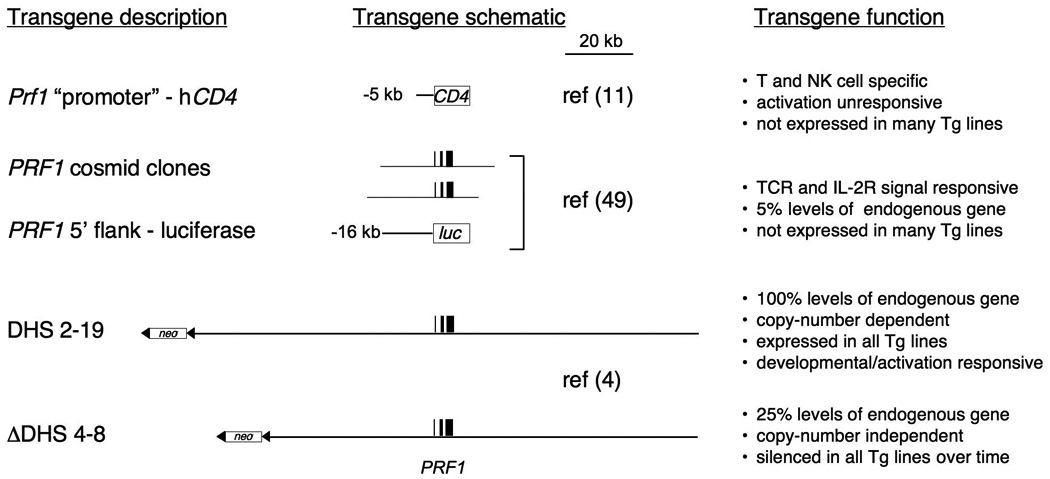
The left column describes the transgene that was analyzed, and is depicted schematically in the center column. To the right, the transgenic control is summarized, and includes the reference for each study. A scale bar is included to give a relative impression of size.
Both PRF1 and Prf1 share a simple genomic structure. Each gene is comprised of three small exons that span a total of less than 6 kb on syntenic regions of human and mouse chromosome 10 (59–61). Both genes possess 5′ and 3′ untranslated regions (UTRs). The nearest 5′ flanking gene is ADAMTS14, which initiates >70 kb upstream of PRF1 and is transcribed in the opposite orientation. On the 3′ flank is Paladin (PALD), which terminates ~30 kb downstream of PRF1 and is also transcribed in the opposite orientation. The murine locus exhibits the same organization, with the Prf1 gene being flanked by the orthologous Adamts14 and Pald genes, although the distances to Prf1 are smaller compared to the human locus. ADAMTS14 is a metalloprotease, and Paladin is a putative phosphatase. PRF1 mRNA expression is not co-regulated with expression of either ADAMTS14 or PALD mRNA (62, MEP, unpublished observations). The relatively compact nature of the Prf1 transcribed region and its distinct expression relative to its flanking genes makes Prf1 particularly amenable to basic transcriptional analyses of gene regulation.
The cis-regulatory regions of the PRF1 locus involved in transcriptional control span at least 150 kb surrounding the PRF1 gene. This estimate was deduced using chromosome transfer to reprogram the perforin locus from an inactive state in non-killer cells to an active conformation in CTLs and by mapping the DNase I hypersensitive sites (DHSs) induced during the process. The core promoter and sites of transcription initiation have been mapped, and cis-acting functional sequences in the proximal regions of the PRF1 gene have been delineated. Analysis of PRF1 transgenes in stable cell lines and in mice has identified and partially characterized the cis-acting function of distal regulatory regions, including two developmentally and activation-responsive enhancers, and a locus control region (LCR). Thus, many of the crucial cis-acting regions in human and murine perforin have been identified. However, very little is known about when these regulatory regions become active and which transcription factors target them to activate and repress PRF1/Prf1 during NK cell development, effector CTL differentiation, and memory CTL reactivation. Furthermore, the basic chromatin level and transcriptional mechanisms that control PRF1/Prf1 remain to be discovered.
The cell-type restricted expression of PRF1 is controlled by its promoter
The perforin promoter is GC-rich, does not possess a TATA-box, and has one major and several minor sites of transcript initiation. Transient transfection analysis localized a core promoter in the murine gene to within 120 bp of perforin’s major transcription initiation site (61). This core fragment is non-specifically active at moderate levels in both killer and non-killer cells. Ubiquitously expressed Sp1 transcription factors transactivate the perforin promoter core through at least three sites in the first 200 bp upstream (61, 63, 64). Nuclear extracts of non-killer cells contain at least two undefined nuclear factors that suppress this basal activity (64). The basal transcriptional activity in killer lymphocytes is repressed in non-killer cells by sequences upstream of −240 bp, and DNase I footprinting suggests this depends on two elements centered near −350 bp and −650 bp. Thus, the cell type-specific perforin promoter activity in vitro is determined by transcription factors in non-killer cells that repress ubiquitous activity derived from the core. Conversely, in killer lymphocytes, the basal core promoter activity is sustained by sequences located between −244 bp and −807 bp. Elements near −500 bp and −650 bp appear to be critical for this function.
Transcription factors from the Ets family support perforin promoter activity in killer cells (61, 63–65). Potential Ets-controlled sites near −500bp comprise a regulatory motif, for which the term NF-P was coined, that activates promoter function in killer cells and is bound by an uncharacterized Ets protein distinct from Ets1, Ets2, or Elf-1 (65). The transcription factor MEF (myeloid elf-1 like factor) might be the unidentified Ets family member that controls the NF-P motif in NK cells in vivo (66). MEF-deficient mice lack detectable perforin protein in NK cells, and purified MEF binds the Ets-binding site in the NF-P motif as well as a second site at −43bp in the murine perforin promoter in vitro. In addition, transient reporter activity driven by the perforin promoter in COS cells could be enhanced by co-transfection with a MEF expression plasmid. Chromatin immunoprecipitation (ChIP) also showed that MEF was associated with the endogenous murine Prf1 gene in CTLL-2 cells, a CTL-derived cell line that expresses high levels of perforin, as opposed to in EL-4 cells, a T-cell line that does not express perforin. However, MEF was apparently not required for perforin expression in normal primary CD8 T cells (66). Thus, MEF appears to be important for programming the perforin promoter in NK cells, whereas the factors that ensure perforin promoter activity in CD8+ T cells have not yet been defined but probably include Runx3 (12).
The activity of the NF-P motif can also be regulated by the basic-helix-loop-helix, leucine zipper transcription factor MITF (micropthalmia transcription factor) encoded by the mouse mi locus (67). A strain of mice bearing mutant mi alleles synthesizes defective MITF proteins that lack the ability to localize to the nucleus, bind DNA and transactivate certain genes. Homozygous mutant mice demonstrate significantly reduced levels of perforin mRNA and protein in their NK cells. Co-transfection of wildtype MITF could enhance basal perforin promoter activity in CTLL-2 cells via the NF-P motif. Reciprocally, the mutant MITF attenuated its basal activity in those cells. However, these functions did not require detectable MITF binding to the NF-P motif in vitro. Thus, a direct role for MITF in regulating perforin promoter function is still unclear.
Sequences upstream of −807 bp in both the murine and human promoters drive significantly enhanced reporter expression in cytotoxic lymphocyte cell lines. In addition, constructs including sequences up to −1100 bp of the murine promoter are highly killer cell specific and are capable of driving reporter expression after stable transfection of the CTL model cell line CTLL-R8 (68). Furthermore, sequences up to −5100 bp sustain high-level killer cell-specific activity in both stable and transient transfections and almost completely squelch promoter activity in non-killer cells (61). Thus, cis-acting sequences upstream of −800 bp of the Prf1 promoter appear to specifically enhance killer-lymphocyte transcription.
In the context of transgenes, the murine Prf1 promoter and its immediate upstream flank exhibit proper tissue-specific activity. Transgenic mice in which 5100 bp of the murine perforin promoter was used to drive expression of a human CD4 reporter gene showed it functions in normal cells that develop in vivo (11) (Fig. 1). In whole tissues, expression of the transgenic mRNA paralleled mRNA derived from the endogenous mouse perforin gene. At the single cell level, the transgenic promoter became active at the DN stage of thymocyte development and remained expressed in all subsequent stages. In mature cells, it was active in NK cells as well as αβ and γδ T-cell lineages but was inactive in B cells and myeloid lineages. Thus, perforin promoter activity was appropriately restricted to the T and NK cell lineages during development.
In contrast, transgenes driven by the Prf1 promoter were unable to mediate the normal transcriptional regulation that is characteristic of endogenous Prf1 gene. Although the transgenic control regions drove constitutive reporter expression in NK cells, they also did so in unstimulated CD8+ and CD4+ αβT cells and γδ T cells, and unlike expression of the endogenous gene, which differs among these four cell types, basal reporter activity driven by these transgenes was equivalent. In addition, in purified T cells, neither TCR signals nor cytokines could upregulate promoter activity. This does not reflect the normal expression of perforin in these lineages. Furthermore, only a fraction of transgenic lines expressed the reporter gene, and in those, expression of the transgenic mRNA was 5–20-fold less than mRNA from the endogenous Prf1 gene. Therefore, additional distal regulatory regions are essential for normal regulation of Prf1 transcription.
PRF1 transcription is regulated by two enhancers at −15 kb and −1 kb
Analysis of a second series of larger transgenes identified two enhancers in the PRF1 locus that respond to stimulation in CTL (Fig. 1). Two partially overlapping cosmid clones that spanned 45 kb of the PRF1 locus were independently transfected into a rodent cell-culture model of CTLs known as SAM-19, and clones derived from single cells were analyzed (50). Unlike the previously analyzed perforin promoter transgenes, IL-2 and TCR-like signals upregulated PRF1 expression of both cosmid transgenes. The response was localized to the 5′ flank of perforin by virtue of a region that was common to both of the clones. The PRF1 promoter and 16 kb of its upstream sequence (the region common to both cosmid clones) was analyzed for its ability to drive a luciferase reporter gene in transgenic mice (50). This construct drove transgenic luciferase expression in T cells purified from the transgenic mice in response to IL-2 and TCR-like stimulation, similar to the response of the full-length cosmid clones when analyzed in the CTL cell line.
IL-2R-activated signal transduction and activator of transcription 5 (Stat5) factors directly stimulate the −15 kb and −1 kb enhancers (50). Both enhancers are controlled by two identically spaced STAT-like elements. In cotransfection assays, both enhancers are activated by constitutively active STAT5 or blocked by a dominant negative STAT5 via the tandem STAT array. The STAT elements in the −1kb enhancer bind activated STAT5 in nuclear extracts from primary NK cells stimulated with IL-2 (69). Moreover, STAT5 binds to the −1 kb region of Prf1 upon ChIP analysis of activated CD8+ T cells differentiated in high but not low concentrations of IL-2, indicating that STAT5 directly regulates Prf1 in primary cells. Consistent with these experiments, Il2rb or Stat5b-deficient splenocytes from mice demonstrate significantly reduced perforin mRNA (47, 70). Thus, the IL-2Rβ/STAT5 signaling pathway leads directly to expression of the PRF1/Prf1 gene, both in NK cells and in differentiating CTLs.
The STAT elements in the −1kb enhancer also link the perforin gene to additional cytokine signals. Electrophoretic mobility shift assay (EMSA) analysis showed that these STAT elements could bind STAT3 from the NK-like cell line YT (69) or STAT4 following activation of the NK line NKL with IL-12 (71). In nuclear extracts from primary NK cells stimulated with IL-6, the −1 kb enhancer bound STAT1a (69). Although the PRF1 gene can respond to multiple cytokines, STAT5 is likely to be a primary factor regulating the −1 kb enhancer, given the requirement for IL-2R signaling in the development and activation of NK cells, the differentiation of effector CTL during acute viral infection, and the maintenance of memory CTLs.
The −1kb enhancer also includes a conserved NF-κB element, and the NF-κB site is activated in response to signals from IL-2Rβ (43). EMSA analysis indicates that NF-κB activity is induced following IL-2 stimulation of primary NK cells, and supershift experiments indicate that the −1kb enhancer binds NF-κB. When the NF-κB site in the −1 kb enhancer was mutated, the enhancer could not modulate a reporter construct driven by the SV40 promoter in response to IL-2. Thus, IL-2Rβ signals are directly linked to the activation of the perforin gene by virtue of STAT5 and NF-κB.
Additional potential regulatory elements are also present in both the −15kb and −1kb enhancers (50). Both enhancers include activator protein-1 (AP-1) sites and Ets cores, while the −15kb enhancer itself includes an E-box and one binding site for the calcium-regulated and cyclosporine A (CsA)-sensitive transcription factor nuclear factor of activated T cells (NFAT). NFAT binding may be the molecular explanation for the calcium-dependent activation and CsA sensitivity of the −15kb enhancer in T cells from transgenic mice (50). In addition, the −1kb enhancer itself contains multiple Ikaros sites and a CREB consensus site. Therefore, both enhancers posses transcription factor-binding sites that allow them to be activated developmentally in lymphocytes and to be regulated in T cells in response to TCR and cytokine signals upon activation.
However, despite the presence of these two enhancers, the data suggested that additional essential cis-regulatory regions were likely to be missing from the cosmid clone transgenes. Although the relatively large PRF1 transgenes (~35 kb each) were regulated similarly to endogenous Prf1 in T cells upon stimulation, their maximum expression remained significantly below the physiological levels of expression of the endogenous gene. Most notably, the absolute amount of transgenic PRF1 mRNA expression in any one clone was ~5% of that of the endogenous murine Prf1 gene. Furthermore, only a fraction of independent clonal lines expressed the transgenes detectably (MGL, unpublished observations). Therefore, at least in transgenic contexts, additional cis-regulatory regions residing outside the nearly 45 kb analyzed were necessary for physiological expression of PRF1.
Identification and transgenic analysis of the PRF1/Prf1 transcriptional ‘territories’
The overall genetic ‘territory’ that is likely to encompass all cognate cis-regulatory regions controlling any individual gene locus is often very large (hundreds of kilobases). In addition, it is likely that multiple individual cis-regulatory regions cooperatively control their target gene’s promoter, and these regions can be scattered at long-distances relative to each other and to the affiliated gene’s promoter. For example, numerous positive and negative cis-regulatory regions control ‘Th2 cytokine’ gene transcription in effector T cells and are interspersed across more than 120 kb of the murine Il5-Il13-Il4 locus in T cells (72–74). Similarly, cis-regulatory regions that control the Ifng locus are scattered throughout at least 130 kb in the murine locus, up to 200 kb in the human IFNg locus, and additional regulatory regions may yet be discovered (75). In the case of PRF1, because reporter transgenes spanning ~45 kb immediately surrounding the PRF1 promoter exhibited both position effect variegation and reduced levels of expression relative to the endogenous gene, a method to map DHSs at very long-range was developed to identify far distal regulatory regions in PRF1 (62).
DHS maps generated from analysis of 230 kb surrounding PRF1 were compared in multiple cell lines that differed sharply in their expression of ADAMTS-14, PRF1, and PALD (62). This study resolved four groups of DHSs: (i) those that were present in all cell-types (i.e. constitutive/ubiquitous), (ii) those that were present only when ADAMTS-14 was expressed, (iii) those only present when PRF1 was expressed, and (iv) those that were present only when PALD was expressed. By ‘subtracting’ the DHSs that correlated specifically with expression of ADAMTS14 and PALD, an approximately 150 kb territory was identified that consisted of 16 DHSs (DHS 2–19) that correlated with PRF1 expression. Of these, 10 were present only when PRF1 was expressed at high levels. The other 6 DHSs were present in all cell types regardless of gene expression, suggesting they could be the boundaries of an active domain that controls PRF1, because they also flanked the 10 PRF1 ‘specific’ DHSs that correlated with PRF1 expression. Most of the DHSs mapped near or within non-coding sequences that are conserved in mice (Table 1), suggesting they might exhibit conserved regulatory function across species. Remarkably, 10 of the DHSs identified within this 150 kb territory were in regions outside previously analyzed PRF1 transgenes, indicating earlier transgenic studies did not assay most potential regulatory regions.
Table 1. Correspondence of human and murine CNS and DHS.
Conserved non-coding sequences (CNS) were identified based on 70% sequence identity within a sliding window of 100 bp upon alignment of human (hg18) and murine (mm9) genomes, with the human locus serving as the base genome. Only CNS in proximity to DNase I hypersensitive sites (DHS) identified in the human locus are shown and their positions are indicated relative to PRF1 transcription initiation (rel to PRF1). Human PRF1 DHS (hpDHS) that might correspond to murine Prf1 DHS (mpDHS) were placed arbitrarily in the same row (see text and Fig. 3 for explanation). The murine Prf1 CNS shown are the aligned sequences derived from the human CNS shown in the same row to left. Certain DHS sites were not originally reported because they were ‘weak’ (weak) or might have been obscured (?) because of proximity to adjacent strong DHS sites.
| Human PRF1 locus (hg18) | Murine Prf1 locus (mm9) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CNS | rel to PRF1 (bp) | hpDHS | cell types/ locations rel to PRF1(kb) | mpDHS in CD8+ CTL | CNS | rel to Prf1 (bp) | ||||||||||
| start | stop | length (bp) |
identity | start | stop | num | HA(10)A | CTL(10) | YT | Jurkat | num | rel to Prf1 (kb) |
start | stop | start | stop |
| 72101985 | 72102291 | 307 | 73.6% | −69448 | −69754 | 2 | −70 | −70 | −70 | 2b weak | 60736123 | 60736406 | −24459 | −24176 | ||
| 72094479 | 72094892 | 414 | 74.6% | −61942 | −62355 | 3 | −61 | ? | −64 | 2 | −21 | 60740663 | 60741212 | −19919 | −19370 | |
| 72090151 | 72090370 | 220 | 73.2% | −57614 | −57833 | 4 | −55 | −55 | −58 | −58 | multiple alignments outside of Prf1 "territory" | |||||
| 72084426 | 72084535 | 110 | 77.3% | −51889 | −51998 | 5 | −52 | 60748107 | 60748632 | −12475 | −11950 | |||||
| 72082371 | 72082574 | 204 | 70.6% | −49834 | −50037 | 6 | −49 | −49 | 3 | −17 | 60747631 | 60748935 | −12951 | −11647 | ||
| 72069503 | 72069757 | 255 | 69.8% | −36966 | −37220 | 7 | −39 | −44 | multiple alingments outside of Prf1 "territory" | |||||||
| 72068546 | 72068879 | 334 | 69.8% | −36009 | −36342 | 8 | −33 | multiple alingments outside of Prf1 "territory" | ||||||||
| 72053717 | 72053849 | 133 | 82.7% | −21180 | −21312 | 9 | −17 | 60690322 | 60690424 | −70260 | −70158 | |||||
| 72046534 | 72046645 | 112 | 70.5% | −13997 | −14108 | 10 | −14 | 60690298 | 60690453 | −70284 | −70129 | |||||
| 72044275 | 72044543 | 269 | 69.9% | −11738 | −12006 | 11 | −12 | −12 | multiple alignments outside of Prf1 "territory" | |||||||
| 72041314 | 72041461 | 148 | 73.6% | −8777 | −8924 | 11a | −9 | multiple alignments outside of Prf1 "territory" | ||||||||
| 72035980 | 72036227 | 248 | 72.6% | −3443 | −3690 | 12 | −3 | −3 | −4 | 60745304 | 60745821 | −15278 | −14761 | |||
| 72033296 | 72033397 | 102 | 70.6% | −759 | −860 | 13 | −1 | 4 | −0.8 | 60759843 | 60759952 | −739 | −630 | |||
| 72032520 | 72032692 | 173 | 74.6% | 17 | −155 | 13 | promoter | 5,6 | −0.2 | 60760345 | 60760499 | −237 | −83 | |||
| 72029744 | 72029885 | 142 | 71.8% | 2793 | 2652 | 13 | intron 2 | 7 | 3.6 | 60764711 | 60764886 | 4129 | 4304 | |||
| 72025042 | 72025174 | 133 | 71.4% | 7495 | 7363 | 8 | 8.6 | 60798585 | 60798730 | 38003 | 38148 | |||||
| 72018584 | 72018714 | 131 | 67.9% | 13953 | 13823 | 14 | 9 | 11 | 11 | 9 | 11 | 60771866 | 60772093 | 11284 | 11511 | |
| 72004911 | 72005112 | 202 | 76.2% | 27626 | 27425 | 15 | 27 | 27 | 26 | 26 | 60734372 | 60734645 | −26210 | −25937 | ||
| 71998001 | 71998268 | 268 | 78.4% | 34536 | 34269 | 15a | 31 | 60782205 | 60788164 | −26210 | −25937 | |||||
| 71995361 | 71995477 | 117 | 70.1% | 37176 | 37060 | 16 | 38 | 38 | 37 | 10 | 21 | 60782205 | 60788164 | 21623 | 27582 | |
| 71992231 | 71992445 | 215 | 67.9% | 40306 | 40092 | 16a | 41 | 60782205 | 60788164 | 21623 | 27582 | |||||
| 71985295 | 71985464 | 170 | 74.1% | 47242 | 47073 | 17 | 44 | 43 | 43 | 43 | 11 | 37 | 60791890 | 60792320 | 31308 | 31738 |
| 71984316 | 71984571 | 256 | 73.4% | 48221 | 47966 | 17a | 45 | 60792348 | 60792606 | 31766 | 32024 | |||||
| 71983895 | 71984127 | 233 | 73.4% | 48642 | 48410 | 17b | 47 | 60754927 | 60755203 | −5655 | −5379 | |||||
| 71979311 | 71979421 | 111 | 71.2% | 53226 | 53116 | 18 | 52 | 52 | 53 | 54 | 12 | 42 | 60799003 | 60802142 | 38421 | 41560 |
| 71978059 | 71978160 | 102 | 70.6% | 54478 | 54377 | 13 | 47 | 60799003 | 60802142 | 38421 | 41560 | |||||
| 71953183 | 71953293 | 111 | 69.4% | 79354 | 79244 | 19 | 78 | 82 | 82 | 80 | 60819702 | 60820969 | 59120 | 60387 | ||
DHSs that are likely to be the regulatory regions involved in PRF1 gene activation were clarified with a functional approach to ‘visualize’ chromatin remodeling of PRF1 upon its transcriptional induction (4). The DHS pattern of PRF1 was assayed in mouse fibroblasts carrying one copy of human chromosome 10 and then was reanalyzed after the chromosome was transferred by microcell fusion to the rodent CTL-like cells SAM-19. The PRF1 gene was silent in the fibroblast cells, whereas both of its neighboring genes were expressed. Upon chromosome transfer into the CTL-like cells, PRF1 was induced and expressed at physiologically relevant levels, while reciprocally the ADAMTS-14 gene was silenced. The PALD gene was expressed both before and after transfer. Comparison of the DHS pattern in each cell type permitted a functional correlation of DHS formation with expressional reprogramming of PRF1 and each of its neighboring genes. These correlations led to the hypothesis of three distinct ‘transcriptional territories’ (Fig. 2).
Fig. 2. The putative transcriptional ‘territories’ in the PRF1 locus.
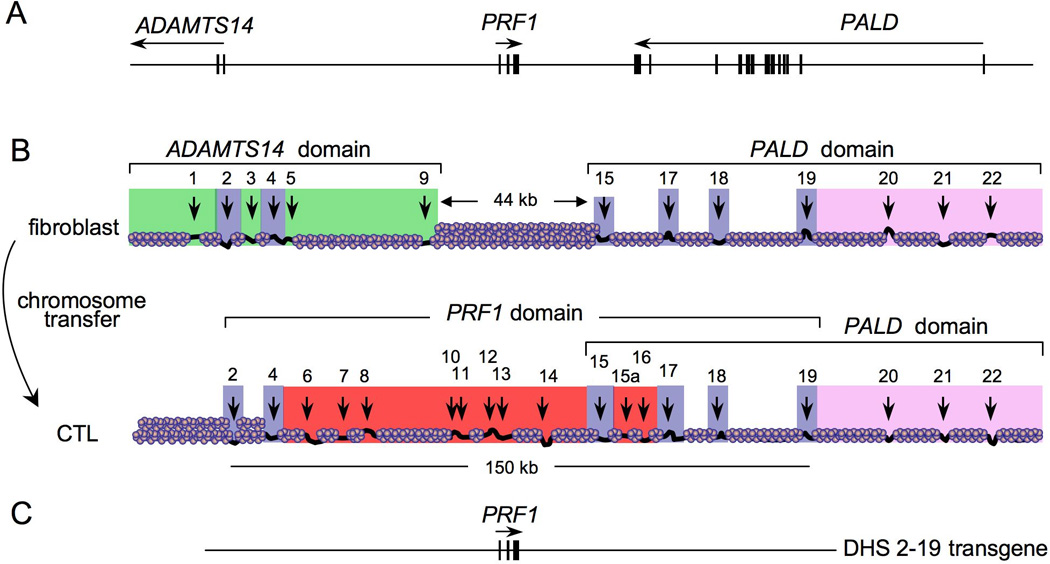
The transcriptional ‘territories’ of PRF1 and its neighboring genes were proposed based upon results from comparative analysis of DHS site formation in different cell-lines and upon reprogramming of the loci during chromosome transfer. (A) A physical map of PRF1 and portions of its neighboring genes. (B) Human chromosome 10 maintained in fibroblasts was transferred by microcell fusion to a recipient CTL line and the chromatin structure was remodeled. Transcriptional territories were deduced based on the reprogramming of DHS sites after transfer. (C) The region of human chromosome 10 analyzed in a BAC transgene denoted DHS 2–19 recapitulated the expression of PRF1 observed from the entire human chromosome 10.
Transgenic analysis of a bacterial artificial chromosome (BAC) from the PRF1 locus that encompassed DHSs 2–19 supported the hypothesis that DHSs 2–19 in the PRF1 locus might constitute an autonomous transcriptional ‘territory’ that regulates PRF1 transcription (Figs 1 and 2). The transgene was specifically expressed in NK cells, activated αβ CD8+ T cells, and γδ T cells, whereas it was not expressed in CD4+ T cells, B cells, or cells in the myeloid lineage (4). All transgenic lines expressed PRF1, were regulated by IL-2 stimulation, and the amount of expression was correlated with the number of transgene copies in each clone. In fact, the amount of expression per individual transgene copy, even from a clone with a single copy integration, was nearly identical to the amount of expression from one allele of the endogenous murine Prf1 locus in the CTL cell line, and comparable to the amount of perforin (mRNA and protein) expressed in purified human CD8+ T cells activated in vitro. Thus, the 150 kb region including DHSs 2–19 drives PRF1 transcription in a physiological manner, arguing that this region contains most or all regulatory regions that normally regulate PRF1.
Comparison of the PRF1 and Prf1 loci
Based on a comparison of overall DHS patterns, transcription of murine Prf1 in CTLs differentiated from naive CD8+ T cells in culture appears to be controlled by a transcriptional territory very similar to that of human PRF1 in SAM19 rodent CTL-like cells carrying one copy of human chromosome 10 (4). Specifically, DHS sites 2–12 in Prf1 appear analogous to DHS sites 2–18 in PRF1, based on the relative positions of the DHSs in each locus and the underlying sequences conserved between each species that are associated geographically with the location of DHS sites. The human and murine perforin loci are schematically compared in Fig. 3, and DHS sites that appear to correspond in each species are indicated in Table 1 along with their associated conserved non-coding sequences (CNS) regions. Depending on cell type and activation condition, additional DHS sites are likely to exist in both the human and murine loci. Indeed, TCR-like restimulation of activated CD8+ T cells from mice induces a number of additional DHS sites (MEP, unpublished observations). It is also possible that certain DHS sites were overlooked during the analyses because of their proximity to the restriction sites used in the experiments. Future work is required to more precisely map the DHSs, to clarify their correlation with expression of perforin and its neighboring genes under different conditions, and to define more stringently the similarities and differences among the regulatory regions in the human and murine perforin genes.
Fig. 3. Comparison of the DHS site patterns of the PRF1 and Prf1 loci.
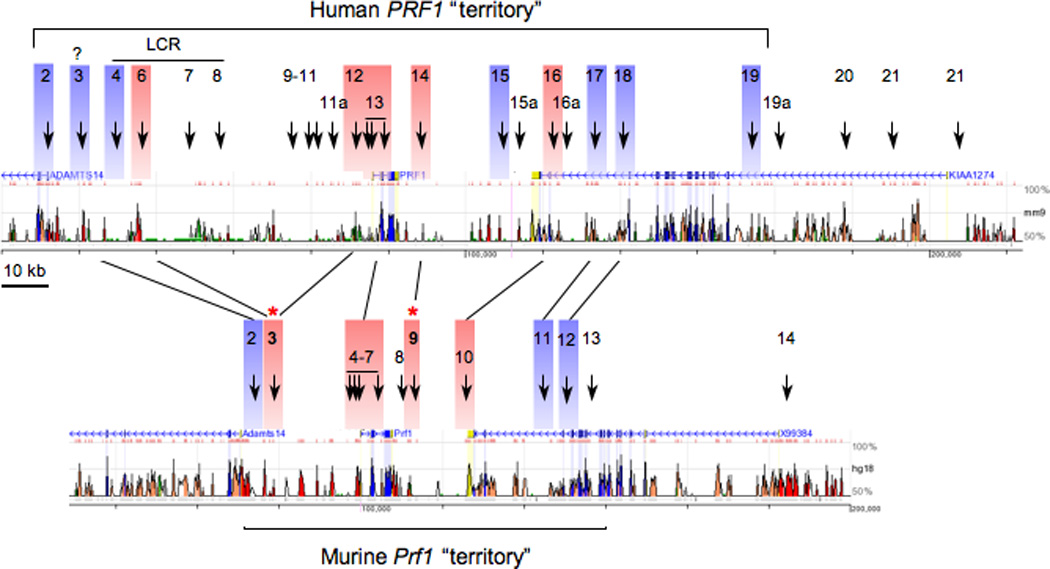
The maps show the relative positions of DHS sites in the human (top) and murine (bottom) loci. Lines indicate DHS sites in the human locus that have been inferred to correspond to DHS sites that form in murine CD8+ CTLs, based upon both CNS in proximity to each DHS site and their relative position in the locus. DHS sites in blue indicate those that are present in most or all cell types, whereas those in red correlate specifically with perforin expression. Bold DHS sites with asterisks indicate CD8+ T-cell-specific sites in murine cells.
The correspondence between murine Prf1 DHS sites (mpDHSs) relative to human PRF1 DHS sites (hpDHSs) is diagrammed in Fig. 3. The relation appears to be (mpDHSs;hpDHSs): (2; 3), (3; 6,12), (4–7; 13), (9; 14), (10; 16), (11; 17) (12; 18), because conserved sequences located near the human DHSs align in proximity to the DHSs mapped in the mouse loci. Notably, the mapped position of each DHS does not always fall exactly within conserved sequences, as also observed when comparing DHS sites in the human and murine interferon-γ gene loci, which were mapped at near base pair resolution (76). In fact, cis-regulatory regions can lack strong sequence conservation yet still retain their function, as found by comparing functions of corresponding enhancers that nevertheless diverge dramatically in their sequence identity between different species of Drosophila (77). Therefore, although some of the conserved sequences associated with particular DHS sites in the human locus do not strictly correlate with the presumably analogous DHS sites in the murine locus, they might still represent the same cis-regulatory region, hypotheses that will need to be tested functionally and validated by ChIP analysis of transcription factor and careful sequence inspection. In summary, additional work is needed to resolve the function of the sequences marked by individual DHS sites in the perforin loci of mice and human.
The known and hypothetical function of DHSs in the perforin locus
A current simplified view of the function of different DHS sites in the PRF1 locus is that hpDHSs 2 and 4 on the distal 5′ end of PRF1, and hpDHSs 15, 17, 18, and 19 on the distal 3′ end of PRF1, act as either boundaries or as constitutive regulators of the locus, because they are present ubiquitously, regardless of whether PRF1 or its neighboring genes are expressed (4, 62). The remaining hpDHSs between these putative boundaries, which correlate specifically with PRF1 expression, are presumed to promote the transcription of PRF1. At least some of these latter hpDHSs are indeed transcriptional enhancers (i.e. −1 kb and −15 kb regions) (50). The hpDHS 13 is a very broad site that most likely encompasses at least the −1 kb enhancer, the promoter, and a potential DHS site in intron 2. A similar logic is applied to the murine Prf1 locus based on the arrangement of DHS sites, with the important extension that mpDHSs 3 and 9 (noted in Fig. 3 by asterisks) are specific to CD8+ CTLs, because they are not formed in primary CD4+ T cells after 1 week of differentiation into Th1 effector cells (4). Notably, in murine cells, CD8+ CTLs expressed greater than 20-fold more perforin mRNA than the Th1 cells. The correlation of mpDHS 3 with high expression of perforin in murine CD8+ CTLs also supports its functional correspondence with hpDHSs 6 (or potentially hpDHS 12 based on sequence conservation), which were specifically present in NK and CTL-like cells, relative to CD4+ like Jurkat T cells, and were necessary for physiological levels of transgenic PRF1 expression (4, 62). The specific functions of mpDHSs are currently under investigation.
The locus control region of PRF1
The distal 5′ flanking regions of both the murine and human perforin genes are likely to be essential for physiological expression. Reporter transgenes driven by the PRF1 promoter on its own, or in the context of large (35 kb) cosmid clone transgenes, conferred cell type specificity and activation responsiveness but did not mediate copy number-dependent expression and were ultimately silenced in all transgenic clones. In contrast, the 150 kb ‘DHS 2–19’ PRF1 BAC transgene mediated copy number-dependent expression at all integration sites, indicating that this larger transgene contains a LCR (4). LCRs are unique cis-acting domains that confer copy number-dependent expression of linked transgenes at all sites of integration (78).
The probable location of the LCR in the PRF1 locus was discovered by comparison of the PRF1 DHS pattern in cells that expressed high versus low amounts of perforin mRNA. Jurkat T cells express basal amounts of perforin mRNA, 20–100 fold lower than the amounts expressed in the CTL or YT cell lines (4, 62), and manifest some but not all PRF1-specific DHSs that are present in the CTL-like cells and YT cells. Four of these DHSs in particular (DHS 4–8) are in the distal 5′ flank of PRF1, a region that was not within the cosmid clone transgenes previously analyzed (50). This far upstream region is required for physiological perforin expression in both chromosomal and transgenic contexts (4).
In further support of the hypothesis that this distal region contains an LCR, a spontaneously fragmented transchromosome that retained all centromeric sequences but had lost all chromosomal sequence upstream (telomeric) of −15 kb in PRF1, did not support PRF1 expression after chromosome transfer into the CTL cell line (4). More specifically, engineered deletion of DHSs 4–8 from the DHS 2–19 BAC transgene reduced PRF1 transcription dramatically, eliminated copy-number dependent expression, and ultimately resulted in the transgenes becoming silenced. Thus, regulatory sequences upstream of −15 kb are necessary for PRF1 transcription in its chromosomal context, and DHSs 4–8 include sequences that are required for LCR activity in transgenes.
The basic functional components of the PRF1 LCR have not yet been thoroughly delineated. However, individual fragments containing conserved sequences corresponding to DHSs 4–8 each confer some degree of enhanced transcriptional activity to a 240 bp minimal PRF1 promoter in cloned reporter constructs, indicating that they might have a transcriptional function specific to PRF1 (MEP and MGL, unpublished observations). DHSs 4 and 6 had the strongest effect when assayed in conjunction and so were tested together for their ability to drive transcription after stable integration. The minimal PRF1 promoter linked to DHSs 4 and 6 drives reporter expression in all derived clones, unlike transgenes driven only by the PRF1 promoter that are not expressed (MEP and MGL, unpublished observations). Thus, conserved regions proximal to DHS 4 and 6 drive integration site-independent expression of PRF1, fulfilling at least one criterion for an LCR. It will be important to continue to dissect the regulatory regions within the area required for LCR activity to determine which sequences are necessary and sufficient for LCR function and to determine the transcription factors and chromatin-modifying enzymes that mediate LCR activity.
It is a reasonable hypothesis that mpDHS 3 is a component of the LCR in the murine Prf1 locus. Its location generally correlates with the location of hpDHS 6, although the sequence conservation of hpDHS 6 aligns approximately 4 to 5 kb downstream from the location of the mpDHS in the murine genome (Table 1). In addition, mpDHS 3 is developmentally specific to CD8+ CTLs relative to CD4+ Th1 effector cells, which argues that it is specifically active in cells that express physiologically high amounts of perforin. Furthermore, secondary TCR-like stimulation of previously primed naïve murine CD8+ T cells induces de novo DHSs in regions in the vicinity of mpDHS 3, suggesting that this region has additional regulatory qualities that have not yet been identified (MEP, unpublished observations). In addition, it is possible that mpDHS 2 and its potential human counterpart (hpDHS 3) could be affiliated with the LCR. In murine CD8+ T cells, the region near mpDHS 2 strongly associates with the transcription factor Runx3, a protein that is absolutely required for perforin mRNA expression in activated CD8+ T cells from mice (12). Thus, mpDHS 2 and 3 might be core domains in the murine Prf1 LCR, and future transgenic experiments will be essential to dissect the specific function of the sequences near mpDHS 2 and 3.
Regulation of the Prf1 locus in response to IL-2R signaling
Strong IL-2R signaling in activated CD8+ T cells leads to induction of perforin expression. Because DHS sites 3 and 9 were found to form specifically in CTLs expressing large amounts of perforin (relative to Th1 cells), it was presumed that they might only be active (i.e. DNase I hypersensitive) in stimulated CD8+ T cells that express high amounts of perforin, as opposed to those that do not. The large difference in perforin expression in TCR-activated CD8+ T cells in response to IL-2 was used to investigate whether the DHS site pattern of Prf1 also diverged in response to IL-2 (49). However, essentially the same pattern of DHS sites was found in activated CD8+ T cells irrespective of the concentration of IL-2 the cells had experienced during differentiation or the amount of perforin they expressed. This unexpected result suggested that the high-level expression of perforin in cells cultured in high IL-2 could not be explained simply by long-range chromatin remodeling of the Prf1 locus. Furthermore, it indicated that DHS 3 and DHS 9 might be formed developmentally in all CD8+ T cells or be induced early after CD8+ T-cell activation.
Increased IL-2R signaling leading to induced perforin expression by activated CD8+ T cells was found to correlate with a dramatic increase in RNA polymerase II (RNA Pol II) loading at the Prf1 promoter (49) (Fig. 4). Thus, a major function of IL-2 during CTL differentiation appears to involve enhancing transcription initiation. Although RNA Pol II density also increased slightly over the transcribed portion of Prf1, the majority of Pol II remained restricted to the transcription start site, suggesting that IL-2 mainly affected RNA Pol II recruitment rather than RNA Pol II elongation. This interpretation is consistent with earlier studies that used nuclear run-on assays to examine how Prf1 is induced transcriptionally in nuclei prepared from CTL cell lines activated with IL-2 (50). Notably, the recruitment of RNA Pol II to the gene was coordinated with the binding of the transcription factors Stat5 and Eomesdermin to the −1 kb enhancer and promoter, respectively. This observation suggests that one of the important functions of the −1 kb enhancer is to increase the rate of transcription initiation in response to signals from the IL-2R. It is still unclear how the CTL-specific DHS sites 3 and 9 participate in this process.
Fig. 4. Model of Prf1 gene activation in stimulated CD8 T cells.
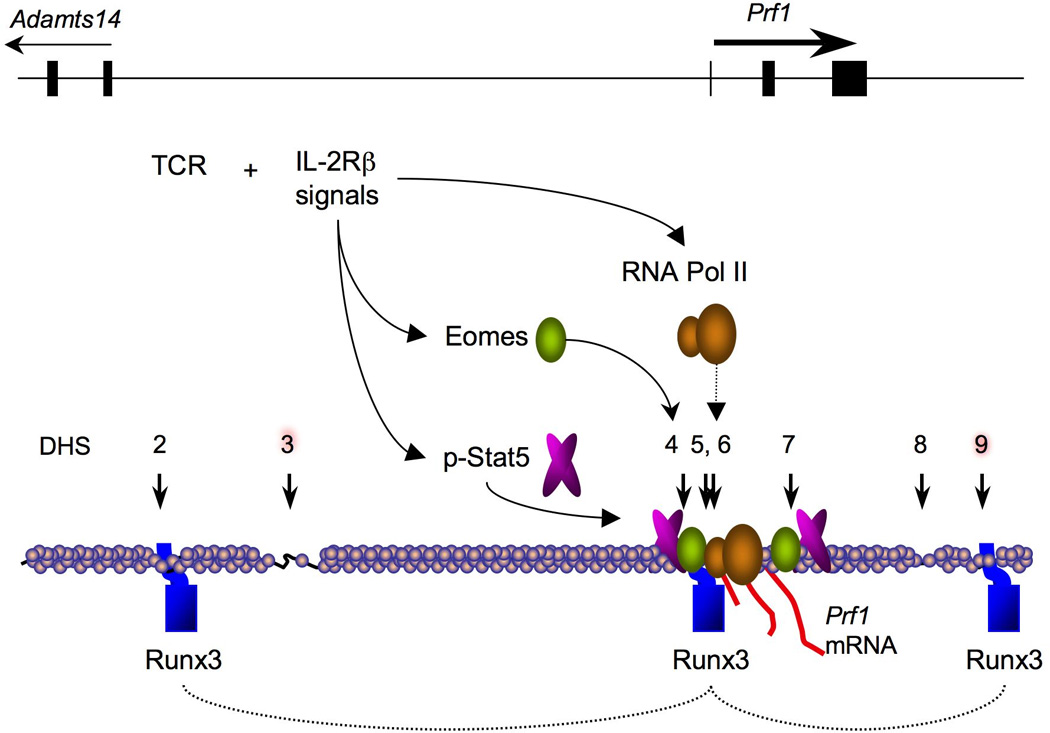
The schematic depicts some key events during perforin gene activation in activated CD8+ T cells. Runx3 transcription factors (blue) are bound to several cis-regulatory regions near DHS sites (vertical arrows) and might function to bring these distal sites into proximity with the promoter, and this could be established in the thymus. CTL-specific DHS sites are highlighted in red. After activation of naive CD8+ T cells with antigen and persistent IL-2R signaling, p-Stat5 and Eomes are induced and both bind the −1 kb enhancer and promoter region (indicated with curved arrows) as well as an intronic DHS site. Binding of p-Stat5 and Eomes is coordinated with increased recruitment of RNA Pol II (dashed arrow) to the Prf1 transcription start site and increased perforin mRNA expression (red lines) in response to IL-2.
Transcription factor networks that regulate CTL differentiation and the PRF1 locus
Since the most recent review of transcription factors that control the cytotoxic program in lymphocytes (79), additional important insights have been gained. Now that most potential cis-regulatory regions in the PRF1/Prf1 genes have been identified, we can begin to examine whether transcription factors previously implicated in driving cytotoxic lymphocyte differentiation do so by acting directly at the Prf1 gene. We now know that Prf1 transcription is controlled by sequential activation of a network of transcription factors during development and activation of CD8+ T cells. In addition, most previously identified factors have not been shown to bind near the newly discovered DHS sites, especially those that appear to be CTL-specific. Thus, additional transcription factors that have not been implicated in the differentiation of CTLs are very likely to be important for perforin expression.
The transcription factor Runx3 is necessary for perforin expression and normal CTL differentiation (12). Although only a few mature single positive CD8+ T cells develop in the absence of Runx3, perforin mRNA expression was nearly undetectable by Northern blotting in cells activated with TCR signals and high concentrations of IL-2 (12). Thus, Runx3 is required for perforin gene expression in activated CD8+ T cells. Interestingly, Runx3 mRNA expression closely parallels expression of perforin message in DP thymocytes undergoing positive selection into the CD8 lineage (10), whereas both transcripts are downregulated in cells inducing the CD4+ lineage fate.
It is not yet known how Runx3 induces the Prf1 gene; however, it appears to serve at least two roles. First, ChIP analysis revealed that Runx3 directly binds several regulatory regions in the Prf1 locus, namely DHS 2, 6 and 9. Second, Runx3 is necessary for the induction of another factor, the T-box transcription factor Eomesodermin (Eomes), which also binds to the Prf1 locus and contributes to perforin expression (12, 80, 81). However, overexpression of Eomes is unable to restore perforin expression in the absence of Runx3, indicating that Runx3 might be required for Eomes to function at the Prf1 gene (12, 81). Thus, a genetic pathway involving Runx3 induces Eomes expression, and both factors then ‘feed forward’ to activate the Prf1 gene and drive CTL differentiation.
Given the correlation between expression of Runx3 and perforin during thymocyte development, it is reasonable to postulate that Runx3 might initially be required to configure the PRF1/Prf1 loci into a permissive conformation prior to commitment to full gene activation. Future experiments will be necessary to address how Runx3 acts at the Prf1 locus to establish the locus for high level transcription. However, given that Runx3 binds primarily at three DHS in the locus, which are located at the 5′ and 3′ ends of the locus, and promoter, respectively, it is interesting to speculate that Runx3 might serve as a tether that apposes distal cis-acting sequences adjacent to the Prf1 promoter. Indeed thus far, Eomes seems to mainly bind to the promoter region in ChIP assays, suggesting that its function might depend on the proximity of the additional distal cis-acting domains to induce Prf1 transcription. The two CTL specific DHS sites 3 and 9 seem to be good candidates for participating in this process.
The two T-box factors T-bet and Eomes both drive the differentiation of effector CTL, although each is likely to have distinct functions. Gene knockout studies have shown that perforin expression is strongly reduced in the absence of both factors (80). However, Eomes, rather than T-bet, appears to be the primary inducer of Prf1 transcription in activated naive CD8+ T cells. First, T-bet-deficient CD8+ T cells induce normal amounts of perforin mRNA expression with normal kinetics upon activation in cell culture (12). Second, IL-2 signals strongly induce Eomes but not T-bet expression, and this correlates closely with the upregulation of perforin expression in response to IL-2 (49). Third, Runx3 is essential for both Eomes and perforin expression, but Runx3-deficient cells show no difference in the level of T-bet expression (13). Although T-bet-deficient CD8+ T cells activated by viral infection in vivo do show reduced perforin mRNA expression, they also exhibit reduced expression of IL-2Rβ, unlike cells activated in culture (49, 80, 82). It is clear that IL-2R signals are important for upregulating Eomes in TCR-stimulated CD8+ T cells, and it is known that both T-bet and Eomes can upregulate IL-2Rβ expression. Consequently, T-bet deficiency could indirectly impair perforin expression by reducing IL-2R signaling in activated CD8+ T cells in vivo. Thus, it appears that T-bet and Eomes might also operate in a feed forward loop in CD8+ T cells to promote perforin expression and CTL differentiation.
Concluding remarks
Perforin expression is essential for immune system homeostasis, tumor immunosurveillance, and responses to intracellular microbes. Its importance has been demonstrated not only in experimental animals but also more poignantly in an often fatal uncontrolled inflammatory response in humans, known as familial hemophagocytic lymphohistiocytosis (FHL). FHL can be genetically linked to at least four independent loci, but inherited mutations in the PRF1 locus and attendant perforin deficiency causes FHL in a large fraction of cases (83, 84). Most PRF1-linked FHL cases are due to mutations identified in the transcribed region of PRF1 that cause a lack of protein expression or defective biological activity. However, some PRF1-linked FHL cases have been identified that do not possess any coding or splice-site junction mutations, leaving the reason for perforin deficiency in question (85). A reasonable hypothesis is that distal defects exist in these patients, perhaps in essential cis-regulatory regions that affect PRF1 transcription (4). Indeed, the degree of perforin deficiency or its biological activity correlates with disease onset or severity in FHL (84), suggesting that the mechanisms which regulate perforin expression levels can be critical. From this perspective, achieving concise understanding of perforin transcriptional (and posttranscriptional) regulatory mechanisms is a crucial piece of knowledge in human biology, and might perhaps lead to gene therapeutic strategies to restore defective perforin expression in PRF1-null FHL patients.
Investigating perforin gene regulation is an ideal strategy to gain insight into understanding cytotoxic lymphocyte differentiation and activation. The analysis of the transcriptional control of genes specific to NK cells and CD8+ CTLs has lagged behind the surging interest in the specification of different effector CD4+ T-cell ‘lineages’ and the control of cytokine gene loci (13). However, identification of the locus-wide set of cis-regulatory regions that control perforin transcription is a strong basis to accelerate future identification of the transcription factors and regulatory mechanisms that control perforin and the overall cytotoxic gene program in lymphocytes. A complete understanding of how perforin is regulated will likely be integral information for designing rational immunotherapeutic and vaccination strategies to generate immune protection against tumors and infections.
Acknowledgements
We thank Dr. Fernando Cruz-Guilloty and members of both the Rao and Lichtenheld laboratories for helpful discussions. This work was funded by NIH grants AI44432 and AI707088 (to A.R.), the University of Miami Developmental Center for AIDS Research (D-CFAR): 5P30AI073961-02 (to M.G.L.), and the National Cancer Institute F32CA126247 (to M.E.P).
References
- 1.Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- 2.Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol. 2007;19:301–308. doi: 10.1016/j.coi.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pipkin ME, et al. Chromosome transfer activates and delineates a locus control region for perforin. Immunity. 2007;26:29–41. doi: 10.1016/j.immuni.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev. 2006;214:56–72. doi: 10.1111/j.1600-065X.2006.00451.x. [DOI] [PubMed] [Google Scholar]
- 6.Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol. 2007;7:703–714. doi: 10.1038/nri2154. [DOI] [PubMed] [Google Scholar]
- 7.Jacobs R, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol. 2001;31:3121–3127. doi: 10.1002/1521-4141(2001010)31:10<3121::aid-immu3121>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Di Santo JP. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol. 2001;31:1900–1909. doi: 10.1002/1521-4141(200106)31:6<1900::aid-immu1900>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, et al. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol. 2002;3:523–528. doi: 10.1038/ni796. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Taylor BJ, Sun G, Bosselut R. Analyzing expression of perforin, Runx3, and Thpok genes during positive selection reveals activation of CD8-differentiation programs by MHC II-signaled thymocytes. J Immunol. 2005;175:4465–4474. doi: 10.4049/jimmunol.175.7.4465. [DOI] [PubMed] [Google Scholar]
- 11.Lichtenheld MG, Podack ER, Levy RB. Transgenic control of perforin gene expression. Functional evidence for two separate control regions. J Immunol. 1995;154:2153–2163. [PubMed] [Google Scholar]
- 12.Cruz-Guilloty F, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. 2009;206:51–59. doi: 10.1084/jem.20081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pipkin ME, Rao A. SnapShot: effector and memory T cell differentiation. Cell. 2009;138:606 e1–606 e2. doi: 10.1016/j.cell.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 15.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rufer N, et al. Ex vivo characterization of human CD8+ T subsets with distinct replicative history and partial effector functions. Blood. 2003;102:1779–1787. doi: 10.1182/blood-2003-02-0420. [DOI] [PubMed] [Google Scholar]
- 17.Hamann D, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 19.Kelso A, Costelloe EO, Johnson BJ, Groves P, Buttigieg K, Fitzpatrick DR. The genes for perforin, granzymes A-C and IFN-gamma are differentially expressed in single CD8(+) T cells during primary activation. Int Immunol. 2002;14:605–613. doi: 10.1093/intimm/dxf028. [DOI] [PubMed] [Google Scholar]
- 20.Tomiyama H, Matsuda T, Takiguchi M. Differentiation of human CD8(+) T cells from a memory to memory/effector phenotype. J Immunol. 2002;168:5538–5550. doi: 10.4049/jimmunol.168.11.5538. [DOI] [PubMed] [Google Scholar]
- 21.Roos MT, et al. Changes in the composition of circulating CD8+ T cell subsets during acute epstein-barr and human immunodeficiency virus infections in humans. J Infect Dis. 2000;182:451–458. doi: 10.1086/315737. [DOI] [PubMed] [Google Scholar]
- 22.Takata H, Takiguchi M. Three memory subsets of human CD8+ T cells differently expressing three cytolytic effector molecules. J Immunol. 2006;177:4330–4340. doi: 10.4049/jimmunol.177.7.4330. [DOI] [PubMed] [Google Scholar]
- 23.Johnson BJ, et al. Single-cell perforin and granzyme expression reveals the anatomical localization of effector CD8+ T cells in influenza virus-infected mice. Proc Natl Acad Sci USA. 2003;100:2657–2662. doi: 10.1073/pnas.0538056100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peixoto A, et al. CD8 single-cell gene coexpression reveals three different effector types present at distinct phases of the immune response. J Exp Med. 2007;204:1193–1205. doi: 10.1084/jem.20062349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaiss DM, Sijts AJ, Mosmann TR. Enumeration of cytotoxic CD8 T cells ex vivo during the response to Listeria monocytogenes infection. Infect Immun. 2008;76:4609–4614. doi: 10.1128/IAI.00563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Rosa SC, Andrus JP, Perfetto SP, Mantovani JJ, Herzenberg LA, Roederer M. Ontogeny of gamma delta T cells in humans. J Immunol. 2004;172:1637–1645. doi: 10.4049/jimmunol.172.3.1637. [DOI] [PubMed] [Google Scholar]
- 27.Smyth MJ, et al. Interleukin 2 induction of pore-forming protein gene expression in human peripheral blood CD8+ T cells. J Exp Med. 1990;171:1269–1281. doi: 10.1084/jem.171.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol. 2002;2:336–345. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 29.Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol. 2006;177:2888–2898. doi: 10.4049/jimmunol.177.5.2888. [DOI] [PubMed] [Google Scholar]
- 30.Brown DM, Kamperschroer C, Dilzer AM, Roberts DM, Swain SL. IL-2 and antigen dose differentially regulate perforin- and FasL-mediated cytolytic activity in antigen specific CD4+ T cells. Cell Immunol. 2009;257:69–79. doi: 10.1016/j.cellimm.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Gumperz JE, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med. 2002;195:625–636. doi: 10.1084/jem.20011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Assarsson E, et al. CD8+ T cells rapidly acquire NK1.1 and NK cell-associated molecules upon stimulation in vitro and in vivo. J Immunol. 2000;165:3673–3679. doi: 10.4049/jimmunol.165.7.3673. [DOI] [PubMed] [Google Scholar]
- 34.Kambayashi T, et al. Emergence of CD8+ T cells expressing NK cell receptors in influenza A virus-infected mice. J Immunol. 2000;165:4964–4969. doi: 10.4049/jimmunol.165.9.4964. [DOI] [PubMed] [Google Scholar]
- 35.Ohkawa T, et al. Systematic characterization of human CD8+ T cells with natural killer cell markers in comparison with natural killer cells and normal CD8+ T cells. Immunology. 2001;103:281–290. doi: 10.1046/j.1365-2567.2001.01248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakata M, et al. Expression of perforin and cytolytic potential of human peripheral blood lymphocyte subpopulations. Int Immunol. 1992;4:1049–1054. doi: 10.1093/intimm/4.9.1049. [DOI] [PubMed] [Google Scholar]
- 37.Berthou C, et al. Cord blood T lymphocytes lack constitutive perforin expression in contrast to adult peripheral blood T lymphocytes. Blood. 1995;85:1540–1546. [PubMed] [Google Scholar]
- 38.Salcedo TW, Azzoni L, Wolf SF, Perussia B. Modulation of perforin and granzyme messenger RNA expression in human natural killer cells. J Immunol. 1993;151:2511–2520. [PubMed] [Google Scholar]
- 39.Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol. 2004;172:2048–2058. doi: 10.4049/jimmunol.172.4.2048. [DOI] [PubMed] [Google Scholar]
- 40.Gaddy J, Broxmeyer HE. Cord blood CD16+56- cells with low lytic activity are possible precursors of mature natural killer cells. Cell Immunol. 1997;180:132–142. doi: 10.1006/cimm.1997.1175. [DOI] [PubMed] [Google Scholar]
- 41.Gamero AM, Ussery D, Reintgen DS, Puleo CA, Djeu JY. Interleukin 15 induction of lymphokine-activated killer cell function against autologous tumor cells in melanoma patient lymphocytes by a CD18-dependent, perforin-related mechanism. Cancer Res. 1995;55:4988–4994. [PubMed] [Google Scholar]
- 42.Smyth MJ, Ortaldo JR. Comparison of the effect of IL-2 and IL-6 on the lytic activity of purified human peripheral blood large granular lymphocytes. J Immunol. 1991;146:1380–1384. [PubMed] [Google Scholar]
- 43.Zhou J, Zhang J, Lichtenheld MG, Meadows GG. A role for NF-kappa B activation in perforin expression of NK cells upon IL-2 receptor signaling. J Immunol. 2002;169:1319–1325. doi: 10.4049/jimmunol.169.3.1319. [DOI] [PubMed] [Google Scholar]
- 44.Kawasaki A, Shinkai Y, Yagita H, Okumura K. Expression of perforin in murine natural killer cells and cytotoxic T lymphocytes in vivo. Eur J Immunol. 1992;22:1215–1219. doi: 10.1002/eji.1830220516. [DOI] [PubMed] [Google Scholar]
- 45.Fehniger TA, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 46.Liu CC, Rafii S, Granelli-Piperno A, Trapani JA, Young JD. Perforin and serine esterase gene expression in stimulated human T cells. Kinetics, mitogen requirements, and effects of cyclosporin A. J Exp Med. 1989;170:2105–2118. doi: 10.1084/jem.170.6.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malek TR, Yu A, Scibelli P, Lichtenheld MG, Codias EK. Broad programming by IL-2 receptor signaling for extended growth to multiple cytokines and functional maturation of antigen-activated T cells. J Immunol. 2001;166:1675–1683. doi: 10.4049/jimmunol.166.3.1675. [DOI] [PubMed] [Google Scholar]
- 48.Alves NL, Hooibrink B, Arosa FA, van Lier RA. IL-15 induces antigen-independent expansion and differentiation of human naive CD8+ T cells in vitro. Blood. 2003;102:2541–2546. doi: 10.1182/blood-2003-01-0183. [DOI] [PubMed] [Google Scholar]
- 49.Pipkin MG, Sacks J, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. IL-2 and inflammation induce distinct transcriptional programs that promote the differentiation of CD8+ T cells into effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Scordi I, Smyth MJ, Lichtenheld MG. Interleukin 2 receptor signaling regulates the perforin gene through signal transducer and activator of transcription (Stat)5 activation of two enhancers. J Exp Med. 1999;190:1297–1308. doi: 10.1084/jem.190.9.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu P, Garcia-Sanz JA, Lichtenheld MG, Podack ER. Perforin expression in human peripheral blood mononuclear cells. Definition of an IL-2-independent pathway of perforin induction in CD8+ T cells. J Immunol. 1992;148:3354–3360. [PubMed] [Google Scholar]
- 52.Janas ML, Groves P, Kienzle N, Kelso A. IL-2 regulates perforin and granzyme gene expression in CD8+ T cells independently of its effects on survival and proliferation. J Immunol. 2005;175:8003–8010. doi: 10.4049/jimmunol.175.12.8003. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Sanz JA, Podack ER. Regulation of perforin gene expression in a T cell hybrid with inducible cytolytic activity. Eur J Immunol. 1993;23:1877–1883. doi: 10.1002/eji.1830230822. [DOI] [PubMed] [Google Scholar]
- 54.Liu K, Catalfamo M, Li Y, Henkart PA, Weng NP. IL-15 mimics T cell receptor crosslinking in the induction of cellular proliferation, gene expression, and cytotoxicity in CD8+ memory T cells. Proc Natl Acad Sci USA. 2002;99:6192–6197. doi: 10.1073/pnas.092675799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smyth MJ, Ortaldo JR, Bere W, Yagita H, Okumura K, Young HA. IL-2 and IL-6 synergize to augment the pore-forming protein gene expression and cytotoxic potential of human peripheral blood T cells. J Immunol. 1990;145:1159–1166. [PubMed] [Google Scholar]
- 56.Smyth MJ, Norihisa Y, Gerard JR, Young HA, Ortaldo JR. IL-7 regulation of cytotoxic lymphocytes: pore-forming protein gene expression, interferon-gamma production, and cytotoxicity of human peripheral blood lymphocytes subsets. Cell Immunol. 1991;138:390–403. doi: 10.1016/0008-8749(91)90163-6. [DOI] [PubMed] [Google Scholar]
- 57.Smyth MJ, Strobl SL, Young HA, Ortaldo JR, Ochoa AC. Regulation of lymphokine-activated killer activity and pore-forming protein gene expression in human peripheral blood CD8+ T lymphocytes. Inhibition by transforming growth factor-beta. J Immunol. 1991;146:3289–3297. [PubMed] [Google Scholar]
- 58.Uellner R, et al. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J. 1997;16:7287–7296. doi: 10.1093/emboj/16.24.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fink TM, Zimmer M, Weitz S, Tschopp J, Jenne DE, Lichter P. Human perforin (PRF1) maps to 10q22, a region that is syntenic with mouse chromosome 10. Genomics. 1992;13:1300–1302. doi: 10.1016/0888-7543(92)90050-3. [DOI] [PubMed] [Google Scholar]
- 60.Lichtenheld MG, Podack ER. Structure of the human perforin gene. A simple gene organization with interesting potential regulatory sequences. J Immunol. 1989;143:4267–4274. [PubMed] [Google Scholar]
- 61.Lichtenheld MG, Podack ER. Structure and function of the murine perforin promoter and upstream region. Reciprocal gene activation or silencing in perforin positive and negative cells. J Immunol. 1992;149:2619–2626. [PubMed] [Google Scholar]
- 62.Pipkin ME, Lichtenheld MG. A Reliable Method for Mapping DNaseI Hypersensitive Sites in Locus-Wide Domains. Nucl Acids Res. 2006;34:e34. doi: 10.1093/nar/gkl006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Youn BS, Kim KK, Kwon BS. A critical role of Sp1- and Ets-related transcription factors in maintaining CTL-specific expression of the mouse perforin gene. J Immunol. 1996;157:3499–3509. [PubMed] [Google Scholar]
- 64.Zhang Y, Lichtenheld MG. Non-killer cell-specific transcription factors silence the perforin promoter. J Immunol. 1997;158:1734–1741. [PubMed] [Google Scholar]
- 65.Koizumi H, et al. Identification of a killer cell-specific regulatory element of the mouse perforin gene: an Ets-binding site-homologous motif that interacts with Ets-related proteins. Mol Cell Biol. 1993;13:6690–6701. doi: 10.1128/mcb.13.11.6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lacorazza HD, et al. The ETS protein MEF plays a critical role in perforin gene expression and the development of natural killer and NK-T cells. Immunity. 2002;17:437–449. doi: 10.1016/s1074-7613(02)00422-3. [DOI] [PubMed] [Google Scholar]
- 67.Ito A, Kataoka TR, Kim DK, Koma Y, Lee YM, Kitamura Y. Inhibitory effect on natural killer activity of microphthalmia transcription factor encoded by the mutant mi allele of mice. Blood. 2001;97:2075–2083. doi: 10.1182/blood.v97.7.2075. [DOI] [PubMed] [Google Scholar]
- 68.Trapani JA, Dupont B. Novel putative promoter/enhancer sequences are shared by the mouse and human perforin (Pfp) genes. Tissue Antigens. 1990;36:228–234. doi: 10.1111/j.1399-0039.1990.tb01833.x. [DOI] [PubMed] [Google Scholar]
- 69.Yu CR, Ortaldo JR, Curiel RE, Young HA, Anderson SK, Gosselin P. Role of a STAT binding site in the regulation of the human perforin promoter. J Immunol. 1999;162:2785–2790. [PubMed] [Google Scholar]
- 70.Imada K, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. 1998;188:2067–2074. doi: 10.1084/jem.188.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto K, Shibata F, Miyasaka N, Miura O. The human perforin gene is a direct target of STAT4 activated by IL-12 in NK cells. Biochem Biophys Res Commun. 2002;297:1245–1252. doi: 10.1016/s0006-291x(02)02378-1. [DOI] [PubMed] [Google Scholar]
- 72.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 73.Fields PE, Lee GR, Kim ST, Bartsevich VV, Flavell RA. Th2-specific chromatin remodeling and enhancer activity in the Th2 cytokine locus control region. Immunity. 2004;21:865–876. doi: 10.1016/j.immuni.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 74.Lee GR, Fields PE, Griffin TJ, Flavell RA. Regulation of the Th2 cytokine locus by a locus control region. Immunity. 2003;19:145–153. doi: 10.1016/s1074-7613(03)00179-1. [DOI] [PubMed] [Google Scholar]
- 75.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 76.Schoenborn JR, et al. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol. 2007;8:732–742. doi: 10.1038/ni1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ho MC, et al. Functional evolution of cis-regulatory modules at a homeotic gene in Drosophila. PLoS Genet. 2009;5:e1000709. doi: 10.1371/journal.pgen.1000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987;51:975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 79.Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat Rev Immunol. 2004;4:900–911. doi: 10.1038/nri1490. [DOI] [PubMed] [Google Scholar]
- 80.Intlekofer AM, et al. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science. 2008;321:408–411. doi: 10.1126/science.1159806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pearce EL, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 82.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci USA. 2009;106:9809–9814. doi: 10.1073/pnas.0903815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 85.Stepp SE, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. [PubMed] [Google Scholar]