

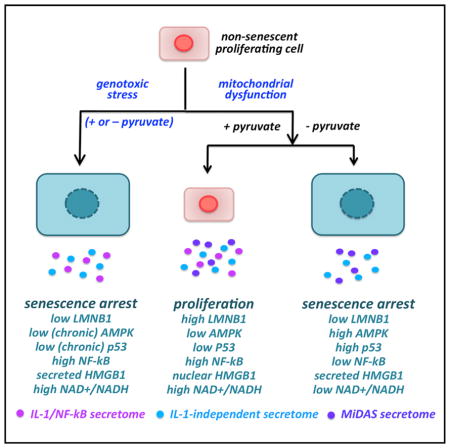
SUMMARY
Cellular senescence permanently arrests cell proliferation, often accompanied by a multi-faceted senescence-associated secretory phenotype (SASP). Loss of mitochondrial function can drive age-related declines in the function of many post-mitotic tissues, but little is known about how mitochondrial dysfunction affects mitotic tissues. We show here that several manipulations that compromise mitochondrial function in proliferating human cells induce a senescence growth arrest with a modified SASP that lacks the IL-1-dependent inflammatory arm. Cells that underwent mitochondrial dysfunction-associated senescence (MiDAS) had lower NAD+/NADH ratios, which caused both the growth arrest and prevented the IL-1-associated SASP through AMPK-mediated p53 activation. Progeroid mice that rapidly accrue mtDNA mutations accumulated senescent cells with a MiDAS SASP in vivo, which suppressed adipogenesis and stimulated keratinocyte differentiation in cell culture. Our data identify a distinct senescence response and provide a mechanism by which mitochondrial dysfunction can drive aging phenotypes.
Graphical Abstract
INTRODUCTION
Age is the largest risk factor for myriad pathologies, ranging from neurodegeneration to cancer. These pathologies likely arise from a loss of tissue homeostasis driven by one or more basic aging process, together with stochastic, genetic, and environmental factors (Vijg and Campisi, 2008). Mitochondria are potential drivers of aging phenotypes. Dysfunctional mitochondria accumulate with age, best documented in tissues comprised largely of post-mitotic cells (e.g., muscle cells and neurons) (Herbst et al., 2007; Lee et al., 2010; Safdar et al., 2010; Wallace, 2010). Less is known about how dysfunctional mitochondria drive aging in mitotically active tissues, despite evidence from murine models that these tissues experience age-related degeneration when mitochondria are compromised (Kang et al., 2013; Kujoth et al., 2005; Trifunovic et al., 2004).
One consequence of mitochondrial dysfunction is cellular senescence, a complex stress response by which proliferative cells permanently lose the ability to divide (Braig and Schmitt, 2006; Campisi and d’Adda di Fagagna, 2007). The senescence response suppresses the development of cancer (Campisi, 2003; Campisi, 2013), but there is mounting evidence that senescent cells can accumulate with age and cause or contribute to aging phenotypes and pathologies. The permanent growth arrest can deplete progenitor or stem cell pools, thereby compromising tissue repair and regeneration (Kuilman et al., 2010; Sousa-Victor et al., 2014; Velarde et al., 2015). Further, senescent cells secrete molecules with potent paracrine effects (Coppé et al., 2006, 2008). This senescence-associated secretory phenotype (SASP) comprises pro-inflammatory cytokines, proteases, and growth and angiogenesis factors (Coppé et al., 2008) that can disrupt tissue microenvironments and compromise tissue structure and function.
Dysfunctional mitochondria can induce cellular senescence in culture (Moiseeva et al., 2009; Wang et al., 2003) and in vivo (Dai et al., 2010; Kang et al., 2013). However, little is known about the mechanisms that mediate this effect. Some studies implicate mitochondrial reactive oxygen species (ROS) as causal (Jiang et al., 2013b; Moiseeva et al., 2009; Passos et al., 2010; Velarde et al., 2012), but other outcomes of mitochondrial dysfunction are also likely. For example, sustained activation of 5′AMP-activated protein kinase (AMPK), a major bioenergetic sensor, is a hallmark of senescence (Moiseeva et al., 2009) and can induce a senescence arrest (Jiang et al., 2013b; Jones et al., 2005; Wang et al., 2003). Unlike the growth arrest and markers such as senescence-associated β-galactosidase (SA-Bgal) (Dimri et al., 1995), little is known about how mitochondria affect the SASP.
Because mitochondria oxidize NADH to NAD+ (Lehninger et al., 2013), mitochondrial dysfunction can decrease the NAD+/NADH ratio. While mitochondria oxidize NADH generated by the tricarboxylic acid (TCA) cycle or fatty acid oxidation, they also oxidize the cytosolic NAD +/NADH pool through the α-glycerophosphate and malate-aspartate shuttles (Houtkooper et al., 2010). Inhibition of the latter by depletion of malate dehydrogenase lowers the NAD+/NADH ratio and induces a senescence arrest (Lee et al., 2012), suggesting that elevated cytoplasmic NADH can drive cells into senescence. Notably, NAD+ declines with age in several tissues (Braidy et al., 2011; Gomes et al., 2013; Stein and Imai, 2014; Yoshino et al., 2011), linking NAD to both senescence and aging.
In a screen of sirtuins (SIRTs)—protein deacetylases, desuccinylases, demalonylases, deacylases, and ADP-ribose transferases (Hirschey, 2011; Jiang et al., 2013a) that are linked to aging (Haigis and Sinclair, 2010; Merksamer et al., 2013)—for ability to regulate senescence, we identified the mitochondrial SIRTs SIRT3 and to a lesser extent SIRT5, as suppressors of senescence and modulators of the SASP. Other mitochondrial perturbations induced a similar senescent phenotype, which we term mitochondrial dysfunction-associated senescence (MiDAS). We show that MiDAS results from a decreased NAD+/NADH ratio, activation of AMPK and subsequently the tumor suppressor p53, which then limits the IL-1 mediated arm of the SASP. Despite lacking IL-1-dependent SASP factors, the MiDAS SASP suppressed adipocyte differentiation and promoted keratinocyte differentiation. These phenotypes occurred in cultured human cells and a murine model of premature aging through mitochondrial dysfunction, thus linking mitochondrial dysfunction and aging through a subset of senescence phenotypes across species and in vivo.
RESULTS
Mitochondrial SIRTs Suppress Senescent Phenotypes
To determine whether SIRTs regulate senescence responses, we used lentiviruses to express shRNAs that target each of the seven human SIRTs in IMR-90 or BJ1 human fibroblasts. Immunoblots confirmed depletion of all seven SIRTs to different extents (Figures 1A and S1A). Surprisingly, depletion of SIRT3 or SIRT5, but not other SIRTs, induced senescence as judged by increased SA-Bgal activity and decreased BrdU labeling 10 days later. SIRT3 showed a stronger response, likely because it was more efficiently depleted (Figures 1A, 1B, S1B, and S1C). Cells induced to senesce by ionizing radiation (IR) (10 Gy), a common senescence inducer (Coppé et al., 2008; Rodier et al., 2009), showed no change in SIRT3 mRNA or protein (Figure S1D), indicating that reduced SIRT3 expression does not comprise the senescence response. Nonetheless, SIRT3 depletion induced three senescence markers: reduced lamin B1 (LMNB1) levels (Freund et al., 2012), decreased nuclear localization of high mobility group protein B1 (HMGB1) (Davalos et al., 2013), and increased levels of the cell-cycle inhibitor p21WAF1 (Campisi and d’Adda di Fagagna, 2007) (Figures 1C and 1D).
Figure 1. Loss of Mitochondrial SIRTs Induce Senescence.

(A) (Left) SA-Bgal and BrdU indices 10 days after IMR-90 cells were infected with control (scrambled sequence), shSIRT3-, or shSIRT5-expressing lentiviruses. (Right) SIRT3, SIRT5, and actin (control) protein levels after infection.
(B) Representative image of cells 10 days after senescence induction by shSIRT3, shSIRT5, and/or 10 Gy IR, stained for SA-Bgal.
(C) Intracellular LMNB1, p21, and HMGB1 protein levels 10 days after IR or infection with control (−) or shSIRT3 (+) lentiviruses.
(D) (Left) Representative image of EdU and HMGB1 staining in control and shSIRT3-expressing cells. (Right) Quantification of nuclear HMGB1 staining intensity.
(E) IL-6, measured by ELISA, in conditioned media from control, shSIRT3-, or shSIRT5-expressing cells 10 days after mock irradiation or 10 Gy IR.
(F) Heatmap of the indicated mRNA levels detected by qPCR in non-senescent (mock) or senescent (IR) cells infected with control or shSIRT3 lentiviruses, normalized to mock cells.
(G) Heatmap of secreted protein levels by above cells, detected by antibody array.
(H and I) Secreted IL-6 levels in response to senescence inducers, normalized to control shRNA cells: IR, NaBu, Bleo, and expression of constitutively active MKK6 (MKK6EE) or oncogenic RAS. Bar graphs indicate mean + SEM.
To determine whether mitochondrial SIRT depletion induces a SASP (Coppé et al., 2008), we measured secretion of IL-6, a major SASP factor, 10 days after SIRT3 or SIRT5 depletion. Despite the induction of other senescence markers, neither SIRT depletion increased IL-6 secretion (Figure 1E). Further, IR, a robust IL-6 inducer in unmodified cells, also failed to induce IL-6 secretion in SIRT3-depleted (and to a lesser extent SIRT5-depleted) cells (Figures 1E and S1E). In contrast, IR induced IL-6 secretion in cells depleted of SIRT2, SIRT4, SIRT6, or SIRT7 (Figure S1E); it induced cell death after SIRT1-depletion (not shown), as reported (Cheng et al., 2003; Yuan et al., 2012). Additional shRNAs confirmed the failure to induce IL-6 secretion in the absence of SIRT3 (Figures S1F and S1G). Thus, senescence resulting from depleted SIRT3, and to a lesser extent SIRT5, failed to include the SASP factor IL-6. Further, mitochondrial SIRT depletion suppressed IL-6 secretion by cells induced to senesce by IR.
SASPs result primarily from increased mRNA abundance (Coppé et al., 2008). We analyzed control and SIRT3-depleted cells, with or without IR, for IL-6 and other SASP mRNAs by qPCR. SIRT3-depleted cells failed to express significant levels of the SASP mRNAs encoding IL1B, CXCL1, CXCL2, IL-6, IL-8 and VEGF, but expressed high levels of mRNAs encoding the SASP factors AREG and IL-10, regardless of IR exposure (Figure 1F). Antibody arrays, which interrogate a different but overlapping set of SASP proteins, confirmed that SIRT3-depleted cells failed to secrete IL-1β, CXCL1, CXCL2, IL-6, IL-8, and VEGF (Figure 1G). Thus, SIRT3 depletion induced senescence but with a SASP that differed from that induced by IR, suggesting a mitochondrial function is required for expression of a subset of SASP factors. Notably, many SASP factors that were not expressed by SIRT3-depleted cells are pro-inflammatory, although the cells uniquely secreted CCL27 (Figure 1G), a pro-inflammatory cytokine that comprises the SASP of mouse, but not human, fibroblasts induced to senesce by IR (Coppé et al., 2010).
To determine whether SIRT3 depletion suppressed SASP factors in response to senescence inducers other than IR, we treated SIRT3-depleted cells with the HDAC inhibitor sodium butyrate (NaBu) or clastogen bleomycin (Bleo), or expressed oncogenic RAS or constitutively active MKK6 (MKK6EE), all of which induce a growth arrest and IR-like SASP (Coppé et al., 2008; Freund et al., 2011; Ogryzko et al., 1996; Orjalo et al., 2009). SIRT3 depletion strongly suppressed IL-6 secretion by cells induced to senesce by IR, NaBu, or Bleo (Figure 1H) and moderately suppressed IL-6 secretion in response to MKK6EE or RAS (Figure 1I), suggesting that multiple SASP inducers require a pathway in which SIRT3 acts. Surprisingly, while SIRT3 shRNA induced senescence in wild-type (WT) mouse embryonic fibroblasts (MEFs), MEFs from SIRT3 knockout mice did not senesce (Figures S1H and S1I), indicating that an absence of SIRT3 throughout embryogenesis and acute loss of SIRT3 after development can have different effects, as reported (Fernandez-Marcos et al., 2012).
Mitochondrial Dysfunction and Senescent Phenotypes
Because SIRT3 and SIRT5 are mitochondrial proteins, the above senescence responses might be a general effect of mitochondrial dysfunction. We therefore depleted mtDNA in IMR-90 cells by passage in ethidium bromide (rho0 cells) (Figures 2A and S2A) (Nass, 1972), treated cells with the electron transport chain inhibitors rotenone (Rot) or antimycin A (Anti A) (Figure 2A) or depleted cells of the mitochondrial chaperone HSPA9, which was shown to induce senescence (Yaguchi et al., 2007) (Figure 2A). All treatments induced senescence as determined by reduced EdU incorporation, increased SA-Bgal activity (Figures 2A and S2B), and loss of nuclear HMGB1 (for rho0 and Rot; Figure 2C), but without an increase in IL6 mRNA levels and IL-6 protein secretion, even after 10 Gy IR (Figures 2B, S2C, and S2D). We conclude that mitochondrial dysfunction causes a senescent phenotype that overlaps yet differs from that caused by other senescence inducers.
Figure 2. Mitochondrial Dysfunction-Induced Senescence.

(A) (Left) IMR-90 cells were cultured in DMSO (control), ethidium bromide (rho0), rotenone (Rot), or antimycin A (Anti A) or (right) infected with control, shSIRT3-, or shHSPA9-expressing lentiviruses and analyzed for SA-Bgal and EdU 10 days later.
(B) Cells from (A) were mock irradiated or irradiated (10 Gy) and analyzed for IL-6 mRNA by qPCR.
(C) Cells cultured in control DMSO, EtBr (rho0), or rotenone (Rot) for 14 days were analyzed for EdU incorporation and nuclear HMGB1 by immunofluorescence and cellular HMGB1 by immunoblotting.
(D) Rho0 IMR-90 (left) or MSC-1 (right) were generated in the presence of pyruvate and uridine. WT and rho0 cells cultured with (Pyr+) or without (Pyr−) pyruvate for 7 days were analyzed for EdU incorporation.
(E) SA-Bgal activity in cells from (D).
(F) WT cells were mock or irradiated to senesce by IR (IR) and compared to rho0 cells cultured + or −pyruvate. Indicated mRNA levels were determined by qPCR. Bar graphs indicate mean + SEM.
To better understand MiDAS, we cultured WT and rho0 IMR-90 cells and human mesenchymal stem cells (MSCs) with uridine and pyruvate, which prevents the rho0 growth arrest (Morais et al., 1994). Upon pyruvate withdrawal, rho0, but not WT, cells rapidly senesced, as measured by low EdU labeling and high SA-Bgal (Figures 2D and 2E). Pyruvate also prevented the Rot-induced growth arrest (Figure S2E). Notably, pyruvate promoted the expression of mRNAs encoding many SASP factors, including IL-1A, IL-1B, IL-6, IL-8 and CXCL1 (Figures 2F and S2F) in unirradiated rho0 cells. Upon pyruvate withdrawal, expression of these factors declined to near WT levels (Figures 2F and S2F), and the cells arrested growth (Figure 2D), suggesting that MiDAS depends on a mitochondrial function for which pyruvate compensates.
ROS are thought to mediate senescence in response to mitochondrial dysfunction (Moiseeva et al., 2009). Although shSIRT3 and rho0 cells cultured without pyruvate had elevated Mitosox fluorescence (Figures S2G and S2H), normalization to Mito-tracker showed roughly equivalent ROS levels between control and SIRT3-depleted cells (Figure S2G). Further, the antioxidant EUK-134 reduced mitochondrial, but not cytoplasmic, ROS in SIRT3- and HSPA9-depleted cells (Figures S2I and S2J) but did not prevent the growth arrest of these or Rot-treated cells (Figures S2K and S2L). Mitochondrial ROS is linked to senescence through nuclear DNA damage (Passos et al., 2010), but rho0 and SIRT3-depleted cells showed no increase in 53BP1 foci (Figures S2M–S2O), a DNA damage marker, and EUK-134 had no effect on these foci in any senescent cell (Figure S2O). Thus, MiDAS phenotypes appear to be independent of ROS and DNA damage.
Decreased NAD+/NADH Ratios Mediate MiDAS
Pyruvate is at the nexus of glycolysis and the TCA cycle. In response to mitochondrial dysfunction or hypoxia, lactate dehydrogenase converts pyruvate to lactate, concomitantly converting cytosolic NADH to NAD+. NAD+ is required for glycolysis, whereas NADH inhibits glycolysis (Lehninger et al., 2013). We tested the idea that the NAD+/NADH ratio regulates MiDAS by measuring each metabolite in lysates from WT and rho0 cells cultured with or without pyruvate. The NAD+/NADH ratio declined in rho0, Rot-treated, and SIRT3-depleted cells cultured without, but not with, pyruvate (Figures 3A, S3A, and S3B). Surprisingly, while the NAD+/NADH ratio declined with MiDAS, total NAD+ actually increased (Figure S3C) along with a rise in nicotinamide phosphoribosyltransferase (NAMPT) mRNA (Figure S3D), suggesting that pyruvate acts by altering NAD redox status.
Figure 3. MiDAS Is Mediated by NAD+/NADH Ratios.

(A) NAD+/NADH ratios in WT and rho0 cells cultured + or −pyruvate for 10 days.
(B) Control (WT or DMSO), rho0, or rotenone-treated (Rot) cells cultured without pyruvate and + or −K3[Fe(CN)6], analyzed for SA-Bgal.
(C) Cells cultured −or + K3[Fe(CN)6], and either DMSO or rotenone (Rot), for 8 days, were fixed and stained with crystal violet.
(D–G) WT cells cultured in PBS or AOA for 10 days: (D) SA-Bgal activity and EdU labeling; (E) (left) representative image of HMGB1 staining and EdU labeling in PBS or AOA-treated cells, (middle) quantification of nuclear HMGB1 staining, (right) LMNB1 and HMGB1 protein in cells treated for 7 days with PBS or AOA; (F) IL-6 ELISAs of CM from mock or irradiated cells 10 days after IR and AOA treatment; (G) cells given PBS or AOA + or −pyruvate (Pyr) for 10 days were stained with crystal violet.
(H) Cells cultured for 21 days + or −20 nM FK866 were analyzed for SA-Bgal and EdU labeling.
(I and J) Cells treated with 40 nM FK866 for 3 days following IR were analyzed for IL-6 secretion by ELISA (I) and NAD (J). Bar graphs indicate mean + SEM.
To determine whether the cytosolic or mitochondrial NAD+/ NADH ratio drives MiDAS, we measured NADH in each compartment using two-photon emission spectrometry. In WT cells, lactate increased NADH levels in the cytosol but not mitochondria (Figures S3E and S3F). Conversely, pyruvate lowered NADH in the cytosol, while increasing NADH in mitochondria, and rotenone elevated NADH in both the mitochondria and cytosol. In rho0 cells, lactate elicited only a small cytosolic response, suggesting that NADH was already reduced in this compartment. Likewise, pyruvate lowered cytosolic NADH to a much larger degree than in WT cells; it also lowered mitochondrial NADH in rho0 cells, but to a lesser extent than in the cytosol. Thus, MiDAS reduced the NAD+/NADH ratio mostly in the cytosol.
If decreased cytosolic NAD+/NADH drives MiDAS, NADH oxidation should prevent MIDAS. We treated pyruvate-free rho0 cells with potassium ferricyanide (K3[Fe(CN)6]), a cell-impermeable electron acceptor and NADH oxidase substrate that transfers reducing equivalents across the plasma membrane and allows rho0 cells to proliferate (Martinus et al., 1993). K3[Fe(CN)6] prevented senescence, as determined by SA-Bgal and arrested growth, in both rho0 and Rot-treated cells (Figures 3B and 3C).
NADH reducing equivalents are transferred across the mitochondrial membrane by the malate-aspartate shuttle (Lehninger et al., 2013), and depletion of the shuttle component malate dehydrogenase-1 induces a senescence arrest (Lee et al., 2012). Aminooxyacetate (AOA) inhibits the shuttle, lowering the cytosolic, but not mitochondrial, NAD+/NADH ratio (López-Alarcón and Eboli, 1986). AOA-treated WT cells underwent senescence (high SA-Bgal, low EdU labeling, loss of nuclear HMGB1; Figures 3D, 3E, S3G, and S3H) but failed to secrete IL-6 following IR, despite slightly higher secretion by unirradiated AOA-treated cells (Figure 3F). Importantly, pyruvate rescued the arrest of AOA-treated cells (Figures 3G and S3H).
We also artificially increased NAD+/NADH ratios by treating rho0 cells with the NAD+ precursor nicotinamide mononucleotide (NMN) without pyruvate. NMN delayed senescence by ~2 day relative to untreated cells (Figure S3I), but both groups senesced within 7 days (Figures S3I and S3J). Further, when we depleted NAD+ by preventing NAD salvage using the NAMPT inhibitor FK866, cell proliferation ceased after ~7 day (Figure S3K) and cells became senescent after ~21 days (low EdU labeling, high SA-Bgal) (Figures 3H and S3L). In addition, FK866 suppressed IR-induced IL-6 secretion (Figure 3I) and lowered total NAD levels (Figure 3J). We conclude that low NAD+/ NADH ratios mediate MiDAS phenotypes, which are prevented by NADH oxidation.
Finally, SIRT3 activity requires NAD+ as a coenzyme, in which case loss of mitochondrial NAD+ and reduced SIRT3 function might account for MiDAS phenotypes. However, pyruvate withdrawal from rho0 cells decreased rather than increased acetylation at lysine 68 of SOD2—a known SIRT3 target (Figures S3M and S3N). Thus, MiDAS cells do not senesce owing to loss of SIRT3 function per se.
Low NAD+/NADH Ratios Drive MiDAS through AMPK
Because NAD+ is required for glycolysis and NADH inhibits glycolysis, the NAD+/NADH ratio regulates energy production. We therefore measured ADP:ATP ratios in WT, rho0, SIRT3-depleted, and Rot- or Anti A-treated cells. All these manipulations elevated the ADP:ATP ratio (Figure 4A), which was lowered by pyruvate (Figure S4A). Similarly, rho0 cells showed elevated AMP:ATP ratios (Figure S4B), and inhibition of glycolysis by 2-de-oxyglucose (2-DG) induced senescence in rho0, but not WT, cells, regardless of the presence of pyruvate (Figures 4B and S4C). 2-DG alone did not alter NAD+/NADH ratios (Figure S4D).
Figure 4. MiDAS Depends on AMPK Activation.

(A) Cells infected with control or shSIRT3-expressing lentiviruses (upper panel), or treated with DMSO, Rot, Anti A, or ethidium bromide (EtBr) (lower panel) for 10 days were analyzed for ADP:ATP ratios.
(B) WT or rho0 cells cultured + or −pyruvate and/or 2-DG for 10 days were analyzed for EdU labeling (upper panel) or SA-Bgal (lower panel).
(C) Control or shSIRT3-expressing cells were cultured for 10 days after infection. WT (mtDNA+) and rho0 (mtDNA−) cells were cultured + or − 1 mM pyruvate for 10 days, lysed, and analyzed by immunoblotting for total and phosphorylated AMPK and p53 and intracellular LMNB1, HMGB1, p21WAF1, p16INK4a, SIRT3, COX2, and actin.
(D) Control (siSIRT3/AMPK−), SIRT3 (siSIRT3+), and AMPKα-1/2 (siAMPK+) siRNAs were transfected into cells, which were analyzed by immunoblotting 4 days later for the indicated proteins.
(E and F) Cells infected with control or dominant-negative AMPK (AMPK-DN)-expressing lentiviruses, then control or shSIRT3-expressing lentiviruses were analyzed 7 days later for (E) SA-Bgal and (F) EdU incorporation. Bar graphs indicate mean + SEM.
Increased ADP:ATP or AMP:ATP ratios activate AMPK (Hardie et al., 2012), which can induce senescence by phosphorylating p53 (Jones et al., 2005) or stabilizing p16INK4a mRNA (Chang et al., 2010), making AMPK activation a plausible candidate for mediating MiDAS. We therefore analyzed by immunoblot cells expressing control or SIRT3 shRNAs, and WT and rho0 cells, all cultured with or without pyruvate. Pyruvate reduced AMPKα phosphorylation (activation) in all cells. However, only MiDAS cells showed marked activation of both AMPKα (thr-172 phosphorylation) and p53 (ser-15 phosphorylation), both of which were blunted by pyruvate (Figure 4C). MiDAS also increased p21WAF1 and p16INK4a and decreased LMNB1 and intracellular HMGB1 protein levels, which were prevented by pyruvate. Similar results were obtained in cells treated with Rot or FK866 (Figures S4E and S4F). Treatment with a combination of 2-DG, pyruvate and Anti-A also activated AMPK (Figure S4G).
We also depleted AMPK using siRNAs, which prevented p53 phosphorylation and p21WAF1 expression in SIRT3-depleted cells (Figure 4D). Because siRNA depletion was too transient to assess other senescence markers, we used a lentivirus to stably express a FLAG-tagged dominant-negative (kinase-dead) AMPK mutant (AMPK-DN). AMPK-DN conferred resistance to senescence induced by SIRT3 depletion, Rot, or Anti-A, as assessed by high SA-Bgal, low EdU labeling (Figures 4E and 4F), p53 phosphorylation, high p16INK4a and p21WAF1 expression, and low intracellular HMGB1 (Figures S4H and S4I). Together, the results indicate that AMPK activation mediates MiDAS.
AMPK Mediates MiDAS through p53
Because p53 initiates the senescence arrest and is an AMPK target (Jiang et al., 2013b; Jones et al., 2005), we assessed the p53 dependence of MiDAS. We expressed control or p53 shRNAs in cells depleted of SIRT3, HSPA9, or mtDNA and analyzed them 7 days later for SA-Bgal, EdU labeling, and p21WAF1 levels (Figure 5A). p53 loss suppressed the induction of these senescence markers (Figures 5B, 5C, S5A, and S5B).
Figure 5. p53 Suppresses SASP Factors in MiDAS.

(A–C) Cells were infected with lentiviruses expressing control, SIRT3 or HSPA9 shRNAs, then a p53 shRNA (shp53) or control virus, were assayed 10 days later for HSPA9, SIRT3, p53 and p21 protein (A), SA-Bgal activity (B), and BrdU incorporation (C).
(D) RNA from WT and rho0 cells, infected with shp53 or control lentiviruses and cultured + or − pyruvate for 10 days was analyzed for IL-1A (upper panel) and IL-1B (lower panel) mRNA by qPCR.
(E) RNA from WT or rho0 cells, mock- or IR-treated, and cultured + or − pyruvate for 10 days was analyzed as in (D).
(F) NF-κB reporter activity in cells cultured as in (E).
(G) ChIP assays for RelA and CBP binding at the IL1A promoter in WT and rho0 cells cultured + or − pyruvate for 7 days.
(H) WT and rho0 cells were cultured + or − pyruvate and infected with control or shRelA-expressing lentiviruses, and mRNA levels were measured by qPCR.
(I) WT and rho0 cells cultured + or − pyruvate were analyzed for IL-10 and TNF-α mRNAs by qPCR. Bar graphs indicate mean + SEM.
Because p53 also limits SASP gene expression (Coppé et al., 2008; Rodier et al., 2009), we depleted p53 in WT and rho0 cells and measured IL1A and IL1B mRNA levels. IL-1α and IL-1β initiate a positive feedback loop that drives expression of SASP factors such as IL-6 and IL-8 (Orjalo et al., 2009). p53 loss restored IL1A and IL1B expression to rho0 cells lacking pyruvate to levels near those of rho0 cells cultured with pyruvate (Figure 5D).
IR robustly activates p53, which subsides to a low chronic level in 7–10 days as the SASP develops (Coppé et al., 2008; Rodier et al., 2009). Although IR induced a SASP in WT cells with or without pyruvate, rho0 cells plus pyruvate expressed an enhanced SASP that was suppressed by IR (Figures 5E and S5C). Since both IR and mitochondrial dysfunction activate p53 (Figure 4C), we conclude that p53 limits the expression of selected SASP factors during MiDAS.
p53 can suppress NF-κB (Dey et al., 2007; Murphy et al., 2011), which promotes transcription of many SASP genes (Freund et al., 2011). An NF-κB reporter showed that NF-κB activity was high in rho0 cells cultured with pyruvate but much lower in rho0 cells without pyruvate (Figure 5F). IR increased NF-κB activity in WT cells, as reported (Freund et al., 2011), but failed to do so in rho0 cells without pyruvate and reduced NF-κB activity to below that of unirradiated rho0 cells cultured with pyruvate (Figure 5F). These responses correlated with IL1A and IL1B mRNA levels (Figure 5E), suggesting that lack of the IL-1 signaling loop in MiDAS is due to suppression of NF-κB activity. Indeed, although the RELA NF-κB subunit increased binding to the IL1A promoter in rho0 cells cultured with or without pyruvate, binding of the NF-κB coactivator CBP increased in rho0 cells with pyruvate, but not without pyruvate (Figure 5G). Since p53 inhibits RELA by competing for CBP binding (Webster and Perkins, 1999), p53 likely limits the IL1 arm of the SASP by reducing NF-κB activity.
To further test this idea, we used shRNAs to deplete RELA from WT and rho0 cells cultured with pyruvate (Figure S5D). RELA depletion reduced several IL-1-responsive mRNAs (Figure 5H). Of interest, it also reduced mitochondrially encoded COX2 protein levels (Figure S5D), implicating RELA in retrograde signaling. Notably, some secreted factors were unaffected by MiDAS and were insensitive to pyruvate. Examples include the pro-inflammatory cytokines CCL27 (Figure 1G) and TNF-α (Figure 5I) and the anti-inflammatory cytokine IL-10 (Figure 5I).
MiDAS in POLG Mice
To determine whether MiDAS occurs in vivo, we analyzed POLGD257A mice, which have a mutation in the proofreading domain of the mtDNA polymerase (PolG) and rapidly accumulate mtDNA mutations and aging phenotypes (Kujoth et al., 2005; Trifunovic et al., 2004).
Inguinal adipose tissue (IAT) from 8-month-old POLGD257A mice had more cells with SA-Bgal (Figures 6A, 6C, S6A, and S6B) and loss of nuclear HMGB1 (Figures 6B and 6D) than age-matched WT IAT, primarily in cells between mature adipocytes. POLGD257A IAT also expressed more p16INK4a mRNA (Figure 6E) than age-matched WT IAT. Further, compared to WT, POLGD257A epidermis had more senescent cells in the stratum corneum as judged by SA-Bgal positivity (Figures S6C and S6D) and loss of HMGB1 in suprabasal epidermal nuclei (Figures S6E and S6F). Thus, senescent cells accumulated in vivo in response to mitochondrial dysfunction caused by the POLGD257A mutation.
Figure 6. MiDAS in POLGD257A Mouse Tissues.

(A) Representative image of SA-Bgal in IAT from 8-month-old WT and POLGD257A mice.
(B) Tissues from (A) were sectioned and immunostained for HMGB1 (representative image). Black arrows indicate nuclei; blue arrow indicates SA-Bgal.
(C and D) HMGB1-positive nuclei and SA-Bgal-positive cells were quantified in tissues from (A).
(E) qPCR analysis for p16INK4a mRNA in WT and POLGD257A tissue.
(F) NAD+/NADH ratios in IAT from WT and POLGD257A tissues.
(G) qPCR analysis for the indicated mRNAs in tissues from (E).
(H) 3T3-L1 cells were induced to differentiate in the presence of conditioned media from WT or rho0 IMR-90 fibroblasts. (Left panel) Representative images of cells stained with oil red O 6 days after treatment. (Right panel) quantitation of cells with <25% or >75% of cell area positive for oil red O. (I) qPCR for markers of adipocyte differentiation in cells described in (H). Bar graphs indicate mean + SEM.
To confirm MiDAS phenotypes in vivo, we measured NAD+/NADH ratios in IAT from 8-month-old WT and POLGD257A mice. The ratios varied among individual mice but were significantly less in POLGD257A compared to WT IAT (Figure 6F). NAD+/NADH ratios were also lower in the colon and small intestine of POLGD257A mice (Figures S6G–S6I). Further, IL1A, IL1B, and IL6 mRNAs were unchanged, while IL10 and TNF mRNAs were higher (Figure 6G), in POLGD257A compared to WT IAT, indicating that the MiDAS a SASP occurs in vivo in POLGD257A mice.
Because MiDAS occurred in POLGD257A fat and skin, which display aging phenotypes in these mice (Trifunovic et al., 2004), we asked whether the MiDAS SASP can influence adipocyte and/or keratinocyte phenotypes. Conditioned medium (CM) from rho0 fibroblasts blocked adipogenesis in 3T3-L1 preadipocytes, as measured by oil red O staining (Figure 6H) and levels of mRNAs encoding the adipogenic markers C/EBPβ, PPARγ, and FABP4 (Figure 6I) in a pyruvate-independent manner. Further, conditioned media from rho0 cells promoted keratinocyte differentiation, as determined by staining for the differentiation markers transglutaminase (TGM1) and loricrin (LOR) (Figure S6J), loss of EdU labeling (Figure S6K) and reduced levels levels of mRNAs encoding CD36 and DGAT2 (Figure S6L). These data support the findings that epidermal-specific SOD2-deficient mice rapidly accumulate senescent keratinocytes, which show enhanced differentiation (Velarde et al., 2015) and that POLGD257A mice have a thicker stratum corneum and lipodystrophy compared to age-matched WT mice (Trifunovic et al., 2004).
DISCUSSION
Mitochondrial dysfunction has been linked to aging (Khrapko and Vijg, 2009), but it is not clear how it drives aging, particularly in mitotic tissues. We show that (1) mitochondrial dysfunction induces a senescence response (MiDAS), including arrested growth and a SASP that is distinct from that caused by genotoxic stress; (2) the MiDAS SASP includes IL-10, TNF-α and CCL27, but lacks the IL-1 signaling arm; (3) pyruvate rescues the growth arrest and IL-1 arm of the SASP in MiDAS cells; (4) MiDAS results from a decreased NAD+/NADH ratio, which causes AMPK and p53 activation; (5) the MiDAS SASP alters the differentiation of preadipocytes and keratinocytes; and (6) MiDAS occurs in a progeroid mouse that rapidly accumulates dysfunctional mitochondria. Our findings provide a basis for how dysfunctional mitochondria can drive distinct aging phenotypes.
Mitochondrial ROS can damage nuclear DNA, thus activating a DNA damage response that induces senescence (Moiseeva et al., 2009; Passos et al., 2010). In MiDAS, pyruvate, but not an antioxidant, prevented MiDAS, which showed no evidence of DNA damage. Rather, decreased NAD+/NADH ratios caused MiDAS. In addition, because pyruvate prevented the MiDAS growth arrest but restored NF-κB activity, pyruvate uncoupled the growth arrest from the SASP in cells with compromised mitochondria. It is possible, however, that ROS activates NF-κB and hence the inflammatory arm of the SASP in pyruvate-treated cells.
Our finding that loss of SIRT3 loss drives senescence appears to contradict a report in which cells from SIRT3 knockout mice were highly susceptible to oncogenic transformation; however, this susceptibility depended on the elevated ROS characteristic of sirt3−/− cells (Kim et al., 2010). We reported that elevated oxygen dampens the SASP in humans and mouse cells (Coppé et al., 2010; Parrinello et al., 2003). Other reports show that inducible SIRT3 knockout mice do not necessarily phenocopy germline knockout animals (Fernandez-Marcos et al., 2012), consistent with our finding that acute SIRT3 depletion induces senescence.
We show that low NAD+/NADH ratios drive MiDAS through the energy sensor AMPK. AMPK can induce senescence (Jiang et al., 2013b; Jones et al., 2005) by phosphorylating p53 on ser-15 (Jones et al., 2005), a modification that drives cell-cycle arrest and senescence in response to genotoxic stress, and by preventing cytoplasmic translocation of the mRNA-stabilization factor HuR, thereby reducing pro-proliferation mRNAs (Wang et al., 2003) and stabilizing p16INK4a mRNA (Chang et al., 2010). Because p53 depletion was sufficient to prevent MiDAS, AMPK most likely mediates MiDAS through p53 activation, and subsequent NF-κB inhibition.
Our data from POLGD257A mice indicate that MiDAS can occur in vivo, although mtDNA mutations accumulate with age at rates well below that seen in POLGD257A mice (Khrapko and Vijg, 2009). However, aging also entails loss of mitochondrial function (Schriner et al., 2005), and we show many forms of mitochondrial dysfunction result in MiDAS. Further, our data from POLGD257A mice indicate that the IL1-regulated arm of the SASP is not active in MiDAS in vivo, suggesting that mitochondrial dysfunction drives aging phenotypes through other SASP components. MiDAS cells secreted the anti-inflammatory cytokine IL-10, but also high levels of the pro-inflammatory cytokines CCL27 and TNF-α. In addition, they secreted HMGB1, a pro-inflammatory alarmin that is a feature of senescent cells (Davalos et al., 2013). While we have not identified all components of the MiDAS SASP, our finding that the secretions from MiDAS cells inhibit adipogenesis and promote keratinocyte differentiation suggest the MiDAS SASP can have potent paracrine effects in vivo.
In conclusion, we describe a distinct senescent phenotype associated with compromised mitochondria (MiDAS) in human cells and mice. MiDAS results from an NADH-AMPK-p53-dependent pathway and elicits a SASP that lacks IL-1-dependent factors but includes others. Our results underscore the plasticity of senescent phenotypes and explain how the age-related increase in dysfunctional mitochondrial might drive aging phenotypes and pathologies.
EXPERIMENTAL PROCEDURES
Cell Culture
IMR-90 human fibroblasts were mycoplasma-free, cultured in 3% O2 as described (Coppé et al., 2008; Rodier et al., 2009), and used between 25 and 40 population doublings. To generate rho0, cells were cultured in 100 ng/ml ethidium bromide, 100 mM sodium pyruvate, and 50 μg/ml uridine for 2 months. Cells were mock irradiated or irradiated with 10 Gy X-rays, as described in Rodier et al. (2009), or treated with 100 nM rotenone, 10 nM anti-mycin A, 400 uM animooxyacetate (AOA), 1 mM NaBu, 2 mM 2-deoxyglucose, or 20–40 nM FK866 for 7–10 days unless noted otherwise. Cells were treated with 50 μg/ml Bleo in 20% O2 as described (Orjalo et al., 2009).
Animals
Protocols were approved by the Institutional Animal Care and Use Committee. POLGD257A homozygotes were derived from heterozygous breeders, fed ad libitum from weaning, and aged to 8 months prior to tissue harvest.
SA-Bgal
SA-Bgal activity was detected as described (Dimri et al., 1995) using a commercial kit (Biovision).
Immunofluorescence
Cells were cultured with BrdU (20 μM) or EdU (10 μM) for 24 or 72 hr, fixed in 4% buffered formalin for 10 min, washed, and permeabilized in 0.5% Triton X-100 for 30 min. For BrdU, cells were incubated with Exonuclease III and DNase I for 1 hr. For EdU, cells were treated as instructed by the manufacturer (Life Technologies Cat #C10337). For HMGB1, cells were processed as described (Davalos et al., 2013).
Immunohistochemistry
Tissues were fixed and stained for SA-Bgal as described above, followed by a second fixation in 10% buffered formalin, embedding in paraffin, and sectioning (5–7 μM). Sections were incubated overnight with HMGB1 antibody, and histochemistry was performed using the Vectastain Elite ABC KIT (Vector Labs) as instructed by the manufacturer.
NAD+/NADH Ratios
Cells were made quiescent by incubating in 0.2% serum for 3 days. NAD and NADH were measured using a commercial kit (Biovision) and 5 × 105 cells homogenized in 500 μl lysis buffer. For tissue, 20 mg were homogenized in 400 μl lysis buffer and clarified at 14,000 × g for 5 min at 4°C. Supernatants were fractionated using 10 kDa cutoff filters (Millipore) and spun at 10,000 × g for 45 min. To measure NADH, NAD+ was decomposed by incubation at 65°C for 30 min. Standard curves (5–200 pg/ml) were generated for quantification.
ADP/ATP Ratios
Cells (5 × 104) in 12-well plates were treated as described in the text. One well/treatment was analyzed for protein content by BCA assay. ADP/ATP ratios were measured using a kit (Biovision) as instructed by the manufacturer, and normalized to protein content.
Immunoblots
Cells were lysed in 5% SDS in 10 mM Tris, pH 7.4, and protein content determined by BCA assay. 40 μg protein was separated by electrophoresis and transferred to PVDF membranes. Membranes were blocked in TBST + 5% BSA, incubated overnight with primary antibody, washed in TBST, incubated with HRP-conjugated secondary antibody for 30 min, and visualized by chemiluminescence. Antibodies are listed in Supplemental Experimental Procedures.
PCR
RNA was isolated from cells using a kit (QIAGEN), and from IAT using Qiazol, as instructed by the manufacturer. RNA (250 ng/μl) was used to synthesize cDNA, which was analyzed by qPCR using the universal probe library (Roche). mRNA levels were normalized to averages of actin and tubulin, unless noted otherwise. Primer sequences and probes are listed in Supplemental Experimental Procedures.
Secreted Factors
3 × 104 cells in 12-well plates were treated as indicated in the text, and cultured in 0.5–1 ml serum-free DMEM for 24 hr. CM were collected and clarified at 2,000 × g for 10 min. Supernatants were transferred to a tube; cells were trypsinized and counted. CM (100 or 2.5 μl) were analyzed by colorimetric (R&D) or bead-based ELISAs (AlphaLISA, Perkin-Elmer) as instructed by the manufacturer and normalized to cell number. For antibody arrays, 2 × 105 cells in 6-well plates were cultured in 0.5–1 ml of serum-free DMEM for 24 hr. CM were collected and cells counted. CM were diluted, applied to the arrays (RayBio), and analyzed as described (Coppé et al., 2008; Freund et al., 2011).
siRNAs
Control, SIRT3, and AMPKa1/2 siRNAs (Santa Cruz) were packaged in Lipofectamine RNAiMAX (Life Technologies), and transfections were performed in Opti-MEM with replenishment each day for 3 days. Scrambled sequences served as controls.
Lentiviruses
SIRT3 (TRCN 0000038892), HSPA9 (TRCN0000029450, TRCN0000029452), and scrambled shRNAs (in pLK0.1) were from Sigma (Cat # SHC002). p53 shRNA was described (Rodier et al., 2009). The AMPK-DN (D157A) (Zhou et al., 2009) cDNA was cloned into the DraI and XbaI sites of lentiviral vector 670-1 and packaged as described (Campeau et al., 2009).
Statistical Analysis
Data are presented either as representative examples or means of 3+ experiments. Data are represented as means + SEM. p values were obtained using unpaired two-tailed Student’s t test or two-way ANOVA. *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
Highlights.
Dysfunctional mitochondria cause cell senescence and a distinct secretory phenotype
This secretory phenotype can influence the differentiation of certain cell types
An NAD-AMPK-p53 pathway controls the secretory and mitotic arrest phenotypes
Mice with dysfunctional mitochondria and premature aging accumulate senescent cells
Acknowledgments
We thank Pierre-Yves Desprez for critically reading the manuscript, Simon Melov for POLGD257A mice and breeding protocols, and Zhijun Luo for the AMPK-DN construct. This work was funded by NIH grants T32-AG00266 (J.C. for C.D.W.), R37-AG009909 (J.C.), K99-AG041221 (M.C.V.), fellowships from the American Federation of Aging Research (C.D.W.), SENS Research Foundation (E.A.S.), and Explo’Ra, PULSE and CROUS from France (P.L.). A.A.G. has a financial interest in Image Analyst Software.
Footnotes
Supplemental Information includes six figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2015.11.011.
AUTHOR CONTRIBUTIONS
C.D.W. and K.S. designed and executed the SIRT screen with guidance from J.C. and E.V. H.W.L. created SIRT shRNAs. C.D.W. and A.F. performed and analyzed antibody arrays. M.C.V., E.A.S., and C.D.W. designed and executed ROT, Anti A, and K3[Fe(CN)6] experiments. M.C.V., S.L., and C.D.W. performed differentiation assays. C.D.W. and A.A.G. designed and executed ROS and NADH experiments. S.S.D. and A.R. measured AMP, ADP, and ATP. C.D.W. and P.L. conducted 2-DG and NMN experiments. C.D.W. designed and performed other experiments. C.D.W., M.C.V., and J.C. wrote the paper.
The other authors declare no conflicts of interest.
References
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Braig M, Schmitt CA. Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res. 2006;66:2881–2884. doi: 10.1158/0008-5472.CAN-05-4006. [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer. 2003;3:339–349. doi: 10.1038/nrc1073. [DOI] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Chang N, Yi J, Guo G, Liu X, Shang Y, Tong T, Cui Q, Zhan M, Gorospe M, Wang W. HuR uses AUF1 as a cofactor to promote p16INK4 mRNA decay. Mol Cell Biol. 2010;30:3875–3886. doi: 10.1128/MCB.00169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Kauser K, Campisi J, Beausé jour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–29574. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Krtolica A, Beausé jour CM, Parrinello S, Hodgson JG, Chin K, Desprez PY, Campisi J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201:613–629. doi: 10.1083/jcb.201206006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Wong ET, Bist P, Tergaonkar V, Lane DP. Nutlin-3 inhibits the NFkappaB pathway in a p53-dependent manner: implications in lung cancer therapy. Cell Cycle. 2007;6:2178–2185. doi: 10.4161/cc.6.17.4643. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Jeninga EH, Canto C, Harach T, de Boer VC, Andreux P, Moullan N, Pirinen E, Yamamoto H, Houten SM, et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci Rep. 2012;2:425. doi: 10.1038/srep00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J Gerontol A Biol Sci Med Sci. 2007;62:235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD. Old enzymes, new tricks: sirtuins are NAD(+)-dependent deacylases. Cell Metab. 2011;14:718–719. doi: 10.1016/j.cmet.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, et al. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013a;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013b;493:689–693. doi: 10.1038/nature11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kang S, Louboutin JP, Datta P, Landel CP, Martinez D, Zervos AS, Strayer DS, Fernandes-Alnemri T, Alnemri ES. Loss of HtrA2/Omi activity in non-neuronal tissues of adult mice causes premature aging. Cell Death Differ. 2013;20:259–269. doi: 10.1038/cdd.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrapko K, Vijg J. Mitochondrial DNA mutations and aging: devils in the details? Trends Genet. 2009;25:91–98. doi: 10.1016/j.tig.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Dho SH, Ju SK, Maeng JS, Kim JY, Kwon KS. Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology. 2012;13:525–536. doi: 10.1007/s10522-012-9397-0. [DOI] [PubMed] [Google Scholar]
- Lehninger AL, Nelson DL, Cox MM. Lehninger principles of biochemistry. New York: W.H. Freeman; 2013. [Google Scholar]
- López-Alarcón L, Eboli ML. Oxidation of reduced cytosolic nicotinamide adenine dinucleotide by the malate-aspartate shuttle in the K-562 human leukemia cell line. Cancer Res. 1986;46:5589–5591. [PubMed] [Google Scholar]
- Martinus RD, Linnane AW, Nagley P. Growth of rho 0 human Namalwa cells lacking oxidative phosphorylation can be sustained by redox compounds potassium ferricyanide or coenzyme Q10 putatively acting through the plasma membrane oxidase. Biochem Mol Biol Int. 1993;31:997–1005. [PubMed] [Google Scholar]
- Merksamer PI, Liu Y, He W, Hirschey MD, Chen D, Verdin E. The sirtuins, oxidative stress and aging: an emerging link. Aging (Albany, NY) 2013;5:144–150. doi: 10.18632/aging.100544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Bourdeau V, Roux A, Deschênes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais R, Zinkewich-Péotti K, Parent M, Wang H, Babai F, Zollinger M. Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA. Cancer Res. 1994;54:3889–3896. [PubMed] [Google Scholar]
- Murphy SH, Suzuki K, Downes M, Welch GL, De Jesus P, Miraglia LJ, Orth AP, Chanda SK, Evans RM, Verma IM. Tumor suppressor protein (p)53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc Natl Acad Sci USA. 2011;108:17117–17122. doi: 10.1073/pnas.1114420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass MM. Differential effects of ethidium bromide on mitochondrial and nuclear DNA synthesis in vivo in cultured mammalian cells. Exp Cell Res. 1972;72:211–222. doi: 10.1016/0014-4827(72)90583-6. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol. 1996;16:5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Hamadeh MJ, Kaczor JJ, Raha S, Debeer J, Tarnopolsky MA. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE. 2010;5:e10778. doi: 10.1371/journal.pone.0010778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Velarde MC, Flynn JM, Day NU, Melov S, Campisi J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany, NY) 2012;4:3–12. doi: 10.18632/aging.100423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde MC, Demaria M, Melov S, Campisi J. Pleiotropic age-dependent effects of mitochondrial dysfunction on epidermal stem cells. Proc Natl Acad Sci USA. 2015;112:10407–10412. doi: 10.1073/pnas.1505675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454:1065–1071. doi: 10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- Wang W, Yang X, López de Silanes I, Carling D, Gorospe M. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J Biol Chem. 2003;278:27016–27023. doi: 10.1074/jbc.M300318200. [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaguchi T, Aida S, Kaul SC, Wadhwa R. Involvement of mortalin in cellular senescence from the perspective of its mitochondrial import, chaperone, and oxidative stress management functions. Ann N Y Acad Sci. 2007;1100:306–311. doi: 10.1196/annals.1395.032. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Luo K, Liu T, Lou Z. Regulation of SIRT1 activity by genotoxic stress. Genes Dev. 2012;26:791–796. doi: 10.1101/gad.188482.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Huang W, Tao R, Ibaragi S, Lan F, Ido Y, Wu X, Alekseyev YO, Lenburg ME, Hu GF, Luo Z. Inactivation of AMPK alters gene expression and promotes growth of prostate cancer cells. Oncogene. 2009;28:1993–2002. doi: 10.1038/onc.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.