

Abstract
Gerstmann-Sträussler-Scheinker Disease (GSS) is a familial neurodegenerative disorder characterized clinically by ataxia, parkinsonism, and dementia, and neuropathologically by deposition of diffuse and amyloid plaques composed of prion protein (PrP). The purpose of this study was to evaluate if [11C]Pittsburgh Compound B (PiB) positron emission tomography (PET) is capable of detecting PrP-amyloid in PRNP gene carriers. Six individuals at risk for GSS and eight controls underwent [11C]PiB PET scans using standard methods. Approximately one year after the initial scan, each of the three asymptomatic carriers (two with PRNP P102L mutation, one with PRNP F198S mutation) underwent a second [11C]PiB PET scan. Three P102L carriers, one F198S carrier, and one non-carrier of the F198S mutation were cognitively normal, while one F198S carrier was cognitively impaired during the course of this study. No [11C]PiB uptake was observed in any subject at baseline or at follow-up. Neuropathologic study of the symptomatic individual revealed PrP-immunopositive plaques and tau-immunopositive neurofibrillary tangles in cerebral cortex, subcortical nuclei, and brainstem. PrP deposits were also numerous in the cerebellar cortex. This is the first study to investigate the ability of [11C]PiB PET to bind to PrP-amyloid in GSS F198S subjects. This finding suggests that [11C]PiB PET is not suitable for in vivo assessment of PrP-amyloid plaques in patients with GSS.
Keywords: [11C]Pittsburgh compound B (PiB) positron emission tomography (PET), Gerstmann-Sträussler-Scheinker disease (GSS), prion disease, neuroimaging, amyloid
Introduction
Gerstmann-Sträussler-Scheinker disease (GSS) is an autosomal dominant neurodegenerative disorder associated with prion protein (PrP) gene (PRNP) mutations [1,2]. Onset occurs most frequently between the fourth and the sixth decades of life, with an average disease duration of five years. GSS is clinically characterized by ataxia, extrapyramidal signs, and cognitive impairment [3,4]. The neuropathologic diagnosis is based on the presence of unicentric and multicentric PrP-amyloid plaques in the cerebrum and cerebellum; however, the distribution and severity of PrP deposits vary substantially among GSS phenotypes [2].
In vivo imaging of amyloid has become a valuable tool for evaluating amyloid plaque burden. [11C]Pittsburgh compound B (PiB) is a positron emission tomography (PET) radioligand with a high affinity for beta-amyloid (Aβ) fibrils [5]. [11C]PiB retention is strongly correlated with the presence of amyloid deposits made of Aβ protein in the post-mortem brain of patients with Alzheimer disease. Thus, [11C]PiB is capable of discriminating Alzheimer disease from non-amyloidogenic neurodegenerative diseases [6,7]. [11C]PiB binds with high affinity to the β-sheet structure of Aβ plaques and fails to show retention with other abnormally conformed proteins such as tau of neurofibrillary tangles and α-synuclein of Lewy bodies [5,7]. However, one case study showed cerebellar [11C]PiB retention in a subject with BRI-amyloid (ABRI) deposition, suggesting that [11C]PiB may be capable of binding non-Aβ proteins found in plaques [8].
Like Aβ and ABRI, PrP-amyloid contains a higher-order β-sheet secondary structure. Neuropathological dyes such as the Congo Red derivative (trans, trans),-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy) styrylbenzene (BSB) and the Thioflavin T derivative [4’-(methylamino)phenyl] benzothiazole (BTA-1) bind these three types of amyloid, both in vitro and in vivo in mouse and human brains [9,10]. Given that [11C]PiB is a Thioflavin T derivative, we hypothesized that it may also bind to other amyloid proteins in addition to Aβ and ABRI. Previous [11C]PiB PET studies have demonstrated that [11C]PiB is not specifically retained in individuals affected by a prion disease; however, the majority of these reports were based on single case studies and a heterogeneous sample of prion diseases [11-15].
The purpose of this study was to evaluate if [11C]Pittsburgh Compound B (PiB) positron emission tomography (PET) is capable of detecting PrP-amyloid in PRNP mutation carriers. We evaluated cross-sectional and longitudinal [11C]PiB PET scans in asymptomatic carriers of either the PRNP P102L or the PRNP F198S point mutations (proline to leucine at codon 102 (P102L) or phenylalanine to serine at codon 198 (F198S)) relative to non-carriers. Furthermore, in a symptomatic GSS F198S patient, we investigated the neuropathologic changes and correlated these findings with the [11C]PiB PET data that was obtained 46 months prior to death. This is the first study to investigate the sensitivity of [11C]PiB PET for binding PrP-amyloid plaques in the F198S GSS mutation.
Determining the ability of [11C]PiB PET to detect PrP-amyloid would provide evidence about the suitability of this tracer in trials of novel therapeutics or in early detection of PrP amyloidosis and differentiating them from other prion disease.
Materials and methods
Participants
Six participants at risk for GSS were included in this study. Three were PRNP P102L asymptomatic carriers (AC), one F198S non-carrier (NC), one F198S AC and one F198S symptomatic carrier (SC). Investigators were initially blinded to the participant’s genetic status related to the PRNP gene. Investigators were later unblinded after obtaining all neuropsychological and imaging data reported in the present paper. Neuropsychological testing, cognitive assessments, and [11C]PiB PET imaging of one NC, four ACs, and one SC (Table 1) was completed at the Indiana Alzheimer Disease Center (IADC). In addition, neuropsychological, clinical, and [11C]PiB PET data from eight cognitively healthy NCs from a separate study on aging and dementia at the IADC (the Indiana Memory and Aging Study (IMAS)) were included. All non-carriers and asymptomatic carriers displayed no clinically relevant cognitive impairment or significant cognitive complaints. The symptomatic carrier showed an initial onset of symptoms approximately 5-6 years prior to the [11C]PiB PET scan and was diagnosed with mild dementia 2 years prior to the [11C]PiB PET scan. At the time of the scan, the symptomatic carrier presented with severe neurologic and motor impairments, as well as mild dementia (Table 1). All participants or their legally authorized representative provided written informed consent according to the Declaration of Helsinki and all procedures were approved by the Indiana University Institutional Review Board.
Table 1.
Demographic characteristics and baseline neuropsychological and motor performance
| NC (n = 9) | AC (n = 4) | SC (n = 1) | p | |
|---|---|---|---|---|
| Age | 66.1 (4.2) | 44.3 (11.7) | 41 | < 0.001 |
| Gender (Male, Female) | 3, 6 | 1, 3 | 0, 1 | 0.312 |
| Education | 16.7 (1.7) | 14 (1.4) | 13 | < 0.001 |
| Mutation Status (F198S, P102L) | 0, 0 | 1, 3 | 1, 0 | n.a. |
| MMSE * | 28.9 (1.0)** | 29.5 (0.6) | 27 | 0.364 |
| CDR-SB | 0.1 (0.2) | 0 (0.0) | 6 | 0.933 |
| COWA * | 45.7 (8.0)*** | 44 (6.1) | 22 | 0.968 |
| Finger Tapping (Dom) | n.a. | 49.5 (8.8) | 26 | n.a. |
| Finger Tapping (Non-Dom) | n.a. | 43.3 (8.8) | 24 | n.a. |
| Grooved Pegboard (Dom) | n.a. | 70.3 (8.1) | 154 | n.a. |
| Grooved Pegboard (Non-Dom) | n.a. | 80.3 (17.0) | 149 | n.a. |
Mean (standard deviation). NC = non-carrier; AC = asymptomatic carrier; SC = symptomatic carrier; MMSE = Mini-Mental State Examination; CDR-SB = Clinical Dementia Rating-Sum of Boxes; COWA = Controlled Oral Word Association; n.a. = not available; Dom = dominant hand; Non-Dom = non-dominant hand).
Covaried for age, gender, and education;
Missing one non-carrier value;
Missing three non-carrier values.
Genotyping
Identification of point mutations in the PRNP gene was done as previously described [16].
Briefly, genomic DNA was isolated from fresh blood. The complete PRNP gene was amplified and direct sequencing performed using the DTCS quick start kit (Beckman Coulter, Fullerton, CA). The products were loaded onto a CEQ 8000 GeXP Genetic Analysis System (Beckman Coulter). DNA sequences were compared with the published PRNP sequence (www.ncib.nlm.nih.gov).
Acquisition and assessment of [11C]PiB PET scans
All participants received a baseline [11C]PiB PET scan, acquired on a Siemens EXACT HR+ scanner. Three asymptomatic carriers (2 PRNP P102L, 1 PRNP F198S) also received a second [11C]PiB PET scan approximately one year after the initial scan. Prior to each scan, a 10-minute transmission scan using three internal rod sources was acquired for attenuation correction. For the 6 individuals at risk for GSS (1 NC, 4 AC, 1 SC), injection of [11C]PiB was followed by a 40 min uptake period. Dynamic data acquisition protocol was then initiated with the following frame sequence: 6 × 5 min. Two participants from the IMAS study received a similar protocol as those at risk for GSS (40 min uptake period, dynamic acquisition, frame sequence: 10 × 5 min). In the other 6 participants from IMAS (6 NC), data acquisition was initiated with the injection of [11C]PiB and the frame sequence was: 10 × 30 sec, 9 × 1 min, 2 × 3 min, 8 × 5 min, 3 × 10 min. The average [11C]PiB injected across all participants (those at risk for GSS and IMAS) was 11.17±2.0 mCi. Scans were reconstructed using manufacturer’s software (Siemens; Knoxville, TN) for filtered back-projection. Corrections for scatter, randoms, and attenuation were applied.
Reconstructed [11C]PiB scans were processed using standard techniques. Using Statistical Parametric Mapping 8 (SPM8; Welcome Department of Cognitive Neuroscience, London, UK), scans were converted from ECAT to NiFTI format, coregistered to the structural magnetic resonance (MR) scan from the same visit (3T MPRAGE scan; Siemens Tim Trio), spatially aligned on a frame-by-frame basis, and normalized to Montreal Neurologic Institute (MNI) space using matrices from segmentation of the same time-point MRI. A static [11C]PiB image from 40-70 minutes was created from the appropriate frames (depending on scan acquisition type (see above)) and was intensity normalized using a pons reference region to create a standardized uptake value ratio (SUVR) image for each participant. SUVR images were smoothed with an 8 mm FWHM Gaussian kernel.
Visualization of the average group [11C]PiB images served as a primary qualitative assessment. Mean baseline [11C]PiB SUVR images and mean normalized baseline structural MR images for each diagnostic/mutation status group were created using SPM8. To visualize [11C]PiB retention by group, mean [11C]PiB SUVR images were overlaid onto the respective group mean MR images in MRIcron (http://www.mccauslandcenter.sc.edu/mricro/mricron/). For each participant, mean [11C]PiB SUVR values were extracted from a global cortical grey matter (GM) region of interest (ROI) and a whole cerebellum ROI defined from the AAL atlas and extracted using MarsBaR [17]. SUVR values from each ROI were compared between diagnostic/mutation status groups as described below (Statistical Analysis). In the three AC subjects with follow-up scans, the extracted mean [11C]PiB SUVR values from the ROIs at both time-points were graphed to complement qualitative visual observations.
Neuropathologic assessment
The symptomatic individual (PRNP F198S) expired 46 months after the [11C]PiB scan. Tissue was harvested at Indiana University School of Medicine. The fresh brain was hemisected along the mid-sagittal plane. The left hemibrain was fixed in formalin and the right hemibrain was sliced, frozen, and stored at -70°C.
Following fixation in a 10% formalin solution, brain tissue samples were dehydrated in graded alcohols, cleared in xylene, and embedded in paraffin. Eight-micrometer-thick sections from multiple brain areas were stained with the histological and immunohistochemical methods described below. Hematoxylin and eosin (H&E) and luxol fast blue with hematoxylin & eosin (LFB-H&E) were used to survey gray and white matter for neuronal losses, gliosis, vascular pathology, and other possible pathologic lesions. Thioflavin S method was used to visualize amyloid deposits and neurofibrillary tangles. Neurodegenerative pathology was analyzed using antibodies raised against amyloid β (Aβ) (21F12), tau (AT8), and PrP (3F4).
The signal from polyclonal antibodies was visualized using avidin-biotin, with goat anti-rabbit immunoglobulin as the secondary antibody, followed by horseradish peroxidase-conjugated streptavidin and the chromogens diaminobenzidine or tetramethylbenzidine. The signal from monoclonal antibodies was detected using avidin-biotin, with goat anti-mouse immunoglobulin as the secondary antibody, followed by alkaline phosphatase-conjugated streptavidin and the chromogen diaminobenzidine or tetramethylbenzidine. Immunohistochemical sections were counterstained with hematoxylin.
A semi-quantitative analysis of anatomical brain regions was carried out using a Leica DMLB microscope (Leica Wetzlar, Germany) from the following regions: superior frontal gyrus, middle frontal gyrus, anterior cingulate cortex, posterior cingulate cortex, superior and middle temporal gyri, superior parietal lobule, insular cortex, occipital cortex, amygdala, hippocampus, subiculum, parahippocampus, caudate nucleus, putamen, globus pallidus, thalamus, cerebellar hemisphere, dentate nucleus, substantia nigra, locus coeruleus, basis pontis, and inferior olivary nucleus. The severity of neuropathologic changes were evaluated using a subjective scale: none = 0, mild = 1 (+), moderate = 2 (++), and severe = 3 (+++).
Statistical analysis
Differences between NC and mutation carriers in continuous demographic variables and baseline neuropsychological test performance were evaluated using an ANCOVA model. Differences in gender by diagnostic/mutation status were tested using a chi-square test. Age and years of education were used as covariates when evaluating neuropsychological test scores. An ANCOVA model was also used to test for association of mutation status (carrier vs. non-carrier) with mean [11C]PiB SUVR values from the global cortical GM and cerebellar ROIs, covaried for age and gender. All statistical analyses employed SPSS (version 22.0, Chicago, IL) and graphs were created with SigmaPlot (version 10.0).
Results
The demographics, genetic status, and neuropsychological testing results at baseline are displayed in Table 1. Significant differences were observed for age and education between NC and mutation carriers. Although not statistically significant, more female than male participants were available for this study across all groups. As expected, the SC showed lower cognition and significantly impaired manual motor performance compared to ACs and NCs.
No apparent visual differences in baseline [11C]PiB PET uptake were observed between the NCs, the ACs, and the SC individual, suggesting minimal [11C]PiB binding in ACs and even in the symptomatic individual (Figure 1A-C). MRI of the symptomatic carrier revealed a mild degree of atrophy of the cerebellar cortex. Quantitative regional analysis did not reveal any significant difference in [11C]PiB retention in either global cortical GM or cerebellum between mutation carriers (SC or ACs) and NCs (Figure 2D). Qualitative observation of the baseline and longitudinal follow-up scans for the 3 ACs did not show any appreciable increase in [11C]PiB signal over time (Figure 2A-C). This observation was supported by minimal longitudinal change in mean [11C]PiB SUVR in cortical GM and cerebellum ROIs (Figure 2D).
Figure 1.

Baseline [11C]PiB PET in F198S and P102L carriers and non-carriers. [11C]PiB SUVR images for (A) a symptomatic carrier, (B) asymptomatic carriers (n = 4), and (C) non-carriers (n = 9). Note that (B and C) are the average image across participants within each group, while A is a single participant. No notable visual differences in [11C]PiB SUVR images were observed between the symptomatic carrier, asymptomatic carriers, and non-carriers. (D) [11C]PiB SUVR levels in the global cortical grey matter and the whole cerebellum in non-carriers and mutation carriers are displayed. No significant difference was observed in global cortex and whole cerebellum [11C]PiB SUVR between non-carriers and mutation carriers.
Figure 2.

Baseline [11C]PiB PET in F198S and P102L asymptomatic carriers. Baseline (left) and follow-up (right) [11C]PiB SUVR images for 3 asymptomatic carriers (2 P102L (A, B), 1 F198S (C)). No appreciable increase in [11C]PiB from the baseline to the follow-up scan was observed in any of the participants. (D) Quantitation of mean [11C]PiB SUVR in the cortical grey matter and whole cerebellum supported this qualitative observation.
Moderate (++) to severe (+++) neuronal loss was observed in the superior frontal and cingulate gyri, putamen, caudate nucleus, superior and middle temporal gyri, hippocampus, subiculum, entorhinal cortex, superior parietal lobule, calcarine cortex, cerebellum, midbrain, and spinal cord (anterior horn cells; data not shown). In addition, moderate (++) axonal and myelin loss was observed in the superior frontal and cingulate gyri, caudate nucleus and putamen, superior parietal lobule, superior and inferior temporal gyri, occipital lobe, cerebellar white matter, and spinal cord (dorsal spinocerebellar tract, anterior spinocerebellar tract, lateral corticospinal tract, posterior columns; data not shown).
PrP (Figure 3A, 3C) and Thioflavin S positive (Figure 3E) unicentric and multicentric plaques (severe, +++) were observed in the superior frontal and cingulate gyri, putamen, caudate nucleus, superior and middle temporal gyri, hippocampus, entorhinal cortex, superior parietal lobule, calcarine cortex, cerebellum, and midbrain.
Figure 3.
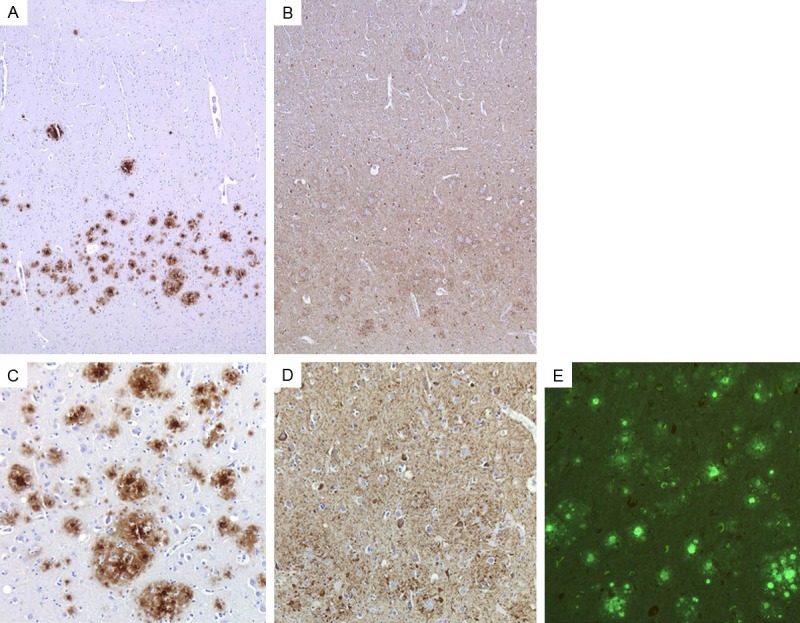
Extracellular PrP deposits and intracellular tau aggregates shown by immunohistochemistry and Thioflavin S in the frontal cortex of the PRNP F198S carrier. 3F4 immunohistochemistry reveals unicentric and multicentric PrP immunopositive plaques (A, C). AT8 immunohistochemistry reveals tauimmunopositive neurons and neuropil threads (B, D). Plaques and neurofibrillary tangles are fluorescent with Thioflavin S. PrP deposits and tau aggregates are shown at two different magnifications (A, B = 50 × total magnification and C-E = 100 × total magnification).
Tau-immunopositive neurofibrillary tangles (+++) (Figure 3B, 3D) and neuropil threads (+++) (Figure 3B, 3D) were observed in the superior frontal and cingulate gyri, entorhinal cortex, amygdala, substantia innominata, superior and middle temporal gyrus, thalamus, superior parietal lobule, middle frontal gyrus, posterior cingulate gyrus, precuneus, inferior parietal lobule, and substantia nigra. Figure 3 is from frontal cortex but is representative of the cerebral cortex. PrP and tau pathologies were more prominent in the deeper layers of the cerebral cortex. In the cerebellum, there was a severe (+++) loss of neuronal perikarya which was associated with numerous (+++) PrP-immunopositive plaques. The white matter showed extensive (+++) axonal loss. Plaques were observed throughout all layers of the cerebellar cortex (Figure 4A-C). No Aβ immunopositive deposits were seen.
Figure 4.
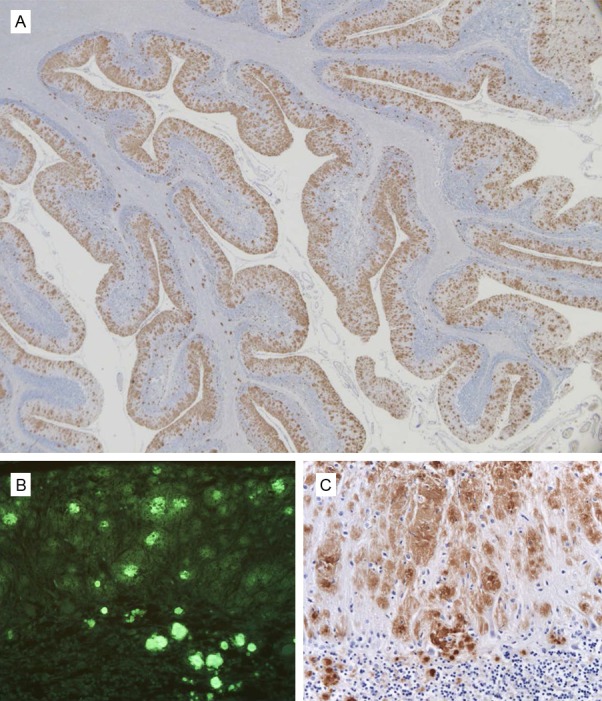
Extracellular PrP deposits shown by immunohistochemistry and Thioflavin S in the cerebellum of the PRNP F198S carrier. 3F4 immunohistochemistry reveals unicentric and multicentric PrP immunopositive plaques throughout the cerebellar cortex (A, C). Plaques are fluorescent with Thioflavin S (B). PrP deposits are shown at two different magnifications (A = 10 × total magnification and B = 100 × total magnification). Thioflavin S fluorescent plaques are shown at 100 × total magnification.
Discussion
[11C]PiB PET imaging is highly informative for the in vivo early detection of deposits of fibrillar Aβ and possibly for the differential diagnosis of Alzheimer disease versus other dementias. The aim of the present study was to determine the feasibility of using [11C]PiB PET to detect PrP-amyloid deposits in individuals at risk for developing GSS, who are carriers of the PRNP P102L or F198S mutations. To our knowledge, this is the largest PET study investigating [11C]PiB in individuals at risk for GSS and the first to evaluate [11C]PiB in GSS subjects with the F198S mutation. Furthermore, this is the first study evaluating individuals at risk for a hereditary prion disease longitudinally using [11C]PiB PET. Our results show that there was no appreciable [11C]PiB retention in one symptomatic and four asymptomatic mutation carriers. Considering the fact that the asymptomatic mutation carriers were close to the age of disease onset of their parents, it is conceivable that PrP-amyloid might have been already present in some of the AC individuals and particularly in the PRNP F198 symptomatic case.
The findings in our sample are consistent with other reports that also show no [11C]PiB retention in participants with a PRNP mutation. Most of these studies investigated [11C]PiB PET in a single subject, with one study reporting a slightly larger sample size [11]. Our data extend these preliminary findings in a larger cohort of participants. However, other studies have suggested that alternative amyloid tracers, including [18F]FDDNP and [11C]BF-227, show some sensitivity for GSS PrP-amyloid, opening up the possibility that the various amyloid radioligands have unique binding properties with respect to amyloid type [15,18].
The apparent lack of [11C]PiB retention was not likely due to an absence of PrP-amyloid in the PRNP mutation carriers studied. No [11C]PiB retention was observed in the symptomatic individual, who had significant PrP-amyloid deposition in cortical and cerebellar regions, as shown in the neuropathologic specimen obtained 46 months after the PET study. The impairment in neuropsychological and motor performance of this patient at the time of the PET scan also suggested a significant alteration of cerebro-cortica, cerebellar, and striatal function likely due to PrP-amyloid deposition. Further, in cases carrying the F198S mutation PrP-amyloid deposition begins prior to significant clinical symptoms (BG unpublished observations), suggesting that the older F198S AC in this study may also have had PrP-amyloid deposits that were not detected by [11C]PiB PET. In addition, in some cases Aβ is observed around the PrP-amyloid plaques [19,20]. However, we were unable to detect this in vivo using [11C]PiB PET despite its known sensitivity for Aβ. MRI data of the SC obtained at the time of the [11C]PiB PET imaging (data not shown), revealed atrophy of the cerebellar cortex, a finding supporting the view that extensive PrP amyloid deposits were already present 46 months before death. Given the lack of appreciable specific signal in the symptomatic GSS patient, we suggest that [11C]PiB may not bind to PrP-amyloid nor is there enough Aβ deposition to be detected by [11C]PiB PET. The apparent lack of ability of [11C]PiB to detect PrP-amyloid plaques in GSS could be due to several factors, including altered tracer distribution and metabolism in this sample. However, the most likely possibility is that the PrP-amyloid plaques possess a different conformational structure from the Aβ plaques, which may not be conducive to [11C]PiB binding despite their sensitivity to thioflavin on neuropathologic assessment.
One limitation of this study is the small sample of symptomatic carriers who are known to have extensive neuropathological PrP-amyloid deposits. Future studies using a larger sample of affected individuals would be desirable, as these participants would have significant amounts of the PrP-amyloid deposits. In addition, evaluation of other amyloid tracers alone or in combination with [11C]PiB may potentially provide valuable insight into the timing and distribution of PrP-amyloid deposition in this population, as well as the relative sensitivity of different amyloid tracers for detection of the various forms of amyloid. Finally, PET studies with recently developed tau radiotracers in individuals affected by GSS associated with the PRNP F198S, which are known to have neurofibrillary tangles, in combination with amyloid tracers could be useful for the in vivo analysis of pathophysiological processes underlying disease symptoms and for tracking of disease progression.
In summary, we found no evidence for significant [11C]PiB retention in participants carrying the PRNP P102L or F198S mutations, regardless of diagnostic status. Our findings agree with previous reports and suggest that [11C]PiB may be clinically useful for ruling out the presence of Aβ plaques in people at risk for GSS, but would provide no utility for detecting PrP-amyloid pathology in these patients.
Acknowledgements
We would like to thank the GSS family members and IMAS participants for their participation in our research. We would also like to thank John D. West and Tamiko R. Magee for their assistance with this project. This research was supported in part by grants from the National Institute on Aging (P30 AG10133 to BG and AJS; R01 AG19771, to AJS), the Alzheimer’s Association (to SLR), the Indiana University Health-Indiana University School of Medicine Strategic Research Initiative (to SLR), and the Indiana Clinical and Translational Science Institute (CTSI; to SLR).
Disclosure of conflict of interest
None.
References
- 1.Gerstmann J, Sträussler E, Scheinker I. Über eine eigenartige hereditär- familiäre Erkrankung des Zentralnervensystems. Zugleich ein Beitrag zur Frage des vorzeitigen lokalen Alterns. Z Neur Psychiat. 1936;154:736–762. [Google Scholar]
- 2.Ghetti B, Tagliavini F, Takao M, Bugiani O, Piccardo P. Hereditary prion protein amyloidoses. Clin Lab Med. 2003;23:65–85. viii. doi: 10.1016/s0272-2712(02)00064-1. [DOI] [PubMed] [Google Scholar]
- 3.Farlow MR, Yee RD, Dlouhy SR, Conneally PM, Azzarelli B, Ghetti B. Gerstmann-Sträussler-Scheinker disease. I. Extending the clinical spectrum. Neurology. 1989;39:1446–1452. doi: 10.1212/wnl.39.11.1446. [DOI] [PubMed] [Google Scholar]
- 4.Unverzagt FW, Farlow MR, Norton J, Dlouhy SR, Young K, Ghetti B. Neuropsychological function in patients with Gerstmann-Sträussler-Scheinker disease from the Indiana kindred (F198S) J Int Neuropsychol Soc. 1997;3:169–178. [PubMed] [Google Scholar]
- 5.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 6.Rabinovici GD, Furst AJ, O’Neil JP, Racine CA, Mormino EC, Baker SL, Chetty S, Patel P, Pagliaro TA, Klunk WE, Mathis CA, Rosen HJ, Miller BL, Jagust WJ. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- 7.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villemagne VL, Pike K, Pejoska S, Boyd A, Power M, Jones G, Masters CL, Rowe CC. 11CPiB PET ABri imaging in Worster-Drought syndrome (familial British dementia): a case report. J Alzheimers Dis. 2010;19:423–428. doi: 10.3233/JAD-2010-1241. [DOI] [PubMed] [Google Scholar]
- 9.Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Mathis CA. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 2001;69:1471–1484. doi: 10.1016/s0024-3205(01)01232-2. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa K, Doh-ura K, Kudo Y, Nishida N, Murakami-Kubo I, Ando Y, Sawada T, Iwaki T. Amyloid imaging probes are useful for detection of prion plaques and treatment of transmissible spongiform encephalopathies. J Gen Virol. 2004;85:1785–1790. doi: 10.1099/vir.0.19754-0. [DOI] [PubMed] [Google Scholar]
- 11.Boxer AL, Rabinovici GD, Kepe V, Goldman J, Furst AJ, Huang SC, Baker SL, O’Neil JP, Chui H, Geschwind MD, Small GW, Barrio JR, Jagust W, Miller BL. Amyloid imaging in distinguishing atypical prion disease from Alzheimer disease. Neurology. 2007;69:283–290. doi: 10.1212/01.wnl.0000265815.38958.b6. [DOI] [PubMed] [Google Scholar]
- 12.Plate A, Benninghoff J, Jansen GH, Wlasich E, Eigenbrod S, Drzezga A, Jansen NL, Kretzschmar HA, Botzel K, Rujescu D, Danek A. Atypical parkinsonism due to a D202N Gerstmann-Straussler-Scheinker prion protein mutation: first in vivo diagnosed case. Mov Disord. 2013;28:241–244. doi: 10.1002/mds.25188. [DOI] [PubMed] [Google Scholar]
- 13.Villemagne VL, McLean CA, Reardon K, Boyd A, Lewis V, Klug G, Jones G, Baxendale D, Masters CL, Rowe CC, Collins SJ. 11C-PiB PET studies in typical sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2009;80:998–1001. doi: 10.1136/jnnp.2008.171496. [DOI] [PubMed] [Google Scholar]
- 14.Hyare H, Ramlackhansingh A, Gelosa G, Edison P, Rudge P, Brandner S, Brooks DJ, Collinge J, Mead S. 11C-PiB PET does not detect PrP-amyloid in prion disease patients including variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2012;83:340–341. doi: 10.1136/jnnp.2010.233692. [DOI] [PubMed] [Google Scholar]
- 15.Okamura N, Shiga Y, Furumoto S, Tashiro M, Tsuboi Y, Furukawa K, Yanai K, Iwata R, Arai H, Kudo Y, Itoyama Y, Doh-ura K. In vivo detection of prion amyloid plaques using [(11)C] BF-227 PET. Eur J Nucl Med Mol Imaging. 2010;37:934–941. doi: 10.1007/s00259-009-1314-7. [DOI] [PubMed] [Google Scholar]
- 16.Hsiao K, Dlouhy SR, Farlow MR, Cass C, Da Costa M, Conneally PM, Hodes ME, Ghetti B, Prusiner SB. Mutant prion proteins in Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Nat Genet. 1992;1:68–71. doi: 10.1038/ng0492-68. [DOI] [PubMed] [Google Scholar]
- 17.Brett M, Anton JL, Valabregue R, Poline JB. Region of interest analysis using an SPM toolbox [abstract]. Presented at the 8th International Conference on Functional Mapping of the Human Brain; June 2-6; Sendai, Japan. 2002. [Google Scholar]
- 18.Kepe V, Ghetti B, Farlow MR, Bresjanac M, Miller K, Huang SC, Wong KP, Murrell JR, Piccardo P, Epperson F, Repovs G, Smid LM, Petric A, Siddarth P, Liu J, Satyamurthy N, Small GW, Barrio JR. PET of brain prion protein amyloid in Gerstmann-Straussler-Scheinker disease. Brain Pathol. 2010;20:419–430. doi: 10.1111/j.1750-3639.2009.00306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyazono M, Kitamoto T, Iwaki T, Tateishi J. Colocalization of prion protein and beta protein in the same amyloid plaques in patients with Gerstmann-Straussler syndrome. Acta Neuropathol. 1992;83:333–339. doi: 10.1007/BF00713522. [DOI] [PubMed] [Google Scholar]
- 20.Bugiani O, Giaccone G, Verga L, Pollo B, Frangione B, Farlow MR, Tagliavini F, Ghetti B. Beta PP participates in PrP-amyloid plaques of Gerstmann-Straussler-Scheinker disease, Indiana kindred. J Neuropathol Exp Neurol. 1993;52:64–70. doi: 10.1097/00005072-199301000-00008. [DOI] [PubMed] [Google Scholar]