

Abstract
Pulmonary fibrosis results from the excessive deposition of collagen fibers and scarring in the lungs with or without an identifiable cause. The mechanism(s) underlying lung fibrosis development is poorly understood, and effective treatment is lacking. Here we compared mouse lung fibrosis induced by pulmonary exposure to prototypical particulate (crystalline silica) or soluble chemical (bleomycin or paraquat) fibrogenic agents to identify the underlying mechanisms. Young male C57BL/6J mice were given silica (2 mg), bleomycin (0.07 mg), or paraquat (0.02 mg) by pharyngeal aspiration. All treatments induced significant inflammatory infiltration and collagen deposition, manifesting fibrotic foci in silica-exposed lungs or diffuse fibrosis in bleomycin or paraquat-exposed lungs on day 7 post-exposure, at which time the lesions reached their peaks and represented a junction of transition from an acute response to chronic fibrosis. Lung genomewide gene expression was analyzed, and differential gene expression was confirmed by quantitative RT-PCR, immunohistochemistry, and immunoblotting for representative genes to demonstrate their induced expression and localization in fibrotic lungs. Canonical signaling pathways, gene ontology, and upstream transcription networks modified by each agent were identified. In particular, these inducers elicited marked proliferative responses; at the same time, silica preferentially activated innate immune functions and the defense against foreign bodies, whereas bleomycin and paraquat boosted responses related to cell adhesion, platelet activation, extracellular matrix remodeling, and wound healing. This study identified, for the first time, the shared and unique genes, signaling pathways, and biological functions regulated by particulate and soluble chemical fibrogenic agents during lung fibrosis, providing insights into the mechanisms underlying human lung fibrotic diseases.
Keywords: Pulmonary fibrosis, Genome-wide gene expression, Inflammation, Silica, Bleomycin, Paraquat
Introduction
Pulmonary fibrosis is an irreversible stage of pathologic development in a variety of lung diseases (Husain and Kumar 2005). Pulmonary fibrosis can occur as a result of an existing lung condition, such as chronic lung Inflammation, or without a known cause, as exemplified by idiopathic pulmonary fibrosis (IPF), but more often is caused by exposure to a fibrogenic agent, such as inhaled particles, fibers, and microbes, as well as soluble chemicals that selectively accumulate in the lungs and damage lung tissues (Meltzer and Noble 2008; Morgan and Seaton 1995; Raghu et al. 2011; Thomas and Kelley 2010). Despite this enormous etiologic diversity, fibrosis in the lungs shares considerable common pathologic features characterized by excessive and progressive deposition of collagen fibers in the lung interstitial space and scarring of the lungs, which lead to destruction of alveolar and airway structures and loss of respiratory capacity (Husain and Kumar 2005; Morgan and Seaton 1995). The mechanism(s) underlying pulmonary fibrosis is poorly understood, which hampers drug discovery and drug development for treating lung fibrosis. As a result, no effective therapy against human lung fibrotic diseases is available.
In the case of IPF, a chronic, progressive, and ultimately lethal human lung fibrotic disease, lung transplant remains to be the only lifesaving treatment and the median survival of IPF patients is merely 2–5 years after diagnosis (Meltzer and Noble 2008; Raghu et al. 2011). The pathologic features of IPF include the presence of mild-to-moderate Inflammation with inflammatory infiltration, injury and hyperplasia of alveolar epithelial cells, enhanced deposition of extracellular matrix, thickened alveolar septa, formation of scars and fibroblastic foci, and temporally heterogeneous fibrotic remodeling of lung structures (Meltzer and Noble 2008; Raghu et al. 2011). Some of these characteristics can be observed in animal models of induced lung fibrosis (Degryse and Lawson 2011; Moeller et al. 2008).
Bleomycin is a glycopeptide anti-tumor antibiotic that effectively treats certain cancers. Exposure to bleomycin, either systemically or locally, induces pulmonary fibrosis as a side effect of anti-cancer therapy. Bleomycin induces lung fibrosis in rodents, which has been used as an animal model of choice for the study of human IPF during the past decade (Moore and Hogaboam 2008). In the murine model, bleomycin-induced fibrosis develops quickly, but is self-limiting after several weeks of treatment; moreover, drug development based on the single-dose bleomycin-induced lung fibrosis model has not produced clinically effective therapeutics for treating IPF (Degryse and Lawson 2011; Moeller et al. 2008).
Silica is an abundant mineral that commonly exists in the earth's crust and surface in the forms of sand, rock, and mineral ore. Pulmonary exposure to crystalline silica dusts causes the human lung disease called silicosis, characterized by the occurrence of lung Inflammation and fibrosis, decline of lung functions, and premature death (Morgan and Seaton 1995). Exposure to silica typically takes place during industrial activities, such as mining, manufacturing, and construction, through inhalation of silica-containing dusts, though non-occupational exposure to silica also takes place (Morgan and Seaton 1995; Thomas and Kelley 2010). Like IPF, no curative treatment for silicosis is available. In animals, inhalation of silica results in pulmonary inflammatory responses, such as activated alveolar macrophages and increased polymorphonuclear leukocytes in the bronchoalveolar space; pulmonary fibrotic responses, such as elevated collagen deposition and fibrotic nodule formation; and prominent lung tissue damage (Ohtsuka et al. 2006; Porter et al. 2004).
Paraquat (N, N-dimethyl-4, 4′-bipyridinium dichloride) is the second most widely used herbicide globally, but is a potent lung toxicant causing lung fibrosis by accumulating in lung epithelial cells and damaging lung structures (Dinis-Oliveira et al. 2008), similarly to bleomycin. The lung toxicity of paraquat is dominant and lethal and is believed to be mediated through redox cycling and intracellular oxidative stress (Bus and Gibson 1984). Formation of intra-alveolar fibrosis is a major cause of paraquat-induced mortality, for which no effective treatment has been found (Dinis-Oliveira et al. 2008; Gawarammana and Buckley 2011).
From a mechanistic point of view, the fibrotic response to exposure of particles (silica), fibers (asbestos and nanotubes), and volumetric biological materials (helminth eggs), resembles the tissue response to foreign body deposition in the lungs. On the other hand, lung fibrosis induced by soluble chemicals, such as bleomycin and paraquat, is believed to be derived directly from damaging the alveolar and airway cells by the chemicals (Bus and Gibson 1984; Dong and Ma 2015; Husain and Kumar 2005; Moore and Hogaboam 2008; Morgan and Seaton 1995). Nonetheless, in both scenarios, the pulmonary lesions include an acute-phase response characterized by inflammatory infiltration and tissue damage, and chronic fibrosis development marked by interstitial fibrosis, granuloma formation, and scarring of the lungs. The acute-phase inflammatory lesion often reaches its peak after 1 week, but reduces in intensity substantially after 14 days post-exposure to a single-dose treatment (Bissonnette and Rola-Pleszczynski 1989; Bus and Gibson 1984; Dong et al. 2015; Huaux et al. 2003; Rabolli et al. 2011), though repeated exposures can result in sustained inflammatory lesions alongside fibrosis (Porter et al. 2004). This prominent morphologic switch from acute-to-chronic pathology suggests the activation of critical cellular and molecular events, including programmed transcription of genes, at the acute-to-chronic junction, to bring about the drastic pathologic transition and disease progression. Elucidating the molecular mechanisms that mediate or control the acute-to-chronic transition in the development of lung fibrosis would reveal new mechanistic aspects of the pathogenesis of both induced and idiopathic lung fibrotic diseases.
We attempted to address these issues by analyzing and comparing the genome-wide gene expression profiles in experimental lung fibrosis models induced by silica, bleomycin, and paraquat, which are prototypical particulate or soluble chemical fibrogenic inducers. We chose day 7 post-exposure, at which time the acute responses appear to have reached their peaks with a similar intensity, for microarray analysis, in order to gain insights into the critical pathways and mediators that govern the transition from acute-to-chronic pathologic development. The analyses identified both shared and distinct pathways and mechanisms for the development of lung fibrosis induced by the inducers. Therefore, our study uncovered the affected molecules and signaling pathways that might function in the onset of lung fibrosis in general or in a fibrogenic agent-specific man-ner, which offer new clues to understanding the molecular mechanisms underlying lung fibrosis in humans.
Materials and methods
Animals and treatment
Eight- to 10-week-old specific pathogen-free male C57BL/6J mice purchased from The Jackson Laboratory (Bar Harbor, ME, USA) were used throughout this study. Mice were maintained in an accredited, specific pathogen-free and environmentally controlled facility at the National Institute for Occupational Safety and Health. Mice were housed in polycarbonate ventilated cages with HEPA-filtered air and 12-h fluorescent lighting, and were fed with Harlan Teklad Rodent Diet 7913 (Indianapolis, IN, USA) with tap water ad libitum. All animal experiments were performed in accordance with the guidelines approved by the Institutional Animal Care and Use Committee.
Mice were treated with the following agents: (a) saline (Baxter Healthcare Corporation, Deerfield, IL, USA) as solvent control; (b) silica (MIN-U-SIL 5, US Silica, Berkeley Springs, WV, USA) at 2 mg/mouse; (c) bleomycin (bleomycin sulfate, Enzo Life Sciences, Farmingdale, NY, USA) at 0.07 mg/mouse; or (d) paraquat (Thermo Fisher Scientific, Rockford, IL, USA) at 0.02 mg/mouse; six to eight mice per group were used depending on the experiments performed. All reagents were prepared in saline. Mice were anesthetized with isoflurane (Piramal Healthcare, Bethlehem, PA, USA) and were given the above dose in 50 μl saline by pharyngeal aspiration following established procedures (Dong et al. 2015; Porter et al. 2010). Mouse pharyngeal aspiration has been shown to produce an even distribution of administered materials throughout the lungs (Rao et al. 2003) and thus represents a noninvasive and physiologically relevant route of exposure to a specific dose in the lungs. Mice were euthanized to collect lung tissues on day 7 post-exposure.
Histopathology
Mice were euthanized by intraperitoneal injection of sodium pentobarbital (Zoetis, Florham Park, NJ, USA) at a dose of >100 mg/kg body weight. The left lung lobe was removed, fixed with 10 % neutral-buffered formalin through intratracheal perfusion, and embedded in paraffin. Sections of 5 μm thickness were subjected to H&E staining, Masson's Trichrome staining, and Picro-Sirius red staining following standard protocols. Six samples in each treatment group were investigated.
Immunohistochemistry
Formalin-fixed, paraffin-embedded lung tissue sections (left lung lobe, 5 μm) were deparaffinized with three washes of xylene for 5 min each, rehydrated gradually with two washes of 100 % ethanol for 10 min each, two washes of 95 % ethanol for 10 min each, one wash of 70 % ethanol for 5 min, and two washes of deionized water for 5 min each. For antigen unmasking, sections were heated to boil in antigen unmasking solution, Citric Acid Based, pH 6.0 (Vector Laboratories, Burlingame, CA, USA) and then maintained at a sub-boiling temperature for 10 min in a microwave oven. The slides were then cooled on bench top for 30 min, washed with deionized water for 5 min, and incubated with BLOXALL Endogenous Peroxidase and Alkaline Phosphatase Blocking Solution (Vector Laboratories) for 10 min at room temperature to block endogenous peroxidase activity. The assays were performed with ImmPRESS Polymer Detection system (Vector Laboratories), following instructions from the manufacturer. Peroxidase activities were detected with the peroxidase substrate ImmPACT NovaRED (Vector Laboratories), which generates positive staining with red color. The slides were then counterstained with Hematoxylin QS (Vector Laboratories), which visualizes nuclei with blue–violet color. The primary antibodies used included anti-collagen I (Abcam, Cambridge, MA, USA), anti-FN1 (Abcam), anti-LCN2 (Abcam), anti-SLPI (Thermo Fisher Scientific), anti-TNC (GeneTex, Irvine, CA, USA), and anti-collagen VI (Abcam). Images were photographed using Olympus Provis AX-70 system (Olympus, Center Valley, PA, USA). Four samples in each treatment group were investigated.
Immunoblotting
Lung tissue samples were homogenized in CelLytic MT Cell Lysis Reagent (Sigma, St. Louis, MO, USA) containing complete Protease Inhibitor Cocktail Tablets (Roche, Basel, Switzerland), followed by incubation on ice for 30 min with shaking, and centrifugation at 14,000 rpm for 20 min at 4 °C to remove cell debris. The supernatant containing the whole protein extract was obtained, of which 20 μg was resolved in SDS-PAGE gel, transferred to PVDF membrane (EMD Millipore, Billerica, MA, USA), blocked with SuperBlock T20 (PBS) Blocking Buffer (Thermo Fisher Scientific) at room temperature for 1 h, incubated with primary antibody at 4 °C overnight, incubated with horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h, and detected using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, UK). GAPDH was detected as an internal control. The primary antibodies used were anti-LCN2 (Abcam), anti-SLPI (Thermo Fisher Scientific), anti-TNC (GeneTex), anti-SPP1 (R&D Systems, Minneapolis, MN, USA), anti-MMP2 (Santa Cruz Biotechnology, Dallas, TX, USA), and anti-GAPDH (Fitzgerald, Acton, MA, USA).
Microarray gene expression analysis
Total RNA was extracted from mouse lung tissue samples using RNeasy Mini Kit (QIAGEN, Valencia, CA, USA), during which genomic DNA was removed with the method of on-column DNase treatment. For each dose group, RNA samples from four mice, which were randomly selected from six treated mice, were used to perform microarray analysis. All the RNA samples used for this assay reached the required quality, including (1) A260/A280 ratio between 1.8 and 2.1; (2) A260/A230 ratio greater than 1.5; and (3) RNA Integrity Number (RIN) of 8 or greater evaluated by an Agilent 2100 Bioanalyzer. The synthesis of cRNA from total RNA using Illumina TotalPrep RNA Amplification Kit (Life Technologies, Carlsbad, CA, USA), hybridization to MouseWG-6 v2.0 Expression BeadChip (Illumina, San Diego, CA, USA), and raw data processing with Illumina HiScan Array Scanner and GenomeStudio software v1.9.0 (Illumina) were performed under a service at Yale Center for Genome Analysis (Yale University School of Medicine, New Haven, CT, USA). Microarray data analysis was carried out under a service at Yale Bioinformatics Resource (Yale University School of Medicine).
Statistical analysis of microarray data
Gene expression data generated by Illumina were imported into Partek® Genomics Suite™ (Partek Incorporated, 6.4). Raw data were processed in steps of background correction of the perfect match values, quintile normalization across all of the chips in the experiment, and median polish summarization. Within each condition, outliers were identified using principle component analysis (PCA) and excluded from all further analysis. Differentially expression analysis of the samples was performed using one-way analysis of variance (ANOVA). p values were adjusted for multiple comparisons using false discovery rate multiple testing correction (Benjamini and Hochberg 1995). Differentially expressed genes were selected with threshold of relative ± twofold change and adjusted p values ≤0.05. The heat map was generated using heatmap.2 function available in “gplots” package in R program. Venn diagrams were generated using VennDiagram package in R program.
Function and pathway analysis of differentially expressed genes
Canonical pathways and gene ontology (GO) biological processes associated with identified differentially expressed genes were disclosed using MetaCore GeneGO server (https://portal.genego.com/). p values were calculated based on hypergeometric distribution and reflected the probability for a pathway or process to arise by chance. Pathways and processes with a Benjamini–Hochberg multiple testing correction p value of ≤0.05 were considered significant.
Transcription factor analysis
The network-building algorithm on transcription regulation from MetaCore was used to examine whether the identified genes were connected to transcription factors. For each candidate transcription factor, a p value was calculated based on hypergeometric distribution, indicating enrichment in the genes of interest. Transcription factors with Benjamini–Hochberg multiple testing correction p value of ≤0.05 were considered significant. Transcription regulation networks were built centering on the most significant transcription factors.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from mouse lung tissue samples using RNeasy Mini Kit and reverse-transcribed to produce cDNA using QuantiTect Reverse Transcription Kit (QIA-GEN). qRT-PCR was performed and analyzed as described previously (Dong et al. 2015). The fold change values for three samples in each treatment group were averaged, and data were presented as the mean ± SD. Statistical evaluation of differences between treatment groups was determined by two-tailed Student's t test. A p value of less than 0.05 was considered statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Pathologic features of lung lesions induced by prototypical particulate and soluble chemical fibrogenic agents
The development of induced lung fibrosis includes an acute-phase response upon fibrogenic agent exposure, followed by progression to chronic fibrosis, with drastically distinct pathologic features between the two phases. Among fibrogenic inducers, soluble chemicals, such as bleomycin and paraquat, selectively accumulate in the lungs and cause marked cytotoxicity to lung cells; on the other hand, agents with a bulky mass, such as inhaled particles, fibers, and microbes, stimulate tissue responses, such as Inflammation, without directly killing lung cells. Nonetheless, both types of inducers cause lung fibrosis in humans and animals, albeit with notable differences in pathologic manifestations. To elucidate the mechanism of lung fbrogenesis at the molecular level, we chose to analyze and compare the molecular events that control the transition from the acute-to-chronic progression during lung fbrosis induced by prototypical fibrogenic inducers, i.e., silica as an example of particulate inducers, and bleomycin and paraquat as examples of soluble chemical inducers. We investigated the genome-wide gene expression patterns on day 7 post-exposure, at which time the acute lesions appear to have reached a climax, and represent a junction for the acute-to-chronic transition and progression.
We first characterized the pathologic features of pulmonary lesions on day 7 post-exposure to silica, bleomycin, and paraquat at a dose of 2, 0.07, and 0.02 mg, respectively, by pharyngeal aspiration, in comparison with saline control. Histological examinations revealed dramatically increased cellularity and accumulation of matrix materials in the lung parenchyma of treated mice compared with saline control (Fig. 1A and data not shown). The lesions appeared similar in intensity among silica, bleomycin, and paraquat treatment groups, which provided a basis for comparison among the treatments at the molecular level in later analyses. In addition to inflammatory infiltration, the data revealed prominent fibrotic alterations in all three treatment groups, as demonstrated by the Masson's Trichrome staining (Fig. 1A, a–d), which distinguishes cells from their collagenous connective surroundings, and Picro-Sirius red staining (Fig. 1A, e–h), which preferentially detects collagens I and III, two major collagen fibers involved in lung fibrosis. The findings indicate that fibrotic changes occurred early and accompanied the acute inflammatory alterations.
Fig. 1.

Induced lung fibrosis and expression of fibrosis marker proteins. Wild-type C57BL/6J mice received saline vehicle control, silica (2 mg), bleomycin (0.07 mg), or paraquat (0.02 mg) and were killed on day 7 post-exposure. A Total collagen fibers were detected by Masson's Trichrome staining of lung tissue sections, with collagen fibers stained blue (a–d, scale bar 50 μm). Collagen I and III fibers were visualized by Picro-Sirius red staining, with collagen fibers stained red (e–h, scale bar 50 μm). B Collagen I (a–d) and FN1 (e–h) levels were determined by immunohistochemistry on lung sections. Red indicates positive staining, and blue indicates nuclear counterstaining (scale bar 20 μm)
Notable differences in the pathologic phenotypes induced by the agents were observed. The lesions resulting from silica exposure were characterized by a low-to-moderate level of inflammatory infiltration, formation of nodule-like fibrotic foci, and intensive deposition of collagen fibers within the fibrotic foci (Fig. 1A, b and f), whereas the lesions induced by bleomycin or paraquat shared a higher degree of similarity between the two treatments than with silica and fell into the other group, featured with severe inflammatory infiltration and diffuse deposition of collagen fibers without prominent nodule-like fibrotic focus formation (Fig. 1A, c and g, d and h). Notably, bleomycin and paraquat induced marked tissue damage and, in the case of paraquat exposure, mild-to-moderate hemorrhagic lesions in early time points (data not shown), which is in agreement with the notion that both chemicals are highly cytotoxic to lung cells.
Collagen and fibronectin (FN1) are two major components of the extracellular matrix (ECM) and, therefore, serve as markers of fibrosis. To further validate the fibrotic responses induced by these fibrogenic agents, the protein levels and distribution patterns of collagen I and FN1 were examined by immunohistochemistry. The levels of both collagen I (Fig. 1B, a–d) and FN1 (Fig. 1B, e–h) were dramatically elevated in fibrotic lungs from mice exposed to silica, bleomycin or paraquat, compared with saline control. Moreover, collagen I and FN1 exhibited a more compact and focal pattern of distribution within the nodule-like fibrotic foci in the lungs exposed to silica (Fig. 1B, b and f), but a more diffusely distributed pattern was observed in the lungs exposed to bleomycin or paraquat (Fig. 1B, c and g, d and h). These findings confirmed that silica, bleomycin, and paraquat potently induced fibrotic responses in the lungs on 7 days post-exposure, with distinct morphologic features, in addition to commonly increased deposition of fibrotic matrix proteins, implicating both distinct and overlapping underlying mechanisms in the development of the induced lung fibrosis.
Genome-wide gene expression induced by fibrogenic agents
To analyze genome-wide gene expression, we performed microarray analysis of RNA samples from lungs of four mice from each of the saline, silica, bleomycin, or paraquat treatment group. Differentially expressed genes were identified using the criteria in which the p values with false discovery rate (FDR) were ≤0.05, and the up-regulated or down-regulated fold changes in comparison with saline control were more than 2.0 following silica, bleomycin or paraquat exposure.
Treatment with silica affected the expression of 143 genes, in which 124 were up-regulated and 19 downregulated as compared with the saline control. Treatment with bleomycin or paraquat affected the expression of significantly more genes than silica treatment. In bleomycin-treated lungs, 247 genes were affected, including 170 up-regulated and 77 down-regulated. Paraquat exposure resulted in modified expression of 299 genes with 205 genes up-regulated and 94 down-regulated. Heat map analysis of these differentially expressed genes is shown in Fig. 2A, and detailed expression levels of the genes are listed in Table S1. Figure 2B listed ten top genes with their fold changes, compared with saline control, from each treatment group, such as lipocalin-2 (Lcn2), secretory leukocyte peptidase inhibitor (Slpi), tenascin-C (Tnc), secreted phosphoprotein 1 (Spp1, osteopontin, OPN), matrix metalloproteinase (Mmp), and tissue inhibitor of metalloproteinases 1 (Timp1), which have all been implicated in the pathogenesis of lung fibrosis and related lung diseases in humans and experimental animals.
Fig. 2.
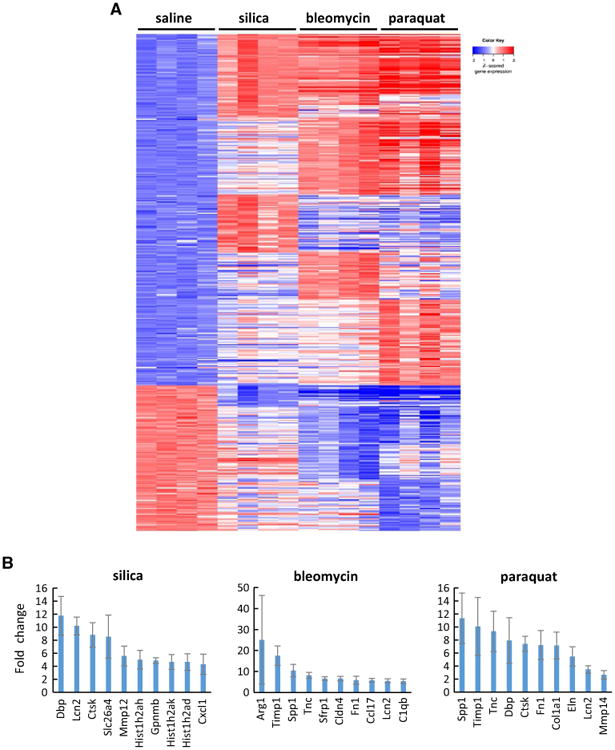
Differential gene expression revealed by microarray. Genome-wide gene transcription profiling was obtained by microarray gene expression analysis on RNA samples isolated from the lungs exposed to fibrogenic agents for 7 days (n = 4 for each group). The genes possessing p value with FDR ≤ 0.05, and the either up-regulated or down-regulated fold change, compared with saline-treated control group, more than 2.0 following silica, bleomycin, or paraquat exposure, were identified and studied. A A heat map was generated for the differentially expressed genes. In the heat map, up-regulated genes were arranged in the upper part, and down-regulated genes in the lower part. Within each of these two parts, the common genes affected by all three agents, i.e., silica, bleomycin, and paraquat, were listed first; the common genes affected by any two agents, i.e., silica and bleomycin, or silica and paraquat, or bleomycin and paraquat, were listed secondly; and the unique genes that were only affected by one agent, i.e., silica, bleomycin, or paraquat, were listed last. Red, white, and blue indicate high, medium, and low expression levels, respectively. B Examples of up-regulated genes for each treatment group were shown with fold changes (mean ± SD, n = 4)
These findings demonstrate that the treatments induced dramatic alterations in gene expression, indicating active transcription during the acute-to-chronic fibrotic transition in the lungs. Moreover, the heat map clearly revealed both shared and distinct gene expression patterns among the three treatment groups. There were apparently more similarities in gene expression between bleomycin and paraquat treatments than with silica treatment, and more genes affected by bleomycin or paraquat than by silica, which are consistent with the pathologic findings qualitatively.
Quantitative comparisons of the regulated genes were presented in Venn diagrams (Fig. 3A). Sixty-five genes were affected by all three treatments; 76 genes were affected by both silica and bleomycin; 80 genes were affected by both silica and paraquat; and 164 genes were affected by both bleomycin and paraquat. Several groups of genes were commonly induced by all three agents. In addition to fibrosis marker genes, such as Col1a1 (collagen 1a1) and Mmp2, genes functioning in cell proliferation and survival, such as Cdc20 and Birc5 (survivin), and genes involved in DNA synthesis, damage, and repair, and chromatin architecture, such as the members of the Hist1h2a gene family, were found induced by silica, bleomycin, and paraquat, implicating proliferation and DNA metabolism as key common mechanisms of regulation at the level of gene transcription for lung fibrosis induced by all three inducers.
Fig. 3.
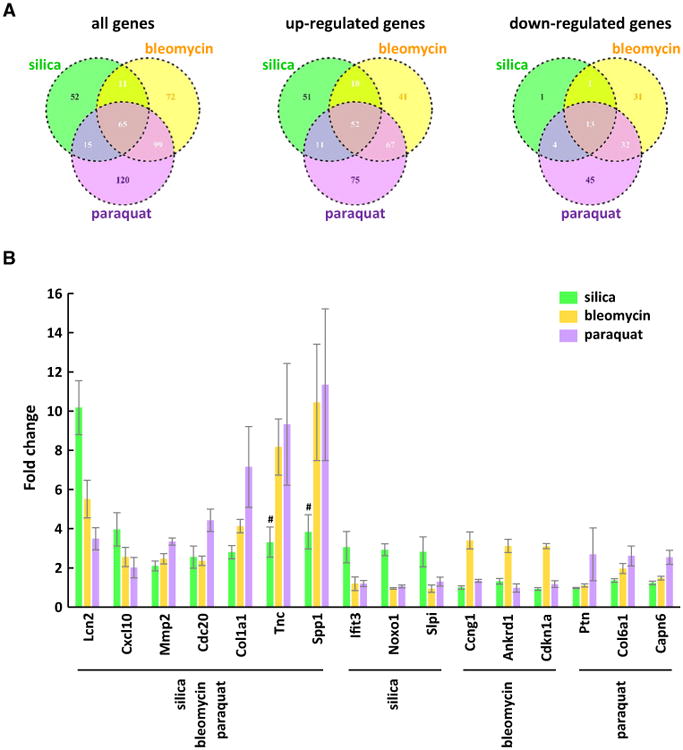
Shared and unique patterns of gene expression in induced lung fibrosis. A Venn diagram analysis determined the similarity and distinctiveness among the genes affected by silica, bleomycin, or paraquat. B Among up-regulated genes, five genes that were commonly induced by three agents, and three genes that were uniquely induced by each specific agent, were selected to present with fold changes as examples (mean ± SD, n = 4). For Tnc and Spp1, the genes were induced by bleomycin and paraquat to much higher levels than by silica, where # indicates p value with FDR > 0.05
There were apparently groups of genes uniquely regulated by each treatment. The number and percentage of unique genes regulated were 52 (36.4 %) for the silica treatment, 72 (29.1 %) for bleomycin, and 120 (40.1 %) for paraquat, respectively. For instance, among the 52 genes distinctively induced by silica, a number of genes are involved in immune regulation and function, including Ccl3, Ccl4, Ccl6, Cxcl1, Il1rn, Il4i1, Csf2, Oas1g, and Oas2. The 72 genes affected by bleomycin treatment only, including Cdkn1a, Ccng1, Egr1, Egr2, Ctsa, Ctsb, Ctsh, Cxcl13, and Ccr5, have been implicated in cell cycle control, mitogenesis, differentiation, protein turnover, or Inflammation. Paraquat regulated the expression of 120 genes uniquely, some of which play important roles in mitosis and DNA replication, such as Aurka and Mcm2; ECM modification, such as Col6a1 and Mmp3; or inflammatory response, such as Ccl2.
Representative, commonly or uniquely induced genes are shown in Fig. 3B. Seven genes were induced by all three fibrogenic inducers; within these genes, induction of Lcn2 by silica was markedly higher than by bleomycin or paraquat, whereas induction of Col1a1 by paraquat was remarkably higher than by silica or bleomycin, and induction of Tnc and Spp1 by bleomycin or paraquat was dramatically higher than by silica. Shown also in Fig. 3B are sets of three genes from the treatment groups that illustrate unique gene induction by each of the treatment agents. Therefore, there were both common and distinct gene expression patterns among the treatments and there was a higher degree of shared gene expression between bleomycin- and paraquat-treated groups than with the silica group.
Differential mRNA and protein expression in induced lung fibrosis
We performed quantitative RT-PCR to further validate differential expression of the genes revealed by microarray analysis. Shown in Fig. 4A are the fold changes of mRNA induction of five representative genes. Col1a1 and Fn1 are fibrosis marker genes, and their mRNAs were induced significantly by all three inducers in qRT-PCR (Fig. 4A, upper panels), which correlated well with the mRNA induction revealed by microarray (Figs. 2B, 3B). Timp1 is a matrix remodeling enzyme secreted into the matrix from cells like myofibroblasts. Timp1 was highly induced during lung fibrosis (Fig. 4A, lower left panel); induction by bleomycin and induction by paraquat were similar to each other but much higher than by silica, which correlated well with the microarray data (Fig. 2B). Lcn2 and Tnc were induced by all three treatments, but with distinct and opposite patterns of induction, as revealed by microarray analysis, wherein induction of Lcn2 was highest by silica, followed by bleomycin, and lastly by paraquat, but induction of Tnc was highest by paraquat, followed by bleomycin, and lastly by silica (Fig. 3B). The qRT-PCR analysis revealed similar patterns of induction of the two genes to those by microarray analysis (Fig. 4A, lower middle and right panels). Therefore, qRT-PCR revealed similar levels and patterns of mRNA induction of the genes to those from microarray analysis.
Fig. 4.
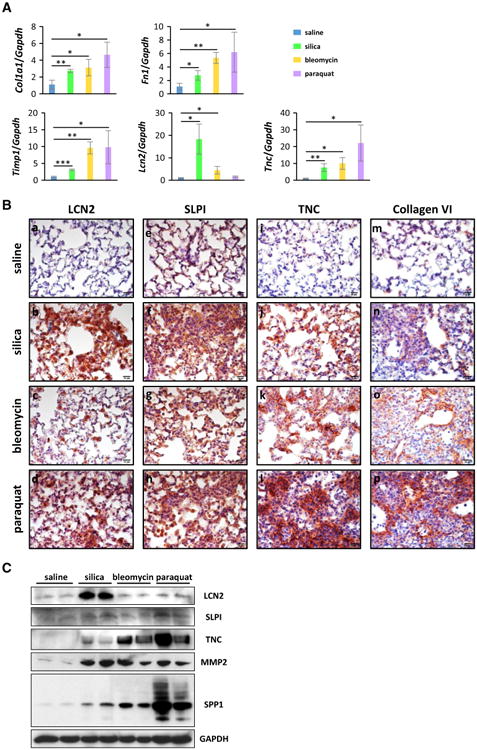
Induction of gene expression in fibrotic lung tissues. A Quantitative RT-PCR analysis was performed and fold changes of mRNA levels relative to that of Gapdh were presented as mean ± SD (n = 3 for each group; *p < 0.05; **p < 0.01, ***p < 0.001). B Levels of LCN2 (a–d), SLPI (e–h), TNC (i–l), and collagen VI (m–p) were determined on lung sections by immunohistochemistry (red, scale bar 20 μm). Blue indicates nuclear staining. C The levels of LCN2, SLPI, TNC, MMP2, and SPP1 in lung tissues were determined by immunoblotting; blotting results for two randomly selected samples from each group were presented as illustration. GAPDH was used as an internal control
To verify whether the identified differential mRNA expression of the genes affected their expression at the protein level in lung tissues undergoing fibrosis, we performed immunohistochemical analysis for representative genes on lung sections from control and treatment groups.
LCN2, also known as neutrophil gelatinase-associated lipocalin (NGAL), is a member of the lipocalin family and functions as an iron-trafficking protein. LCN2 has been implicated in several critical biological processes, including innate immunity, cell proliferation, and apoptosis. Serum and urine LCN2 are considered as biomarkers for early diagnosis of acute kidney injury (Haase-Fielitz et al. 2014; Makris and Kafkas 2012) and cardiovascular disease (Iqbal et al. 2013). As Lcn2 was significantly induced by silica, bleomycin, or paraquat from the microarray analysis (Fig. 2B), we studied LCN2 protein expression in the lungs exposed to these fibrogenic agents. Compared with saline control, the LCN2 protein was induced significantly by silica, followed by paraquat, and by bleomycin at a mild, detectable level (Fig. 4B, a–d), which correlated with the mRNA expression data from microarray and qRT-PCR (Figs. 3B, 4A) qualitatively. The data indicated that the level of LCN2 protein was increased by fibrogenic agents, though at variable levels, and suggest that LCN2 might play a role in the onset of Inflammation and induced fibrosis in the lungs.
SLPI is a potent inhibitor of proteolytic enzymes including the neutrophil proteases and elastase. SLPI plays an important role in anti-protease defense and exhibits anti-inflammatory, anti-bacterial, and anti-viral activities in the lungs (McKiernan et al. 2011; Zani et al. 2011). SLPI inhibits NF-κB-mediated pro-inflammatory gene expression, which may account for its anti-inflammatory function in the lungs (Taggart et al. 2005; Weldon and Taggart 2007). The SLPI protein level was elevated in the lungs exposed to all three fibrogenic agents, but induction was the highest by silica, followed by paraquat, and lastly by bleomycin, compared with saline control (Fig. 4B, e–h), which differed from the microarray data where the mRNA of Slpi was significantly induced by silica, but not much by paraquat or bleomycin comparatively (Fig. 3B).
TNC is an ECM protein highly expressed in a number of pathological conditions, such as chronic Inflammation and cancer, and during tissue repair following tissue injury and infection. TNC functions in ECM remodeling, cell migration and proliferation, and signal transduction, presumably through its interaction with other ECM molecules and cell surface receptors (Midwood et al. 2011; Udalova et al. 2011). TNC expression was not detected in the lungs exposed to saline (Fig. 4B, i), but paraquat induced TNC to a dramatically high amount, and silica and bleomycin induced TNC at lower levels than paraquat (Fig. 4B, j–l), which correlated well with the microarray and qRT-PCR data. This finding is consistent with previous reports in which TNC was induced and was involved in fibrotic diseases (Carey et al. 2010; Estany et al. 2014). Therefore, TNC potentially serves as a candidate for a role in the mechanism of induced lung fibrosis.
Collagen VI was reported to be induced during lung fibrosis, but its regulation and function in fibrosis remain largely unclear (Specks et al. 1995). Collagen VI is an ECM protein and is a major structural component of microfibrils. The expression and accumulation of collagen VI protein were induced at a detectable but mild level in the lungs exposed to silica; at a moderate level by bleomycin; and at a considerably high level by paraquat (Fig. 4B, m–p). Thus, collagen VI induction appeared to be more significantly induced by paraquat treatment than by bleomycin and silica.
Induction of protein expression was also examined by immunoblotting (Fig. 4C). The overall protein level of LCN2 in the lungs was induced dramatically by silica and slightly by bleomycin and paraquat, which is consistent with the results from immunohistochemical staining (Fig. 4B, a–d). The SLPI level was induced by silica and paraquat, but not by bleomycin, in the whole protein extract from lung tissues, indicating a limited effect of bleomycin on SLPI protein expression. The TNC level was significantly elevated in the lungs exposed to bleomycin and paraquat, but was only slightly elevated by silica, which is in agreement with the results detected by immunohistochemistry (Fig. 4B, i–l). MMP2 functions in the breakdown of ECM and has been shown to play an anti-fibrotic role (Giannandrea and Parks 2014; Tan et al. 2006). Mmp2 gene transcription was induced by silica, bleomycin, and paraquat (Fig. 3B). Here, we showed that the level of MMP2 protein was greatly increased by the three inducers. SPP1 (OPN) has been reported to be induced and to function during wound healing and fibrosis in multiple tissues and organs including the skin, liver, and heart (Leung et al. 2013; Matsui et al. 2004; Mori et al. 2008). The SPP1 level was increased in peripheral blood mononuclear cells and the bronchoalveolar lavage fluid from IPF patients (Desai et al. 2011; Pardo et al. 2005), as well as mouse lungs exposed to bleomycin (Berman et al. 2004). We found that the transcription level of Spp1 was induced by bleomycin or paraquat for more than tenfold; the Spp1 level was also increased by silica for 3.839-fold, though the p value with FDR of silica-treated group was higher than 0.05 (Fig. 3B). The SPP1 protein was induced dramatically by paraquat, at a moderate level by bleomycin, and at a mild but detectable degree by silica.
Taken together, these data revealed that, overall, the protein levels of the genes tested in the lungs correlated qualitatively with the expression of the proteins in fibrotic regions revealed by immunohistochemistry and expression of their mRNA revealed by microarray and qRT-PCR. One exception was Slpi, which showed strong induction at the protein level by all three inducers by immunohistochemistry; however, at the mRNA level, it was significantly induced by silica, but only mildly by bleomycin and paraquat. The mechanism(s) responsible for these differential effects of the treatments on the mRNA and protein expression levels of Slpi remains to be studied.
Shared and distinct pathways and functions activated in induced lung fibrosis
To understand the roles of the differentially regulated genes in lung fibrosis, we analyzed the signaling pathways and associated functions of the genes. We first employed canonical pathway identification for each treatment group. The top ten statistically significant pathways from exposure to each fibrogenic agent are listed in Fig. 5A. Several features are notable. First, a number of pathways important for cell cycle control were activated by the treatments (three for silica, three for bleomycin, and six for paraquat). Among these pathways, two pathways involving cell cycle regulation through the anaphase-promoting complex (APC) or spindle assembly and chromosome separation were common to all three treatments (Fig. 5A, marked by a in dark red). One pathway involving metaphase checkpoint regulation was common to silica and paraquat (marked by b in bright blue), and one pathway involving NIMA (never in mitosis gene a)-related kinase (NEK) was identical in the bleomycin and paraquat groups (marked by c in dark green). Second, five pathways of immune responses involving interferon, Toll-like receptor (TLR), alternative complement activation, substance P, and prostaglandin E2 (PGE2) functions and signaling were activated by silica (marked by d in purple), and one pathway of immune response involving C1q was activated by bleomycin (marked by d in purple). Third, several other pathways involving cell adhesion and ECM remodeling, hematopoietic stem cell function, chemokines and adhesion, or platelet activation were activated by bleomycin or paraquat, among which three pathways were activated by both agents (marked by e in bright red).
Fig. 5.

Comparison of signaling pathways and cellular processes activated by fibrogenic inducers. A Identification and comparison of activated canonical pathways. The top ten statistically significant pathways enriched by the exposure to each fibrogenic agent were presented. Shared and unique pathways activated by the agents were labeled with letters in colors as described under “Results”. B gene ontology (GO) enrichment analysis disclosed promoted cellular processes. The top ten statistically significant processes enhanced by each fibrogenic agent were presented. Shared and unique processes were marked with letters in colors as described under “Results”
These findings indicate that, within the top ten pathways activated by the fibrogenic agents, cell cycle control and proliferation were commonly activated by all three agents, which is in agreement with the conclusion from comparative analysis of differentially expressed genes, where Birc5, Cdc20, and the Hist1h2a family of genes were shown to be up-regulated by all three inducers, to impact cell proliferation and survival, DNA synthesis, and chromatin regulation. However, certain pathways and functions were preferentially activated by one or two agents. Silica preferentially stimulated innate immune functions (five out of ten pathways), whereas bleomycin and paraquat displayed more features shared with each other in activating functions relating to cell adhesion, platelet activation, and ECM remodeling. Between bleomycin and paraquat treatments, paraquat activated more cell cycle control functions than bleomycin (six vs. three pathways). Therefore, these three fibrotic agents activated both overlapping and distinct canonical pathways at the junction of acute-to-chronic transition during lung fibrosis.
Next, we performed gene ontology (GO) enrichment analysis to identify the cellular processes in which the proteins encoded by these genes are involved. Figure 5B displays the GO analysis of the up-regulated genes from each treatment group, listing the top ten statistically significant cellular processes activated by each treatment. Unlike the canonical pathway results, in which considerable overlap among the treated groups was observed, the GO cellular process study revealed more distinct patterns of effect from exposure to each agent, with the majority of the cellular processes being unique to a specific agent, and only three processes being shared between any two treatment groups (Fig. 5B, marked by a in dark red, b in bright blue, and c in dark green, respectively).
However, similarly to the canonical pathway analysis, the GO data showed that silica preferentially activated immune functions (four out of ten, marked by d in purple), whereas bleomycin and paraquat preferentially activated processes involved in ECM remodeling and collagen catabolism (marked by e in bright red). Paraquat also preferentially activated cell cycle regulation processes (marked by f in black). In addition, a number of responses related to defense against other organisms or foreign bodies were found activated by silica (five out of ten GO processes, marked by g in bright green) and a response to wounding was activated by bleomycin (marked by h in yellow), which presumably reflect the nature of silica as a foreign body with bulky mass and that of bleomycin as a cell killing and tissue wounding agent. The GO analysis clearly demonstrated that the cellular effects and biological consequences induced by these fibrogenic agents can be diverse and distinctive, which may reflect the unique mode of action by each agent in the lungs.
Upstream transcription factors and signaling networks activated by fibrogenic inducers
We further analyzed the mechanisms by which the inducers modulate the expression of the genes by identifying major upstream transcription factors and signaling networks through a transcriptional network analysis. The biological networks affected by each fibrogenic agent were identified based on p values and z-scores, and the top five networks for each treatment group are shown in Fig. 6. The top five most affected transcriptional networks for silica treatment were CREB1, c-Myc, RelA (p65 NF-κB subunit), STAT1, and SP1 (Fig. 6, top panel); for bleomycin treatment, SP1, CREB1, c-Myc, RelA (p65 NF-κB subunit), and p53 (middle panel); and for paraquat treatment, CREB1, SP1, c-Myc, p53, and E2F1 (bottom panel). Thus, the CREB1, SP1, and c-Myc networks were activated by all three fibrogenic inducers; RelA (p65 NF-κB subunit) was preferentially activated by silica and bleomycin; and p53 was preferentially activated by bleomycin and paraquat. In addition, STAT1 was activated in the silica-exposed group, and E2F1 was activated in the paraquat-treated group.
Fig. 6.
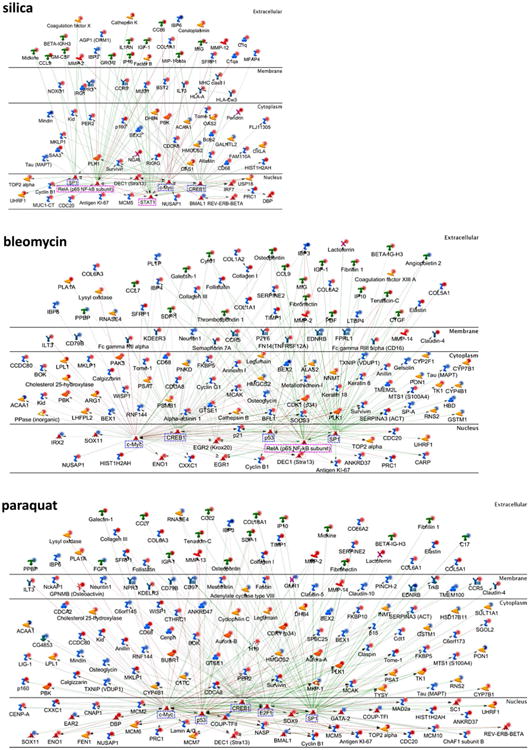
Upstream regulators and networks modulated by fibrogenic inducers. The top five statistically significant transcriptional regulation networks were identified and displayed for each agent. The regulators were presented in the cellular compartments or extracellular space where they play biological functions. The up-regulated molecules with fold changes ≥3 were marked with dark red dots and <3 with light red dots, whereas the down-regulated molecules with fold changes ≤−3 were marked with dark blue dots and >−3 with light blue dots. The transcription factors mainly involved in cell growth, cell differentiation, and cell death were labeled with blue dashed rectangles, whereas those functioning in immune responses were labeled with pink dashed rectangles. The positive/activated relation between two regulators was shown with green line, negative/inhibitory relation with red line, and unspecified relation with gray line
CREB1, SP1, c-Myc, p53, and E2F1 are known to be important regulators of cell growth, differentiation, and survival (marked in blue dashed rectangles), whereas transcription factors RelA (p65 NF-κB subunit) and STAT1 play important regulatory functions in immune responses (marked in pink dashed rectangles). The findings from the top five transcription factor networks activated by each agent suggest that: (a) all three agents activate transcription factors CREB1, SP1, and c-Myc to boost cell proliferation and survival; (b) silica activates the RelA and STAT1 pathways of gene transcription, which may account for its preferential activation of immune functions; and (c) bleomycin and paraquat share more transcriptional pathways than with silica, and both activate more transcriptional networks relating to cell cycle control and DNA damage response than silica, consistent with their apparent cytotoxicity.
In addition to the major transcription factors, Fig. 6 revealed dysregulated upstream signaling networks, the interrelationship among the regulatory factors, and the cellular localization of the factors, for each treatment. In this respect, the analysis has integrated functional molecules and pathways affected by each agent to provide a comprehensive presentation of the mechanisms of gene regulation by particulate and soluble chemical fibrogenic inducers that drive the transition from acute inflammation to chronic fibrosis during the development of lung fibrosis.
Discussion
Induced lung fibrosis in animals has served as experimental models for examining and understanding the occurrence and progression of human lung fibrotic diseases, including induced lung fibrosis like silicosis and idiopathic lung diseases like IPF. IPF has drawn a great attention in the past decades because the onset of this disease leads to a short survival time and a high fatality rate in patients, which is in part due to a lack of effective drug treatment. Several factors have been shown to significantly increase the risk of developing IPF, including cigarette smoke and a number of environmental and occupational exposures such as wood, coal, and hay dusts (Olson and Swigris 2012). In addition to IPF, several other human diseases, such as sarcoidosis and scleroderma, are also idiopathic in origin and have progressive lung fibrosis as a severe form of pathologic development and cause of mortality, though other organ systems may also be involved. Identifying the risk factors and mechanisms involved in the development of induced or idiopathic human lung fibrotic lesions is a necessary step for understanding their pathogenesis and for effective therapy and prevention against the diseases.
Several experimental lung fibrosis models, such as those induced by particles (silica) and fibers (asbestos), and those by soluble chemicals (bleomycin and paraquat), have been commonly used to examine mechanisms of lung fibrosis. The pathologic features of these models differ significantly from one another with each model only partially mimicking IPF. Furthermore, fibrogenic inducers display distinct modes of action upon entering the lungs, wherein soluble chemicals, like bleomycin and paraquat, directly kill lung cells, but insoluble particulate inducers, such as silica, likely stimulate tissue responses as a major mode of action to trigger fibrogenesis. Therefore, comparing among experimental models of lung fibrosis that have distinct pathologic features and are induced by fibrogenic agents with different modes of action would real new aspects of the pathogenesis and development of human lung fibrotic diseases like IPF.
It is known that induced experimental lung fibrosis commonly includes an acute-phase response characterized by acute inflammatory infiltration and tissue damage, and a chronic phase progression marked by interstitial fibrosis and granuloma formation; this finding suggests that common, but as-yet-unidentified, mechanisms or programs may exist and dictate the biphasic responses and the progression from acute-to-chronic fibrosis development, at the molecular level. From these considerations, we used microarray gene expression assay and computational data analysis to dissect and compare lung fibrosis models induced by silica, bleomycin, or paraquat, in order to elucidate the common and inducer-specific mechanisms critical for lung fibrosis development. We chose day 7 post-exposure as the time point for the analyses, because it represents the time point at which the peak acute responses to silica, bleomycin, and paraquat exposures take place and, thus, reflects a junction of transition for the acute-to-chronic pathologic progression.
Differentially expressed genes were identified that discern the effects of each fibrogenic agent (Figs. 2, 3; Table S1). Through comparison among the genes regulated by silica, bleomycin, or paraquat, the genes were separated into three groups: (a) those commonly regulated by three agents; (b) those commonly regulated by two agents; and (c) those uniquely regulated by one agent (Figs. 2A, 3A). Differential gene expression in fibrotic lungs was confirmed at the mRNA level by qRT-PCR and at the protein level by immunohistochemistry and immunoblotting for several selected groups of genes (Fig. 4). Analyses of canonical pathways, gene ontology, and upstream transcription factors and signaling networks confirmed the findings of the gene analyses and, more importantly, provided insightful information on the organization structure, signal transduction, function, and transcriptional regulation of the genes in the context of lung fibrosis (Figs. 5, 6).
A major finding of the gene expression study was the identification of genes and pathways commonly regulated by all three inducers; remarkably, a large number of these genes and functions are critically involved in cell cycle control, cell proliferation and survival, DNA dynamics, and chromatin regulation. Among these genes, Birc5 (survivin), Cdc20, Cdca3, Cdca8, and Cdkn3 regulate cell proliferation and survival, whereas nine members of Hist1h2a gene family that were up-regulated encode histone H2A and thereby play important roles in DNA synthesis, DNA damage and repair, chromatin architecture, cell cycle regulation, and cell proliferation and survival. Consistent with this notion, the canonical pathways involving APC and the spindle assembly and chromosome separation were commonly activated by all three inducers to mediate cell cycle control and cell proliferation. At the transcriptional level, all three fibrosis inducers stimulate the activation of gene transcription by c-Myc, CREB1, and SP1, transcription factors known to boost cell cycle control and proliferation, through specific upstream signaling networks.
During wound healing, fibroblasts are activated to proliferate and differentiate into myofibroblasts to secrete matrix proteins and cause contraction of repaired tissues; other cells, such as lung bronchial and alveolar epithelia and vascular cells, may also undergo vigorous proliferation for tissue repair and remodeling. By analogy with these findings on wound healing, it is believed that the fibroblastic proliferation following a fibrogenic stimulation is a critical mechanism for the development of lung fibrosis. Indeed, many fibrogenic inducers stimulate fibroblast proliferation in vitro. However, data obtained from in vivo study of fibroblast proliferation in lung fibrosis have been inconclusive, which may reflect the fact that, during lung fibrosis, fibroblasts and other lung cells may undertake a dynamic life cycle of proliferation, differentiation, and turnover, in a fibrotic stage-dependent manner during the development of lung fibrosis; therefore, the time point at which cell proliferation is examined after exposure would be critical for detection and quantification of cell proliferation in vivo. In this regard, our finding on the robust gene regulation relating to cell proliferation at the junction of transition from acute inflammation to chronic fibrosis provides a rational explanation to the role of cell proliferation and regulation during lung fibrosis induced by particulate and soluble chemical inducers, which may also be applicable to other forms of lung fibrosis including IPF.
A recent finding reveals that acute DNA damage induced by bleomycin significantly increased the relative nuclear abundance of histone H2A family in primary human HCA2 fibroblasts (Lopez et al. 2012). This observation indicates that fibrogenic agents may confer cytotoxicity through generating genotoxic effects, which is consistent with the previous finding that bleomycin exposure induced single- and double-strand DNA breaks, through which it resulted in cytotoxicity and cell death in the lungs (Huang et al. 1981; Tounekti et al. 1993). Therefore, induction of the Hist1h2a gene family by cytotoxic inducers like bleomycin and paraquat would be part of the response to DNA strand breaks induced by the inducers. How this mechanism of Hist1h2a gene family induction is applied to silica-induced lung fibrosis in animals and silicosis in humans remains to be studied.
The differentially expressed genes uniquely regulated by silica, bleomycin, or paraquat were identified, which facilitates the understanding of the mechanistic pathways that potentially lead to lung fibrosis induced by a specific agent. Among the 52 genes uniquely induced by silica, it is evident that a number of these genes play important roles in immune responses, including Ccl3, Ccl4, Ccl6, Cxcl1, Il1rn, Il4i1, Csf2, Oas1g, and Oas2. This finding is further supported by subsequent analyses, which revealed consistently that the silica treatment preferentially activated several canonical pathways and gene ontogeny processes related to immune response and regulation, as well as the transcriptional networks of NF-κB and STAT1, transcription factors known to play key roles in immune and inflammatory responses. In addition, silica stimulated a number of defense responses and functions, some of which are related to exposure to bulky masses, such as microorganisms and particles.
The above findings may have important implications for silicosis and lung fibrosis in humans. It is known that both innate and adaptive immune responses play important roles in fibrogenesis (Wynn and Ramalingam 2012). Therefore, an early activation of the immune system might be one of the mechanisms mediating silica-induced lung fibrosis. A portion of silicosis patients manifests apparent autoimmune symptoms or diseases, including nephritis, scleroderma, and systemic lupus erythematosus, in addition to lung fibrosis; moreover, in experimental animals, silica exhibits adjuvant effects on immune functions (NIOSH 2002; Parks et al. 1999). Whether the differentially regulated immune response genes by silica identified in the mouse model can be linked to silica's autoimmune effects observed in human patients with silicosis remains to be examined. In addition, silicosis is commonly accompanied by mycobacterial infection, such as tuberculosis, as a major clinical complication (NIOSH 2002). In addition to immune functions, we found silica preferentially activated the defense responses against other organisms and stresses. It remains to be examined how silica-induced alterations of gene expression, signaling pathways, and functions involved in immune response and defense contribute to this altered susceptibility to mycobacterial infection in patients with silicosis. Nevertheless, our findings on the regulation of immune and defense functions by silica provide an experimental approach to studying the effect of silica on immune regulation and its role in the development of lung fibrosis, autoimmune dysfunction, and mycobacterial complication in humans.
Bleomycin and paraquat share more features with each other than with silica in regard to their modes of action and pathologic features in lung fibrosis. Both chemicals selectively accumulate in the lungs, regardless of route of exposure; both are highly cytotoxic to lung cells in which they accumulate; and both induce diffusive fibrosis in the lungs that is progressive and lethal in humans. There are also notable differences between bleomycin- and paraquat-induced lung fibrosis. Namely, bleomycin kills cells by causing DNA strand breaks through an as-yet-unclear mechanism, whereas paraquat undergoes redox cycling upon entering cells to produce a large amount of ROS in lung cells and, thereby, results in oxidative lesions and cell death. Pathologically, paraquat may induce extensive hemorrhage in addition to cell death in the lungs compared with bleomycin treatment.
Our analyses of gene expression in the lungs exposed to bleomycin and paraquat uncovered both overlapping and unique patterns of gene expression and activation of pathways and transcriptional signaling networks, which appeared to be in a good agreement with their modes of action and pathology in the lungs. Firstly, there were apparently more genes affected by bleomycin and paraquat treatments than by silica treatment, which may reflect the fact that both bleomycin and paraquat caused more significant cell death and tissue damage in the lungs than silica, which would provoke more cell proliferation and repair activities, resulting in more active gene transcription. Secondly, there were more shared genes, pathways, gene ontology, and transcription factors activated between bleomycin and paraquat treatments than with silica treatment. For instance, both bleomycin and silica activated pathways implicated in cell adhesion, platelet activation, and ECM remodeling (Fig. 5) and both activated the p53 transcription network (Fig. 6); this latter effect on p53 may reflect the DNA damaging capability of the chemicals. Between bleomycin and paraquat treatments, paraquat activated more cell cycle control functions than bleomycin.
The analyses also revealed considerable numbers of genes uniquely regulated by bleomycin or paraquat. Seventy-two genes were affected by bleomycin alone, including Cdkn1a, Ccng1, Egr1, Egr2, Ctsa, Ctsb, and Ctsh. These genes are involved in certain fundamental processes, such as cell cycle regulation, mitogenesis, differentiation, and cellular protein turnover. Additionally, bleomycin uniquely induced the transcription of Cxcl13 and Ccr5, which function in inflammatory response. One hundred and twenty genes were uniquely regulated by paraquat, such as Aurka, Aurkb, Mcm2, Mcm6, and Mcm10, which play critical roles in mitosis and DNA replication; Col3a1, Col6a1, Col16a1, Col18a1, Mmp3, and Mmp13, which function in ECM modification; and Ccl2 and Ccl7, which are involved in inflammatory response. These findings provide new clues to understanding the molecular mechanisms underlying fibrotic responses specifically to bleomycin or paraquat exposure in the lungs.
In aggregate, our data from this study reveal a number of new and important patterns, pathways, and mechanisms of gene regulation that are commonly or uniquely activated at the junction of transition from acute inflammation to chronic fibrosis during the development of lung fibrosis induced by silica, bleomycin, or paraquat in vivo. The results provide a comprehensive and integrated understanding of the regulation and signal transduction of the pathways and functions affected by the fibrogenic agents to induce fibrosis, especially for the transition of acute-to-chronic pathologic development in the lungs. The findings would aid in the study to understand and treat lung fibrogenesis in human lung fibrotic diseases, including both induced lung fibrosis, such as silicosis and those induced by chemical toxicity or mycobacterial infection, and idiopathic diseases, such as IPF, sarcoidosis, and scleroderma.
Supplementary Material
Acknowledgments
This work was funded to Q.M. by National Institute for Occupational Safety and Health, Health Effects Laboratory Division.
Abbreviations
- APC
Anaphase-promoting complex
- BAL
Bronchoalveolar lavage
- CNT
Carbon nanotubes
- Col
Collagen
- ECM
Extracellular matrix
- FDR
False discovery rate
- FN1
Fibronectin
- GO
Gene ontology
- IL
Interleukin
- IPF
Idiopathic pulmonary fibrosis
- Lcn2
Lipocalin-2
- Mmp
Matrix metalloproteinase
- MWCNT
Multi-walled carbon nanotubes
- NEK
NIMA (never in mitosis gene a)-related kinase
- NF-κB
Nuclear factor-κB
- NLRP3
Nucleotide-binding oligomerization domain- like receptor, pyrin domain-containing 3
- OPN
Osteopontin
- PBS
Phosphate-buffered saline
- PGE2
Prostaglandin E2
- ROS
Reactive oxygen species
- Slpi
Secretory leukocyte peptidase inhibitor
- Spp1
Secreted phosphoprotein 1
- Timp1
Tissue inhibitor of metalloproteinases 1
- TLR
Toll-like receptor
- Tnc
Tenascin-C
Footnotes
Disclaimer The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
Electronic supplementary material The online version of this article (doi:10.1007/s00204-015-1589-3) contains supplementary material, which is available to authorized users.
Compliance with ethical standards: Conflict of interest The authors declare that they have no conflict of interest.
Ethical approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
References
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Berman JS, Serlin D, Li X, et al. Altered bleomycin-induced lung fibrosis in osteopontin-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2004;286(6):L1311–L1318. doi: 10.1152/ajplung.00394.2003. [DOI] [PubMed] [Google Scholar]
- Bissonnette E, Rola-Pleszczynski M. Pulmonary inflammation and fibrosis in a murine model of asbestosis and silicosis. Possible role of tumor necrosis factor. Inflammation. 1989;13(3):329–339. doi: 10.1007/BF00914399. [DOI] [PubMed] [Google Scholar]
- Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environ Health Perspect. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey WA, Taylor GD, Dean WB, Bristow JD. Tenascin-C deficiency attenuates TGF-ss-mediated fibrosis following murine lung injury. Am J Physiol Lung Cell Mol Physiol. 2010;299(6):L785–L793. doi: 10.1152/ajplung.00385.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degryse AL, Lawson WE. Progress toward improving animal models for idiopathic pulmonary fibrosis. Am J Med Sci. 2011;341(6):444–449. doi: 10.1097/MAJ.0b013e31821aa000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai B, Mattson J, Paintal H, et al. Differential expression of monocyte/macrophage- selective markers in human idiopathic pulmonary fibrosis. Exp Lung Res. 2011;37(4):227–238. doi: 10.3109/01902148.2010.538132. [DOI] [PubMed] [Google Scholar]
- Dinis-Oliveira RJ, Duarte JA, Sanchez-Navarro A, Remiao F, Bastos ML, Carvalho F. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Crit Rev Toxicol. 2008;38(1):13–71. doi: 10.1080/10408440701669959. [DOI] [PubMed] [Google Scholar]
- Dong J, Ma Q. Advances in mechanisms and signaling pathways of carbon nanotube toxicity. Nanotoxicology. 2015;9:658–676. doi: 10.3109/17435390.2015.1009187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Porter DW, Batteli LA, Wolfarth MG, Richardson DL, Ma Q. Pathologic and molecular profiling of rapid-onset fibrosis and inflammation induced by multi-walled carbon nanotubes. Arch Toxicol. 2015;89(4):621–633. doi: 10.1007/s00204-014-1428-y. [DOI] [PubMed] [Google Scholar]
- Estany S, Vicens-Zygmunt V, Llatjos R, et al. Lung fibrotic tenascin-C upregulation is associated with other extracellular matrix proteins and induced by TGFbeta1. BMC Pulm Med. 2014;14:120. doi: 10.1186/1471-2466-14-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawarammana IB, Buckley NA. Medical management of paraquat ingestion. Br J Clin Pharmacol. 2011;72(5):745–757. doi: 10.1111/j.1365-2125.2011.04026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Models Mech. 2014;7(2):193–203. doi: 10.1242/dmm.012062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase-Fielitz A, Haase M, Devarajan P. Neutrophil gelatinase-associated lipocalin as a biomarker of acute kidney injury: a critical evaluation of current status. Ann Clin Biochem. 2014;51(Pt 3):335–351. doi: 10.1177/0004563214521795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Mirabelli CK, Jan Y, Crooke ST. Single-strand and double-strand deoxyribonucleic acid breaks produced by several bleomycin analogues. Biochemistry. 1981;20(2):233–238. doi: 10.1021/bi00505a001. [DOI] [PubMed] [Google Scholar]
- Huaux F, Liu T, McGarry B, Ullenbruch M, Phan SH. Dual roles of IL-4 in lung injury and fibrosis. J Immunol. 2003;170(4):2083–2092. doi: 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- Husain AN, Kumar V. The lung. In: Kumar V, Abbas AK, Fausto N, editors. Robbins and Cotran pathologic basis of disease. 7th. Elsevier Saunders; Philadelphia: 2005. pp. 711–772. [Google Scholar]
- Iqbal N, Choudhary R, Chan J, Wentworth B, Higginbotham E, Maisel AS. Neutrophil gelatinase-associated lipocalin as diagnostic and prognostic tool for cardiovascular disease and heart failure. Expert Opin Med Diagn. 2013;7(2):209–220. doi: 10.1517/17530059.2013.763795. [DOI] [PubMed] [Google Scholar]
- Leung TM, Wang X, Kitamura N, Fiel MI, Nieto N. Osteopontin delays resolution of liver fibrosis. Lab Invest. 2013;93(10):1082–1089. doi: 10.1038/labinvest.2013.104. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Tollervey J, Krastins B, et al. Depletion of nuclear histone H2A variants is associated with chronic DNA damage signaling upon drug-evoked senescence of human somatic cells. Aging. 2012;4(11):823–842. doi: 10.18632/aging.100507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris K, Kafkas N. Neutrophil gelatinase-associated lipocalin in acute kidney injury. Adv Clin Chem. 2012;58:141–191. doi: 10.1016/b978-0-12-394383-5.00012-6. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Jia N, Okamoto H, et al. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension. 2004;43(6):1195–1201. doi: 10.1161/01.HYP.0000128621.68160.dd. [DOI] [PubMed] [Google Scholar]
- McKiernan PJ, McElvaney NG, Greene CM. SLPI and inflammatory lung disease in females. Biochem Soc Trans. 2011;39(5):1421–1426. doi: 10.1042/BST0391421. [DOI] [PubMed] [Google Scholar]
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3:8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midwood KS, Hussenet T, Langlois B, Orend G. Advances in tenascin-C biology. Cell Mol Life Sci. 2011;68(19):3175–3199. doi: 10.1007/s00018-011-0783-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40(3):362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L152–L160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- Morgan WKC, Seaton A. Occupational lung diseases. 3rd. W.B. Saunders Company; Philadelphia: 1995. [Google Scholar]
- Mori R, Shaw TJ, Martin P. Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med. 2008;205(1):43–51. doi: 10.1084/jem.20071412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIOSH. Health effects of occupational exposure to respirable crystalline silica. DHHS CDC NIOSH; Cincinnati, OH: 2002. DHHS (NIOSH) Publication 2002-129. [Google Scholar]
- Ohtsuka Y, Wang XT, Saito J, Ishida T, Munakata M. Genetic linkage analysis of pulmonary fibrotic response to silica in mice. Eur Respir J. 2006;28(5):1013–1019. doi: 10.1183/09031936.06.00132505. [DOI] [PubMed] [Google Scholar]
- Olson AL, Swigris JJ. Idiopathic pulmonary fibrosis: diagnosis and epidemiology. Clin Chest Med. 2012;33(1):41–50. doi: 10.1016/j.ccm.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Pardo A, Gibson K, Cisneros J, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2(9):e251. doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks CG, Conrad K, Cooper GS. Occupational exposure to crystalline silica and autoimmune disease. Environ Health Perspect. 1999;107(Suppl 5):793–802. doi: 10.1289/ehp.99107s5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DW, Hubbs AF, Mercer R, et al. Progression of lung inflammation and damage in rats after cessation of silica inhalation. Toxicol Sci. 2004;79(2):370–380. doi: 10.1093/toxsci/kfh110. [DOI] [PubMed] [Google Scholar]
- Porter DW, Hubbs AF, Mercer RR, et al. Mouse pulmonary dose- and time course-responses induced by exposure to multi-walled carbon nanotubes. Toxicology. 2010;269(2–3):136–147. doi: 10.1016/j.tox.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Rabolli V, Lo Re S, Uwambayinema F, Yakoub Y, Lison D, Huaux F. Lung fibrosis induced by crystalline silica particles is uncoupled from lung inflammation in NMRI mice. Toxicol Lett. 2011;203(2):127–134. doi: 10.1016/j.toxlet.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao GV, Tinkle S, Weissman DN, et al. Efficacy of a technique for exposing the mouse lung to particles aspirated from the pharynx. J Toxicol Environ Health A. 2003;66(15):1441–1452. doi: 10.1080/15287390306417. [DOI] [PubMed] [Google Scholar]
- Specks U, Nerlich A, Colby TV, Wiest I, Timpl R. Increased expression of type VI collagen in lung fibrosis. Am J Respir Crit Care Med. 1995;151(6):1956–1964. doi: 10.1164/ajrccm.151.6.7767545. [DOI] [PubMed] [Google Scholar]
- Taggart CC, Cryan SA, Weldon S, et al. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med. 2005;202(12):1659–1668. doi: 10.1084/jem.20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan RJ, Fattman CL, Niehouse LM, et al. Matrix metalloproteinases promote inflammation and fibrosis in asbestos-induced lung injury in mice. Am J Respir Cell Mol Biol. 2006;35(3):289–297. doi: 10.1165/rcmb.2005-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CR, Kelley TR. A brief review of silicosis in the United States. Environ Health Insights. 2010;4:21–26. doi: 10.4137/EHI.S4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tounekti O, Pron G, Belehradek J, Jr, Mir LM. Bleomycin, an apoptosis-mimetic drug that induces two types of cell death depending on the number of molecules internalized. Cancer Res. 1993;53(22):5462–5469. [PubMed] [Google Scholar]
- Udalova IA, Ruhmann M, Thomson SJ, Midwood KS. Expression and immune function of tenascin-C. Crit Rev Immunol. 2011;31(2):115–145. doi: 10.1615/critrevimmunol.v31.i2.30. [DOI] [PubMed] [Google Scholar]
- Weldon S, Taggart CC. Innate host defense functions of secretory leucoprotease inhibitor. Exp Lung Res. 2007;33(10):485–491. doi: 10.1080/01902140701756547. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zani ML, Tanga A, Saidi A, et al. SLPI and trappin-2 as therapeutic agents to target airway serine proteases in inflammatory lung diseases: current and future directions. Biochem Soc Trans. 2011;39(5):1441–1446. doi: 10.1042/BST0391441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.