

SUMMARY
SDHAF1 mutations cause a rare mitochondrial complex II (CII) deficiency, which manifests as infantile leukoencephalopathy with elevated levels of serum and white matter succinate and lactate. Here, we demonstrate that SDHAF1 contributes to iron-sulfur (Fe-S) cluster incorporation into the Fe-S subunit of CII, SDHB. SDHAF1 transiently binds to aromatic peptides of SDHB through an arginine-rich region in its C-terminus, and specifically engages a Fe-S donor complex, consisting of the scaffold, holo-ISCU, and the co-chaperone-chaperone pair, HSC20-HSPA9, through an LYR motif near its N-terminal domain. Pathogenic mutations of SDHAF1 abrogate binding to SDHB, which impairs biogenesis of holo-SDHB and results in LONP1-mediated degradation of SDHB. Riboflavin treatment was found to ameliorate the neurologic condition of patients. We demonstrate that riboflavin enhances flavinylation of SDHA, and reduces levels of succinate and Hypoxia-Inducible Factor (HIF) -1α and -2α, explaining the favorable response of patients to riboflavin.
Graphical Abstract
INTRODUCTION
Succinate dehydrogenase (SDH) or complex II (CII) is a tetrameric enzyme composed of two membrane-embedded subunits (SDHC and SDHD), anchoring the catalytically active core of the complex (SDHA and SDHB) to the inner mitochondrial membrane (Sun et al., 2005). CII contains one FAD cofactor covalently attached to SDHA and four additional prosthetic groups containing iron, in the form of a heme b moiety bound by the SDHC/SDHD membrane anchors, and three Fe-S clusters coordinated by SDHB (Hagerhall and Hederstedt, 1996).
Fe-S clusters are versatile cofactors involved in numerous biological processes that are crucial for normal human physiology (Rouault, 2015). During their biogenesis, nascent Fe-S clusters are assembled on a dedicated scaffold protein, ISCU, contained within a multimeric complex (Maio and Rouault, 2014). Subsequent binding of a chaperone/co-chaperone system facilitates transfer of the nascent Fe-S cluster to recipient proteins (Rouault, 2015). We recently identified a mechanism by which the co-chaperone HSC20 guides Fe-S cluster acquisition by specific recipients (Maio et al., 2014). The tripeptide Leu-Tyr-Arg (LYR) was identified as the binding site for the co-chaperone HSC20, which is a component of the Fe-S transfer complex together with ISCU and the HSP70 protein, HSPA9. SDHB was identified as an HSC20 binding partner which contains two highly conserved L(I)YR motifs that are essential for Fe-S cluster acquisition, and therefore, for SDH assembly and activity (Maio et al., 2014).
Isolated CII deficiency is a relatively rare cause of mitochondrial respiratory chain defects, accounting for 2–8% of OXPHOS defective cases (Ghezzi et al., 2009; Munnich and Rustin, 2001). Two main clinical phenotypes have been reported for mutations in CII-related genes: mitochondrial encephalomyopathy and cancer (Ghezzi and Zeviani, 2012; Van Vranken et al., 2014). The neurodegenerative phenotype was found to be associated with mutations in genes encoding the structural subunits SDHA, SDHB, SDHD and the assembly factor SDHAF1 (Alston et al., 2012; Ghezzi et al., 2009; Ghezzi and Zeviani, 2012; Jackson et al., 2014; Ohlenbusch et al., 2012). SDHAF1 is an LYR motif protein (also known as LYRM8), which was shown to be important for SDH activity and for assembly of the holo-complex in fibroblasts (Ghezzi et al., 2009). Homozygous mutations in SDHAF1 cause a distinctive early-onset leukoencephalopathy in which accumulations of lactate and succinate in the white matter are associated with selective loss of SDH activity (Ghezzi et al., 2009; Jain-Ghai et al., 2013; Ohlenbusch et al., 2012); however the molecular role of SDHAF1 mutations in disease pathogenesis remains undefined. We previously found that the co-chaperone HSC20 binds to SDHAF1 (Maio et al., 2014). Furthermore, SDHAF1 interacts with the C-terminus of SDHB, raising the possibility that it participates in biogenesis of the SDH complex by providing nascent Fe-S clusters to SDHB (Maio et al., 2014). Previous studies in S. cerevisiae concluded that Sdh6, the SDHAF1 ortholog, exerts its function on SDH assembly by binding to and protecting an Sdh1-Sdh2 intermediate (SDHA-SDHB in human, respectively) from the deleterious effects of oxidants (Na et al., 2014). However, the human protein failed to rescue the respiratory defect of a ΔSdh6 yeast strain (Ghezzi et al., 2009).
In the present study, we investigated the molecular role of SDHAF1 in SDH assembly by performing biochemical and functional analyses on cell lines derived from patients with reported SDHAF1 missense mutations that impair CII assembly, and from a patient with a novel nonsense mutation. We found that SDHAF1 has an active role in recruiting the Fe-S cluster transfer machinery at the C-terminus of SDHB through direct binding of its LYR motif to the co-chaperone HSC20. Moreover, we have identified the distinct regions of SDHAF1 involved in binding either HSC20 or SDHB, and have characterized the complementary interaction sites on HSC20 and SDHB. No satisfactory treatment is currently available for the majority of mitochondrial disorders and most treatment approaches have focused on supplementation with antioxidants and vitamins to counteract reactive oxygen species generation (Mahoney et al., 2002). Oral administration of riboflavin, a FAD precursor, has been shown to ameliorate the neurologic condition of patients affected by CII deficiencies and of SDHAF1 deficient patients (Bugiani et al., 2006; Jain-Ghai et al., 2013) though the mechanism behind the beneficial effects of the treatment has not been elucidated. Here, we characterize the molecular role of SDHAF1 in SDHB biogenesis and the mechanism by which riboflavin supplementation ameliorates disease progression in SDHAF1 deficient patients.
RESULTS
Pathogenic mutations in SDHAF1 impair binding to SDHB and to the Fe-S transfer complex, leading to defective CII biogenesis
We previously demonstrated that direct binding of specific targets to the co-chaperone HSC20 is mediated by affinity of its C-terminus for proteins that contain the LYR tripeptide motif (Maio et al., 2014). The LYR sequence is the distinctive feature of a family of proteins, named LYRM (LYR motif. Conserved Domains Accession: cl05087) (Angerer, 2015), that has at least ten entries in the human proteome, including SDHAF1, which has been characterized as a bona fide CII assembly factor (Ghezzi et al., 2009). We found that HSC20 interacts with SDHAF1. Here, we pursued the physiological role of the interaction of SDHAF1 with HSC20, and we found that endogenous SDHAF1 interacted with the entire Fe-S transfer complex formed by HSC20, HSPA9 and ISCU, and it also binds to SDHB (Figure 1A, HEK293 mitochondrial lysates).
Figure 1. SDHAF1 pathogenic mutations lead to defective biogenesis of CII by impairing binding to SDHB and to the Fe-S transfer complex.

(A) Immunoprecipitation (IP) of endogenous SDHAF1 from HEK293 mitochondrial lysates, followed by immunoblots (IBs) to SDHB, HSC20, HSPA9, ISCU and SDHA. CYT, cytosolic fraction; MIT, mitochondrial extracts. (B) IP of FLAG-tagged SDHAF1 wild type or the mutants, as indicated, expressed in HEK293 cells. The G57R mutation (SDHAF1G57R-F) abolished binding to SDHB and to the components of the Fe-S transfer complex. The first LYR of SDHAF1 mediates binding to HSC20. SDHAF1-F represents FLAG/MYC-tagged wild type SDHAF1; SDHAF1ILYR-F and SDHAF1IILYR-F represent constructs that lead to replacement of either the first or the second LYR sequence by alanines; SDHAF1R55P-F and SDHAF1G57R-F represent mutations found in patients of Turkish and Italian descents, respectively (Ghezzi et al., 2009). (C) In gel succinate-ubiquinone oxidoreductase (SQR) activity and native immunoblot to SDHB on mitochondrial membrane extracts from MCH46 cells silenced for 5 days to knock down SDHAF1 expression, and test of complementation by recombinant SDHAF1 wild type or by the mutants. SDHB protein levels were drastically reduced in cells lacking a functional SDHAF1 (immunoblot to SDHB), whereas UQCRFS1 (complex III Rieske protein), UQCRC2 (complex III core subunit), NDUFS1 (complex I Fe-S subunit), and ACO2 levels (mitochondrial aconitase) did not change. VDAC1 was used as a loading control. NT, non-targeting si-RNAs. (A–C, n = 6 biological replicates). See also Figure S1.
Next, we analyzed the molecular basis of CII deficiency caused by two reported SDHAF1 pathogenic missense mutations, namely substitution of Arg55 into Pro (R55P) and of Gly57 into Arg (G57R) (Ghezzi et al., 2009). Interestingly, human SDHAF1 has two LYR sequences, but only the first one, which is the canonical motif, is very highly conserved (Figure S1A). The R55P mutation affects the arginine of the second LYR sequence. To assess the molecular role of the proximal and distal LYR sequences in SDHAF1, we also generated mutants in which either the first L14Y15R16 or the second L53Y54R55 motif was replaced by triple alanines (SDHAF1ILYR-F, SDHAF1IILYR-F, respectively). Our results showed that SDHAF1ILYR-F did not interact with HSC20, HSPA9 and ISCU, and had reduced binding to SDHB (Figure 1B, HEK293 mitochondrial lysates). Since SDHAF1ILYR-F retained a wild type distal L53Y54R55, but no interaction with the Fe-S transfer apparatus was detected, we concluded that the first LYR of SDHAF1 recruits the HSC20-HSPA9-ISCU complex. SDHAF1IILYR-F and SDHAF1R55P-F partially retained the ability to interact with HSC20 and with SDHB (Figure 1B). Importantly, the G57R substitution dramatically prevented interaction with SDHB and with the Fe-S transfer complex. SDHAF1G57R-F also migrated slowly on the gel compared to wild type SDHAF1 and to the other mutants (Figures 1B and 1C). However, mass spectrometric analysis of the protein did not reveal post-translational modifications in SDHAF1G57R, suggesting that the shift in mobility is due to the mutation itself, which introduces an additional positively charged amino acid in an arginine rich region of the protein. A functional assay of the SDHAF1 mutant proteins showed that none of them restored CII succinate-ubiquinone oxidoreductase (SQR) activity, or facilitated assembly of the mature SDH complex (Figure 1C, MCH46 mitochondrial lysates). Interestingly, knock down of SDHAF1 specifically affected stability of SDHB (Figure 1C), whereas there was ample expression of the SDHA subunit, which was not found in complex with SDHAF1 (Figures 1A and 1B). Our data suggest that SDHAF1 participates directly in biogenesis of SDHB, assisting full assembly of complex II. In contrast, complex I activity was not affected (Figure S1B, MCH46 mitochondrial lysates). To determine whether SDHAF1 interacted directly with HSC20 and SDHB, we performed pull-down assays with purified S35-labeled proteins. Substitution of the first, but not of the second, LYR of SDHAF1 into triple alanines abolished interaction with HSC20 in vitro (Figure S1C), demonstrating that the first motif mediates direct binding to HSC20. Interestingly, while the G57R mutation adversely affected the interactions with SDHB and HSC20 in vivo, only its ability to bind to SDHB was impaired in vitro (Figure S1D), whereas the interaction with HSC20 was preserved (Figure S1C). The distinctive behavior of the G57R mutant in in vivo and in vitro assays suggests that binding of SDHAF1 to SDHB in vivo might be needed for recruitment of the Fe-S transfer complex by the first LYR motif of SDHAF1. Experimental support for this hypothesis appears later in this paper.
The first LYR motif of SDHAF1 directly interacts with HSC20
We further investigated the molecular determinants in the SDHAF1 sequence that mediate direct binding to HSC20 and to SDHB. Five truncated mutants of SDHAF1 were generated and used in pull down assays in vitro to test their ability to bind HSC20 and/or SDHB. We found that clones 1 and 3 of SDHAF1 (residues 1–39 and 1–35 respectively), containing the first LYR motif, interacted with HSC20 in vitro (Figure 2A). Clones 2 (residues 40–115), 4 (residues 50–115) and 5 (residues 61–115), which lacked the first LYR motif, did not interact with HSC20 (Figure 2A), though the L53Y54R55 sequence was present in clones 2 and 4. Interestingly, clones 2, 4, and 5 bound to SDHB in vitro, whereas clones 1 and 3 did not (Figure 2B). By further examining the molecular determinants that mediated binding of clone 1 of SDHAF1 to HSC20, we found that the LYR tripeptide was the major molecular determinant of the interaction, as substitution of L14Y15R16 with alanines abrogated binding to HSC20 (Figure S2A).
Figure 2. SDHAF1 binds HSC20 through its L14Y15R16 motif and to SDHB through the region L53-R65.

In vitro pull-down assay of S35-labeled FLAG-tagged SDHAF1 wild type or of five truncated clones, as indicated, in the presence of S35-HSC20 (A) or of S35-SDHB (B). Clone 3 (residues 1–35 of SDHAF1) was the smallest domain of SDHAF1 that retained binding to HSC20, whereas clone 5 (residues 61–115) was the shortest clone that bound to SDHB. In mutational mapping analyses in vivo (C) and in vitro (D), clones 8 and 12 had reduced binding to SDHB, and mutagenesis of additional amino acid residues within the region L53-R65 of SDHAF1 (clones 10 and 13) caused complete loss of the interaction with SDHB. Overall, mutations in the region L53-R65 abrogated interactions with SDHB and with the Fe-S transfer complex (HSC20, HSPA9, ISCU). (AD, n = 5 biological replicates). Immunoprecipitations in vivo shown in (C) were performed on HEK293 lysates. See also Figures S2 and S3.
Molecular features of the first LYR of SDHAF1 that enable binding to HSC20
We previously reported that some features of the LYR motif remain to be defined, as not all LYR-containing peptides bind the HSC20 Fe-S transfer complex (Maio et al., 2014). SDHAF1 sequence provides a notable example, as only the first motif binds HSC20, whereas the second LYR does not. We postulated that the steric interference or charge from adjacent residues might interfere with binding. By mutating Ser13 and Asp17 adjacent to LYR in clone 1 (S13L14Y15R16D17), into Tyr and Arg, respectively, to mimic the context of the second L53Y54R55, we found that the corresponding peptide (clone 6) did not bind HSC20 (Figure S2B). Conversely, SDHAF1 in which Tyr52 and Arg56 adjacent to L53Y54R55, were replaced by Ser and Asp, respectively (clone 7) interacted with HSC20 (Figure S2B).
Arg55 and Gly57, which are mutated in reported cases of infantile leukoencephalopathy, are present within the region L53-R65 of SDHAF1 that interacts with SDHB
Deletional analyses of the C-terminus of SDHB, which contains two Fe-S clusters, narrowed down the domain of SDHB that binds SDHAF1 to the region 146–218 of SDHB (Figure S2C). Alanine scanning mutagenesis of SDHAF1 identified that residues L53-R65 are involved in binding SDHB (Figures 2C and S2D, HEK293 lysates). Interestingly, mutations of SDHAF1 that abrogated binding to SDHB also interfered with interaction to the Fe-S transfer complex in vivo (Figure 2C), but not in vitro, where binding to HSC20 was preserved (Figure S2E), similarly to the G57R mutant (Figures 1B, 2C, S1C and S1D). This result is again consistent with the possibility that binding of SDHAF1 to SDHB in vivo precedes and is required for recruitment of the FeS transfer complex by the first LYR motif of SDHAF1, whereas the intrinsic ability of SDHAF1 to bind HSC20 in vitro depends exclusively on the presence of the first LYR sequence (see further in the paper).
Pathogenic mutations in SDHB affect aromatic amino acid residues involved in the interaction with SDHAF1
Alanine scanning mutational analyses of SDHB146-218 identified three distinct SDHB peptides that were important for binding to SDHAF1, F146-I153, L183Y184E185, and S198Y199W200W201N202 both in vitro (Figure 3A) and in vivo (Figure 3B, HEK293 lysates). Interestingly, two reported SDHB pathogenic mutations affect residues involved in the interaction with SDHAF1, p.Phe146Leu (Fokkema et al., 2011) and p.Trp200Cys (Lodish et al., 2010). These mutations changed two aromatic amino acids of SDHB (Phe and Trp) into hydrophobic (in SDHBF146L) or polar (SDHBW200C) residues and greatly diminished binding to SDHAF1 (Figures 3C and 3D, HEK293 lysates), leading to possible insights into the nature of the interaction between SDHB and SDHAF1. Arginines and hydrophobic residues are present on SDHAF1 in the region that binds SDHB, which, in turn, contains the aromatic residues Tyr, Phe, and Trp at the interface. It therefore appears likely that binding of SDHAF1 to SDHB depends on cation-π interactions (Ma and Dougherty, 1997), that enhance binding between arginines and aromatic side chains (Burley and Petsko, 1986; Karlin et al., 1994).
Figure 3. SDHAF1 interacts with three regions of SDHB enriched in aromatic amino acid residues.

(A) Alanine scanning mutational analysis of the region 146–218 of SDHB was performed to identify amino acid residues involved in binding SDHAF1. Mutations harbored by clones 2, 5 and 6 had the most detrimental effect on the interaction with SDHAF1 in vitro. (B) Co-immunoprecipitations (Co-IPs) of FLAG-tagged SDHB wild type or the alanine mutants in the region 146–218 were followed by immunoblots to SDHB, SDHAF1, HSC20, HSPA9, ISCU and SDHA. Overall, SDHB mutations, which were found to affect the interaction with SDHAF1 in vitro (panel A, clones 2, 5 and 6), were confirmed to be detrimental for binding in vivo (panel B, SDHBFYQYKI-F; SDHBLYE-F; SDHBSYWWN-F) (A and B, n = 5 biological replicates). (C and D) The pathogenic mutations SDHBF146L and SDHBW200C impaired binding to SDHAF1 in vivo. The region L171-D181 of SDHB was not involved in binding SDHAF1, as replacement of the most highly conserved amino acids with alanines did not affect interaction with SDHAF1 (panel C, SDHBLQS-RKLD-F). Immunoprecipitations in vivo shown in (B–D) were carried out on HEK293 mitochondrial lysates. (C and D, n = 4 biological replicates). See also Figure S3.
Binding of SDHAF1 to SDHB is necessary for recruitment of the Fe-S transfer complex
By testing several truncations of SDHAF1 in co-immunoprecipitation experiments in vivo, we found that binding of SDHAF1 to SDHB precedes and is necessary for recruitment of the Fe-S transfer complex by the first LYR motif of SDHAF1. Clones that lacked the SDHB binding region (L53-R65) failed to interact with the components of the Fe-S transfer complex (see Supplemental Figures 3A and 3B, lysates from HEK293 cells). To prove that by restoring binding to SDHB we could regain interaction of SDHAF1 with the Fe-S transfer complex, we generated and tested a truncated clone of SDHAF1 from which the 45 final C-terminal residues were removed, named Clone (1–70), which contained both the first LYR motif of SDHAF1 and the newly defined SDHB binding region (Figure S3C, lysates from HEK293 cells). Clone 1–70 interacted with both SDHB and with the components of the Fe-S transfer complex in vivo (Figure S3D, lysates from HEK293 cells), as expected.
Pathogenic mutations in SDHAF1 cause defective Fe-S cluster incorporation into SDHB
The data presented thus far point to an active role of SDHAF1 in the biogenesis of Fe-S clusters of SDHB because of its ability to physically interact with the Fe-S transfer complex. We addressed whether patients affected by infantile leukoencephalopathy caused by SDHAF1 mutations have defects in Fe-S cluster incorporation into SDHB, by analyzing in vivo iron content into SDHB in two distinct patient-derived cell lines. One of the SDHAF1-deficient cell lines was derived from an Italian patient harboring the SDHAF1G57R mutation (Ghezzi et al., 2009). The second cell line was obtained from a patient of Bangladeshi origin who has a novel nonsense mutation, c.103G>T in SDHAF1, predicted to prematurely truncate the protein at amino acid 35 (p.E35*) (see “Characteristics of the p.E35* SDHAF1 patient” in the Experimental Procedures). Fibroblasts from the patient homozygous for the E35* mutation showed markedly reduced SQR activity, low SDHB and undetectable SDHAF1 protein levels (Figures 4A and S4A). SDHA levels were also slightly reduced in the patient fibroblasts, whereas levels of other subunits of respiratory complexes were not diminished (Figures 4A and S4A). The phenotype of the E35* fibroblasts was successfully rescued by lentiviral-mediated transduction of wild type SDHAF1 (Figures 4A and S4A). Loss of SDHB stability is a feature of SDHAF1-deficient cells (Ghezzi et al., 2009) that also occurs when its biogenesis is impaired (Maio et al., 2014). We studied the turnover of SDHB in patient fibroblasts harboring the E35* mutation, by performing an import assay of S35-labeled SDHB into mitochondria isolated from the patient cells. SDHB was rapidly degraded in cells lacking a functional SDHAF1 (Figure 4B), whereas stable restoration of SDHAF1 expression in the patient cells stabilized SDHB. Further experiments established that the mitochondrial serine protease LONP1 was responsible for SDHB degradation (Figure 4C).
Figure 4. Impaired biogenesis of SDHB in patient cells leads to CII deficiency and rapid LONP1-mediated degradation of SDHB.

A) SQR activity and protein blots on mitochondrial lysates from fibroblasts harboring the c.103G>T mutation in SDHAF1, predicting a premature protein truncation at amino acid residue 35 (p.E35*), and from fibroblasts derived from a patient harboring the SDHAF1G57R mutation (Pt5 G57R). The phenotype of the two patient-derived cell lines was rescued by lentiviral transduction of FLAG-tagged SDHAF1 (two stable clones are shown for the SDHAF1E35* cell line, pLenti-SDHAF1-F(1)/(2)). (pLenti-C-Myc-DDK, empty vector). Re-expression of SDHAF1-F in patient cells restored CII activity and SDHB protein levels to normal (compared to control fibroblasts, MCH46). Immunoblots to NDUFS1 (complex I Fe-S subunit), and total OXPHOS showed no change in the levels of subunits of respiratory chain complexes other than CII (immunoblot to SDHB) in the patient cells. (A, n = 5 biological replicates). (B) Import of S35–labeled SDHB-F precursor protein (P-[S35]SDHB-F) into mitochondria isolated from patient fibroblasts harboring the SDHAF1E35* mutation and transduced with SDHAF1 (pLenti-SDHAF1) or with the empty vector (pLenti-C-Myc-DDK). Oligomycin (−Δψ/−Trypsin) abolished mitochondrial translocation. (C) Turnover of [S35]SDHB-F in patient cells upon knock down of CLPP/X (CLPP/X siRNAs) or LONP1 (LONP1 siRNAs). (NT siRNAs, non-targeting siRNAs). Knock down of LONP1 in SDHAF1-deficient mitochondria stabilized [S35]SDHB-F. (B and C, n = 3 biological replicates). See also Figure S4.
The cell line derived from the Italian patient harbors the SDHAF1G57R mutation and has a profound CII deficiency, which was restored by lentiviral-mediated transduction with wild type SDHAF1 (Figure 4A, Pt5 G57R). Our studies provide a molecular mechanism to explain the pathogenicity of the mutation, which interferes with binding of SDHAF1 to SDHB and to the Fe-S transfer complex. Moreover, we found that SDHAF1G57R was rapidly degraded (Figure S4B, mitochondria isolated from HEK293 cells), and it underwent LONP1-mediated degradation in mitochondrial uptake assays (Figure S4C).
We used the G57R and E35* cell lines to analyze if SDHAF1 contributes to iron incorporation into SDHB. We stably expressed SDHB-FLAG in patients’ fibroblasts, as well as in the same cell lines after restoration of SDHAF1 expression to directly compare cells that lacked or expressed SDHAF1 (Figure 5A). Over-expression of SDHB-F in the absence of a functional SDHAF1 did not restore CII activity (Figure 5B, lanes 2, 3, 8 and 9), indicating that SDHAF1 exerts its function by assisting biogenesis of SDHB rather than simply stabilizing a SDHB pool. Lower levels of recombinant SDHB-F were detected in the patient cells (Figure 5B). To compare equal amounts of SDHB in the Fe55 incorporation assay, we increased lysates from SDHAF1-patient cells approximately three-fold (Figure 6A), and equal amounts of SDHB-F were immunoprecipitated from patient and control cells (Figure 6B). A representative Fe55 autoradiogram of the eluted complex from the SDHB-F immunoprecipitation showed significant reduction of iron incorporation into SDHB synthesized in cells lacking functional SDHAF1 (Figure 6B), which was also confirmed by scintillation counting (Figure S5). Fe55 labeling of ferritin on cytosolic fractions of samples indicated that overall iron homeostasis was unperturbed by SDHAF1 deficiency (Figure 6B). These results demonstrate that SDHAF1 has an active role in assisting biogenesis of Fe-S clusters for SDHB, as modeled in Figure 6C.
Figure 5. Overexpression of SDHB in the absence of a functional SDHAF1 does not restore CII activity.

(A) Schematic of the protocol used to analyze iron incorporation into SDHB in vivo (see text). (B) Patient-derived fibroblasts and control cells (patient cells after stable restoration of SDHAF1 expression) were transduced with C-terminally FLAG-tagged SDHB (pLenti-SDHB-F), as indicated. In-gel SQR activity of complex II and protein levels are shown for the cell lines tested, including NDUFS1 (complex I subunit) and MTCO1, a mitochondrial complex IV subunit, which served as controls (B, n = 5 biological replicates).
Figure 6. SDH deficiency in infantile leukoencephalopathy caused by mutations in SDHAF1 is due to impaired Fe-S cluster incorporation into SDHB.

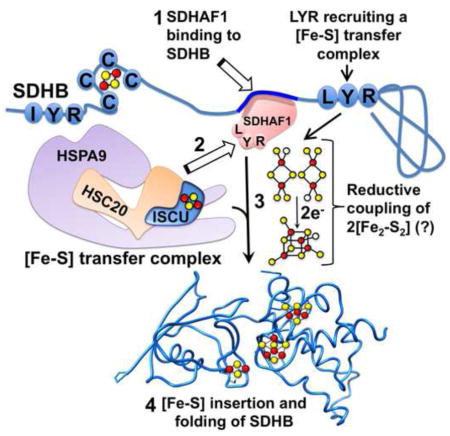
(A) SDHB-F steady state protein levels in SDHAF1-deficient fibroblasts (Fibros E35* or Pt5 G57R) were detected by immunoblot to FLAG. Since SDHB levels were lower in fibroblasts lacking a functional SDHAF1, lysate inputs were increased to compensate and equalize amounts of SDHB-F immunoprecipitated with anti-FLAG antibody. (B) Autoradiograms of SDHB-F immunoprecipitated from Fe55-labeled patient-derived and control cells. Fe55-ferritin (Fe55-Ft) levels of the corresponding cytosolic fractions indicated that iron labeling was even among different samples. (A and B, n = 5 biological replicates). (C) Schematic representation of the proposed role of SDHAF1 in the biogenesis of Fe-S clusters for SDHB. (1) Fe-S clusters are initially assembled on the main scaffold protein ISCU and subsequently transferred to recipients by the Fe-S transfer complex. (2) SDHAF1 binds to SDHB in a region enriched in aromatic amino acid residues close to the second LYR motif of SDHB that recruits a Fe-S transfer complex. (3) The LYR motif of SDHAF1 interacts with a Fe-S donor complex through direct binding to the co-chaperone HSC20. (4) Two holo-ISCU/HSC20/HSPA9 complexes may be brought into close proximity at the C-terminus of SDHB. (5) The reductive coupling of the two [Fe2-S2] clusters (Chandramouli et al., 2007) from two proximal holo-ISCU molecules may lead to Fe-S cluster formation and insertion in the C-terminus of SDHB, which folds into the mature holo protein (6). See also Figure S5.
HIFα levels are elevated in SDHAF1-deficient patient-derived cells and correlate with succinate accumulation
CII deficiency caused by mutations in SDHAF1 leads to bilateral leukoencephalopathy characterized by a succinate peak in the patients’ white matter (Ghezzi et al., 2009; Ohlenbusch et al., 2012). Accumulation of succinate can lead to Hypoxia-Inducible Factor (HIF)-1α stabilization (Selak et al., 2005), due to inhibition of prolyl hydroxylases (PHDs) involved in HIF1α hydroxylation for subsequent degradation (Maxwell et al., 1999). Besides the ubiquitous HIF1α, there is also a more cell-specific HIF2α (Iyer et al., 1998). We found that both HIF1α and HIF2α protein levels were increased in patient fibroblasts (Figures 7A–C), and also in control fibroblasts (MCH46) upon silencing of SDHAF1 (Figures 7A, 7B and S6A). Upon expression of wild type SDHAF1, HIF levels returned to basal, whereas mutant SDHAF1 proteins that were unable to restore CII activity did not reverse increased expression of HIF (Figures 7A–C and S6A). High HIF levels in the patient cells correlated with accumulation of succinate (Figure 7D), which promotes HIF stabilization (Selak et al., 2005). Succinate was, in fact, elevated in the patient cells to levels comparable to those generated by treatment of control fibroblasts with nitropropionic acid (3-NPA, Figure 7D), an irreversible inhibitor of SDH activity (Huang et al., 2006).
Figure 7. Riboflavin enhances flavinylation of SDHA and reduces succinate and HIF levels in patient cells.

(A-B) HIF1α and HIF2α levels in nuclear extracts from MCH46 cells upon knock down of SDHAF1 and in patient cells (Fibros p.E35*). SDHAF1 expression in the patient cells was restored by lentiviral transduction of wild type SDHAF1 (pLenti-SDHAF1), or by transient transfection with FLAG-tagged SDHAF1 in MCH46 cells silenced for endogenous SDHAF1. CREB, nuclear marker, and α-Tubulin were used as loading controls. (C) HIF1α/2α levels decreased in SDHAF1-deficient fibroblasts treated for 14 days with 2mg/L riboflavin. Levels of flavinylated SDHA (Flavo-SDHA) increased after treatment with riboflavin. (A–C, n = 5 biological replicates). (D) Succinate levels in SDHAF1-deficient fibroblasts and in patient cells treated for 14 days with 2mg/L riboflavin. Succinate levels in the patient fibroblasts after stable restoration of SDHAF1 expression (Fibros E35*_SDHAF1 and Pt5 G57R_SDHAF1) are also reported, as controls. Treatments of MCH46 with 3 or 6 μM nitropropionic acid (3-NPA) were performed to provide the experiment with a positive control by irreversibly inhibiting CII activity. Values are expressed as nmol of succinate per mg of proteins and given as mean ± SD (n = 6 biological replicates). (E) Schematic of the mode of action of riboflavin. Flavinylation of SDHA is enhanced by riboflavin and FAD-SDHA is able to perform conversion of succinate into fumarate; however it remains untethered in the matrix because its partner SDHB is absent due to degradation. See also Figures S6 and S7.
Riboflavin enhances SDHA flavinylation and reduces succinate and HIF levels in patient cells
Treatment with riboflavin, a FAD precursor, was previously found to be effective on patients affected by leukoencephalopathy caused by mutations in SDHAF1 (Bugiani et al., 2006; Ghezzi et al., 2009). We added riboflavin in vitro to cultured patient-derived fibroblasts in increasing concentrations to assess dose-response, and found that a dose of 2 mg/L significantly reduced HIF levels (Figures 7C and S6B) and decreased succinate (Figure 7D and S6C). To explain why levels of succinate diminish upon treatment with riboflavin, we checked CII activities. We did not find changes in CII succinate-ubiquinone reductase (SQR) activity after treatment with riboflavin (Figure S6D), as it requires a full complex, which cannot form in these cells because of impaired biogenesis of SDHB. Interestingly, following riboflavin supplementation, we could document a 2-fold increase of succinate dehydrogenase (SDH) activity (Figure S7A). Consistent with increased enzymatic activity, we found that levels of SDHA and, importantly, of flavo-SDHA were significantly increased in patient cells treated with riboflavin (Figures 7C, S6B, S7B and S7C). Increased flavinylation of SDHA accounts for the decreased succinate levels (Figures 7D and S6C), as flavo-SDHA converts succinate to fumarate (Guzy et al., 2008; Lemarie et al., 2011; Senoo-Matsuda et al., 2001; Yankovskaya et al., 2003) (Figure 7E).
DISCUSSION
Mitochondrial disorders are clinical syndromes frequently associated with abnormalities of the oxidative phosphorylation (OXPHOS) system, which consists of five enzymatic complexes of the mitochondrial respiratory chain (MRC). Central to energy metabolism, MRCs are multimeric enzymes that depend on accessory factors for assembly (Ghezzi and Zeviani, 2012). Studies of assembly defects have helped explain the molecular pathogenesis of several mitochondrial disorders and also the biogenesis of MRCs in mammals. However, the mechanism of action of assembly factors has rarely been elucidated, and the definition of an accessory factor for a given MRC is often based on the association between an observed assembly defect and mutations in a particular gene product. Here, we have characterized the role of the assembly factor SDHAF1 in the biogenesis of complex II. We previously demonstrated that the co-chaperone HSC20 of the Fe-S transfer complex interacts with SDHAF1 and LYRM7, likely assisting biogenesis of complexes II and III (Maio et al., 2014). In the present study, we investigated the physiological relevance of the interaction of SDHAF1 with HSC20 for assembly of CII. Pathogenic missense mutations in SDHAF1 cause infantile leukoencephalopathy with selective loss of CII activity by an unidentified mechanism (Ghezzi et al., 2009; Ohlenbusch et al., 2012). Here, we demonstrate that SDHAF1 recruits the Fe-S transfer complex to the C-terminus of SDHB through direct binding of its N-terminal LYR motif to the co-chaperone HSC20. The region L53-R65 of SDHAF1, enriched in arginine residues, interacts with SDHB at three binding sites containing multiple aromatic amino acids. Binding of SDHAF1 to SDHB and recruitment of the HSC20-HSPA9-holo-ISCU complex by its first LYR motif facilitates Fe-S cluster incorporation into SDHB. In vivo iron radiolabeling of SDHB showed defective Fe-S cluster incorporation into the protein synthesized in SDHAF1-deficient patient-derived cells. SDHB levels were drastically reduced in patient cells, where lack of functional SDHAF1 led to impaired biogenesis of SDHB, which was rapidly degraded by the mitochondrial protease, LONP1.
Complex II deficiency caused by mutations in SDHAF1 leads to bilateral leukoencephalopathy with a succinate peak in the white matter of patients (Ghezzi et al., 2009; Ohlenbusch et al., 2012). Succinate is a well-known intermediate of the tricarboxylic acid (TCA) cycle, and it has been characterized as a crucial metabolic signal in tumorigenesis (Selak et al., 2005) and inflammation (Tannahill et al., 2013). We found that impaired CII activity in SDHAF1-deficient patient-derived cells led to elevated intracellular succinate levels, which may have caused the elevation of HIF levels by competitively inhibiting prolyl hydroxylases. Another consequence of succinate accumulation may involve the covalent succinylation of lysine residues in numerous target proteins, including SDHA, isocitrate dehydrogenase (IDH) and pyruvate dehydrogenase (PDH), leading to important metabolic changes that potentially contribute to patients’ phenotype (Higashida et al., 2011; Jalal et al., 2015; Kim et al., 2015). No satisfactory treatment is currently available for most mitochondrial disorders and the therapeutic interventions have been focused on supplementation with antioxidants and vitamins that can reduce reactive oxygen species (Mahoney et al., 2002). Riboflavin administration was found to be an effective treatment for patients affected by SDHAF1 mutations (Bugiani et al., 2006; Ghezzi et al., 2009), and also in clinical cases of Leigh syndrome and leukoencephalopathy associated with selective CII deficiency (Jain-Ghai et al., 2013). Stable or even improved neurological conditions were reported for the SDHAF1 patients taking riboflavin (50–100 mg/day) for up to 80 months, over a monitoring period of 4.5 years; however the mechanism behind the beneficial effects of the treatment was not elucidated (Bugiani et al., 2006; Ghezzi et al., 2010). Riboflavin is a readily absorbed vitamin that is converted to FAD by riboflavin kinase and FAD synthase, both of which exist in cytosol and mitochondria (Ghezzi et al., 2010). We observed that SDHAF1 patient-derived cells had lower SDHA protein levels compared to control cells and drastically reduced levels of flavinylated SDHA. Studies in S. cerevisiae previously demonstrated that deletion of Sdh2 (SDHB ortholog in human) led to significant diminution of covalent flavinylation of Sdh1 (SDHA ortholog) (Kim et al., 2012; Robinson and Lemire, 1996), though the molecular mechanism by which Sdh2 promotes efficient flavinylation in vivo is unclear (Kim and Winge, 2013). Our new results show that impaired biogenesis of SDHB in mammalian cells also correlates with reduced flavinylation of SDHA. Treatment of SDHAF1-patient cells with riboflavin significantly enhanced flavinylation of SDHA by five to six fold compared to untreated cells (Figures 7C, S6B, S7B and S7C). Increased levels of flavo-SDHA correlated with increased SDH activity. Our results are consistent with the conclusion that holo-SDHA is sufficient to mediate SDH activity, as reported in previous studies (Guzy et al., 2008; Lemarie et al., 2011; Yankovskaya et al., 2003). Conversion of succinate into fumarate by flavo-SDHA reduced succinate accumulation in the patient cells and restored the TCA cycle, which is pivotal for much of cellular metabolism. Interestingly, riboflavin is also used in patients affected by a late-onset manifestation of an autosomal disorder caused by defects of a class of flavin-dependent enzymes, known as multiple acyl-CoA dehydrogenase deficiency (MADD). Patients are totally cured by the treatment with riboflavin; hence the clinical phenotype was named riboflavin responsive-MADD (RR-MADD) (Nielsen et al., 2013). It has been speculated that the increased availability of riboflavin, which is converted into FAD, could either compensate for flavin metabolism defects, or improve folding/stability of variant flavoproteins (Ullah et al., 2006). Our results provide a rationale for the favorable response of patients affected by SDHAF1 mutations to treatment with riboflavin. Moreover, our work on the role of SDHAF1 has implications for understanding the role of LYRM proteins that are accessory factors required for functional assembly of Complexes I and III, in addition to Complex II.
EXPERIMENTAL PROCEDURES
Tissue culture procedures
HEK293 cells were purchased from ATCC. The fibroblast cell line from the patient harboring the SDHAF1 G57R mutation (Pt5 G57R) has been reported previously (Ghezzi et al., 2009) and was obtained from the Telethon Network of Genetic Biobanks. Human dermal fibroblasts (MCH46) were a gift from Dr. Jessie Cameron (Hospital for Sick Children, Toronto, Canada). Human fibroblasts harboring the E35* mutation were derived from a 2-mm punch skin biopsy from a patient harboring the homozygous c.103G>T mutation in SDHAF1, predicting a premature protein truncation at amino acid residue 35 (p.E35*) (See patient characteristics for further information). Control and patient cells were propagated in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 2 mM glutamine at 37°C in a humidified incubator containing 5% CO2. Treatment of SDHAF1-deficient fibroblasts or control cells with riboflavin was performed by adding riboflavin (1, 2, 4 mg/L, as indicated) to the culture medium for 14 days, changing the medium every 24 hours. Experiments were carried out avoiding light exposure of the cell cultures because riboflavin is sensitive to light.
Characteristics of the p.E35* SDHAF1 patient
The patient was the second child of Bangladeshi first-degree cousins and was born at term without complications. Developmental milestones were normal until 13-months of age, when she was referred to medical evaluation for a 10-day history of acute psychomotor regression and hypotonia. Brain MRI showed abnormal symmetric T2 hyperintense signaling in periventricular white matter, corpus callosum, cerebellar medial peduncles and brain stem with sparing of U fibers. Magnetic resonance spectroscopy (MRS) demonstrated abnormal succinate and lactate peaks. Analyses of muscle biopsy specimens showed a profound defect of complex II in all fibers. Genomic DNA was purified from blood and cultured skin fibroblasts using standard procedures. The coding exon and exon-intron boundaries of SDHAF1 were amplified by PCR using intronic primers. Mutation screening was performed by bidirectional sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on an ABI3130xl automatic DNA Analyzer. Mutations were confirmed by resequencing newly amplified PCR products. Mutation screening detected a homozygous c.103G>T mutation in SDHAF1, predicting a premature protein truncation at amino acid residue 35 (p.E35*). The mutation was heterozygous in the parents and it was not found in 200 control chromosomes. Segregation of the mutation within the family and analysis of 200 control alleles was performed with ad hoc designed PCR-restriction fragment length polymorphism (PCR/RFLP) strategy.
Lentiviral mediated transduction of patient-derived fibroblasts
Patient cells harboring the E35* or the G57R mutation in SDHAF1 were engineered to stably express C-terminally FLAG/MYC-tagged human SDHAF1 or SDHAF1 without any tag at the C-terminus, by lentiviral mediated transduction with the pLenti-C-MYC-DDK-IRES-Neo (Origene), or with pLenti-IRES-Neo (from which the tags had been removed by site-directed mutagenesis), encoding wild type SDHAF1, using the lenti-vpak packaging system (Origene) (See Table S1 for a complete list of plasmids used in this study). Neomycin resistant clones were isolated, analyzed for SDHAF1 expression by western blot, and tested for CII enzymatic activity. A stable clone with restored SDHAF1 (or SDHAF1-F/M) expression and CII activity was expanded for each patient cell line. In parallel, patient cells were transduced with the empty vector (pLenti-C-Myc-DDK-IRES-Neo or with the same plasmid without tags) and used as controls. Stable expression of FLAG-tagged SDHB was established in SDHAF1-deficient cells and in patient cells after re-expression of SDHAF1, by lentiviral mediated transduction with pLYS5-SDHB-FLAG (Addgene). Hygromycin resistant clones were isolated and analyzed for expression of FLAG-tagged SDHB by western blot. A stable clone for each of the cell lines of interest was expanded and used in further experiments.
In vitro coupled transcription/translation and pull down assay of S35-labeled proteins
The TNT Quick transcription/translation system (Promega), which couples transcription/translation reactions for in vitro synthesis of eukaryotic proteins starting with plasmid DNA as a template (1μg of DNA/reaction), was used to synthesize S35-labeled proteins for pull-down assays (See Supplemental Experimental Procedures).
In vitro transcription/translation and import into isolated mitochondria
The TNT Quick coupled transcription/translation system (Promega) was used to synthesize S35-labeled proteins. The mitochondrial binding and insertion assay was performed as previously described (Maio et al., 2014). (See Supplemental Experimental Procedures).
Iron incorporation assay
The Fe55 incorporation assay into SDHB-FLAG stably expressed in SDHAF1-deficient fibroblasts or in the patient cells after restoration of SDHAF1 expression was done essentially as previously described (Maio et al., 2014). (See Supplemental Experimental Procedures).
Statistical Analyses
Where applicable, data were expressed as the mean ± standard deviation. Pairwise comparisons between two groups were analyzed using the unpaired Student’s t test.
Supplementary Material
Acknowledgments
We thank our colleagues, support by the Eunice Kennedy Shriver NICHD Intramural Research Program, the Cell lines and DNA Bank of Paediatric Movement Disorders and Neurodegenerative Diseases of the Telethon Network of Genetic Biobanks (grant GTB12001J) and the EurobiobanK Network. Supported by the Pierfranco and Luisa Mariani Foundation, Telethon Grant GGP11011 and ERC Advanced Grant FP7-322424. We thank Dr. Ross Tomaino for the mass spectrometric analyses.
Footnotes
AUTHOR CONTRIBUTIONS: N. M. and T. A. R. designed the study and wrote the manuscript. N. M. performed the experiments. D. G. and M. Z. provided G57R patient-derived cells, and T. R. and A. S. assisted in cell culture experiments. E. B. and D. M. diagnosed the E35* SDHAF1 patient. D. V. performed the genetic diagnosis of the E35* patient’s samples. N. M., D. G., M. Z., R. C. and T. A. R. analyzed the data. T. A. R. and R. C. supervised the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alston CL, Davison JE, Meloni F, van der Westhuizen FH, He L, Hornig-Do HT, Peet AC, Gissen P, Goffrini P, Ferrero I, Wassmer E, McFarland R, Taylor RW. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. Journal of medical genetics. 2012;49:569–577. doi: 10.1136/jmedgenet-2012-101146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer H. Eukaryotic LYR Proteins Interact with Mitochondrial Protein Complexes. Biology. 2015;4:133–150. doi: 10.3390/biology4010133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Lamantea E, Invernizzi F, Moroni I, Bizzi A, Zeviani M, Uziel G. Effects of riboflavin in children with complex II deficiency. Brain Dev. 2006;28:576–581. doi: 10.1016/j.braindev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Burley SK, Petsko GA. Amino-aromatic interactions in proteins. FEBS Lett. 1986;203:139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- Chandramouli K, Unciuleac MC, Naik S, Dean DR, Huynh BH, Johnson MK. Formation and properties of [4Fe-4S] clusters on the IscU scaffold protein. Biochemistry. 2007;46:6804–6811. doi: 10.1021/bi6026659. [DOI] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmuller H, D’Adamo P, Gasparini P, Strom TM, Prokisch H, Invernizzi F, Ferrero I, Zeviani M. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nature genetics. 2009;41:654–656. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D’Adamo P, Novara F, Zuffardi O, Uziel G, Zeviani M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. American journal of human genetics. 2010;86:639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Zeviani M. Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv Exp Med Biol. 2012;748:65–106. doi: 10.1007/978-1-4614-3573-0_4. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerhall C, Hederstedt L. A structural model for the membrane-integral domain of succinate: quinone oxidoreductases. FEBS Lett. 1996;389:25–31. doi: 10.1016/0014-5793(96)00529-7. [DOI] [PubMed] [Google Scholar]
- Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, Ding JY, Dornbos D, 3rd, Li X, Guthikonda M, Rossi NF, Ding Y. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. Journal of neurosurgery. 2011;114:92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- Huang LS, Sun G, Cobessi D, Wang AC, Shen JT, Tung EY, Anderson VE, Berry EA. 3-nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. The Journal of biological chemistry. 2006;281:5965–5972. doi: 10.1074/jbc.M511270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes & development. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CB, Nuoffer JM, Hahn D, Prokisch H, Haberberger B, Gautschi M, Haberli A, Gallati S, Schaller A. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. Journal of medical genetics. 2014;51:170–175. doi: 10.1136/jmedgenet-2013-101932. [DOI] [PubMed] [Google Scholar]
- Jain-Ghai S, Cameron JM, Al Maawali A, Blaser S, MacKay N, Robinson B, Raiman J. Complex II deficiency--a case report and review of the literature. Am J Med Genet A. 2013;161A:285–294. doi: 10.1002/ajmg.a.35714. [DOI] [PubMed] [Google Scholar]
- Jalal FY, Yang Y, Thompson JF, Roitbak T, Rosenberg GA. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:1145–1153. doi: 10.1038/jcbfm.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Zuker M, Brocchieri L. Measuring residue associations in protein structures. Possible implications for protein folding. J Mol Biol. 1994;239:227–248. doi: 10.1006/jmbi.1994.1365. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Jeong MY, Na U, Winge DR. Flavinylation and assembly of succinate dehydrogenase are dependent on the C-terminal tail of the flavoprotein subunit. The Journal of biological chemistry. 2012;287:40670–40679. doi: 10.1074/jbc.M112.405704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Winge DR. Emerging concepts in the flavinylation of succinate dehydrogenase. Biochim Biophys Acta. 2013;1827:627–636. doi: 10.1016/j.bbabio.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Choi IY, Dong Y, Wang WT, Brooks WM, Weiner CP, Lee P. Chronic fetal hypoxia affects axonal maturation in guinea pigs during development: A longitudinal diffusion tensor imaging and T2 mapping study. Journal of magnetic resonance imaging: JMRI. 2015;42:658–665. doi: 10.1002/jmri.24825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemarie A, Huc L, Pazarentzos E, Mahul-Mellier AL, Grimm S. Specific disintegration of complex II succinate:ubiquinone oxidoreductase links pH changes to oxidative stress for apoptosis induction. Cell Death Differ. 2011;18:338–349. doi: 10.1038/cdd.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish MB, Adams KT, Huynh TT, Prodanov T, Ling A, Chen C, Shusterman S, Jimenez C, Merino M, Hughes M, Cradic KW, Milosevic D, Singh RJ, Stratakis CA, Pacak K. Succinate dehydrogenase gene mutations are strongly associated with paraganglioma of the organ of Zuckerkandl. Endocr Relat Cancer. 2010;17:581–588. doi: 10.1677/ERC-10-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JC, Dougherty DA. The Cationminus signpi Interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, Parise G, Tarnopolsky MA. Nutritional and exercise-based therapies in the treatment of mitochondrial disease. Curr Opin Clin Nutr Metab Care. 2002;5:619–629. doi: 10.1097/00075197-200211000-00004. [DOI] [PubMed] [Google Scholar]
- Maio N, Rouault TA. Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbamcr.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maio N, Singh A, Uhrigshardt H, Saxena N, Tong WH, Rouault TA. Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell metabolism. 2014;19:445–457. doi: 10.1016/j.cmet.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. American journal of medical genetics. 2001;106:4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- Na U, Yu W, Cox J, Bricker DK, Brockmann K, Rutter J, Thummel CS, Winge DR. The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron-sulfur subunit of succinate dehydrogenase. Cell metabolism. 2014;20:253–266. doi: 10.1016/j.cmet.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen TH, Bindslev TT, Pedersen SM, Toft P, Olsen NV, Nordstrom CH. Cerebral energy metabolism during induced mitochondrial dysfunction. Acta anaesthesiologica Scandinavica. 2013;57:229–235. doi: 10.1111/j.1399-6576.2012.02783.x. [DOI] [PubMed] [Google Scholar]
- Ohlenbusch A, Edvardson S, Skorpen J, Bjornstad A, Saada A, Elpeleg O, Gartner J, Brockmann K. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis. 2012;7:69. doi: 10.1186/1750-1172-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Lemire BD. Covalent attachment of FAD to the yeast succinate dehydrogenase flavoprotein requires import into mitochondria, presequence removal, and folding. The Journal of biological chemistry. 1996;271:4055–4060. doi: 10.1074/jbc.271.8.4055. [DOI] [PubMed] [Google Scholar]
- Rouault TA. Mammalian iron-sulphur proteins: novel insights into biogenesis and function. Nat Rev Mol Cell Biol. 2015;16:45–55. doi: 10.1038/nrm3909. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Yasuda K, Tsuda M, Ohkubo T, Yoshimura S, Nakazawa H, Hartman PS, Ishii N. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. The Journal of biological chemistry. 2001;276:41553–41558. doi: 10.1074/jbc.M104718200. [DOI] [PubMed] [Google Scholar]
- Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. The Journal of biological chemistry. 2006;281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- Van Vranken JG, Na U, Winge DR, Rutter J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit Rev Biochem Mol Biol. 2014:1–13. doi: 10.3109/10409238.2014.990556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.