

Abstract
The nuclear enzyme PARP1 plays a central role in sensing DNA damage and facilitating repair. Tumors with BRCA1/2 mutations are highly dependent on PARP1 as an alternative mechanism for DNA repair, and PARP inhibitors generate synthetic lethality in tumors with BRCA mutations, resulting in cell cycle arrest and apoptosis. Zhou et al. recently synthesized an 18F-labeled PARP1 inhibitor ([18F]FluorThanatrace) for PET, and demonstrated high specific tracer uptake in a xenograft model of breast cancer [1]. In the current study, we characterize the level of baseline PARP expression and activity across multiple human breast cancer cell lines, including a BRCA1 mutant line. PARP expression and activity, as measured by levels of PAR and PARP1, is correlated with in vitro [18F]FluorThanatrace binding as well as tracer uptake on PET in a xenograft model of breast cancer. Radiotracer uptake in genetically-engineered mouse fibroblasts indicates [18F]FluorThanatrace is selective for PARP1 versus other PARP enzymes. This motivates further studies of [18F]FluorThanatrace as an in vivo measure of PARP1 expression and activity in patients who would benefit from PARP inhibitor therapy.
Keywords: PARP1, BRCA mutation, breast cancer
Introduction
Poly (ADP-ribose) polymerase-1 (PARP1) is the most abundant and well-studied member of the PARP family of nuclear enzymes [2]. While PARP1 has a host of cellular functions, including roles in transcription, translation, chromatin remodeling, and telomere maintenance [3], its role in sensing DNA damage and initiating base excision repair of single stranded breaks makes it an attractive anticancer target. PARP1’s DNA binding domain binds to DNA breaks via two zinc finger domains, and the C-terminal catalytic domain sequentially transfers ADP-ribosyl moieties (PAR) from nicotinamide-adenine-dinucleotide (NAD+) to chromatin-associated acceptor proteins involved in repair [2,4]. Given its function in mediating single strand break repair, PARP inhibitors (PARPi) were initially investigated as potentiators of chemotherapy and radiation therapy for a large array of solid malignancies, including breast, pancreatic, gliomas, non-small cell lung, and melanoma [5,6].
More recently, PARPi have demonstrated particular promise in the treatment of tumors that are deficient in DNA repair pathway enzymes and are thus dependent on PARP1 for survival. In tumors with DNA repair deficiencies, such as BRCA1 or BRCA2 (BRCA1/2) mutation breast or ovarian cancers, PARPi offer potential as a stand-alone therapy via the induction of synthetic lethality [6,7]. Functional BRCA1 and BRCA2 are necessary for the repair of double stranded DNA breaks (DSBs) via homologous recombination. Cells with defective BRCA1/2 are unable to localize RAD51 to sites of DNA damage, resulting in impaired homologous recombination and an accumulation of genetic abnormalities, promoting genomic instability [8]. PARPi leads to increased single stranded breaks (SSBs), and the multitude of SSBs eventually leads to DSBs via replication fork collapse [9]. PARPi induced trapping of PARP enzymes onto DNA correlates with toxicity of this class of agents, indicating additional modes of action beyond accumulated SSBs the presence BRCA1/2 mutations, PARPi induced lesions can’t be repaired, resulting in chromosomal aberrations and cell death.
While PARPi offer promise in the treatment of breast cancer, particularly in tumors with DNA repair deficiencies, not all tumors benefit from PARPi therapy. Therefore, a noninvasive imaging procedure that can assess PARP1 levels and/or activity could be useful in determining which patients may benefit from PARPi therapy. Measuring these parameters is of considerable interest given their established relationship to responses to clinically approved PARPi [10,11]. Two 18F-labeled analogs of the PARPi olaparib, [18F]FBO and [18F]PARP-Fl, have been reported and have been used to image tumors in preclinical models of ovarian, pancreatic and glioma [12,13]. Zhou et al. recently synthesized a highly potent (IC50 of 6.3 nM) 18F-labeled PARP1 radiotracer, [18F]FluorThanatrace ([18F]FTT) (Figure 1), which was based on the PARPi AG14699 [1]. Early microPET studies demonstrated high tracer uptake in a xenograft model of human breast cancer, and blockade of radiotracer uptake following pretreatment with olaparib confirmed specificity of [18F]FTT for PARP [1].
Figure 1.
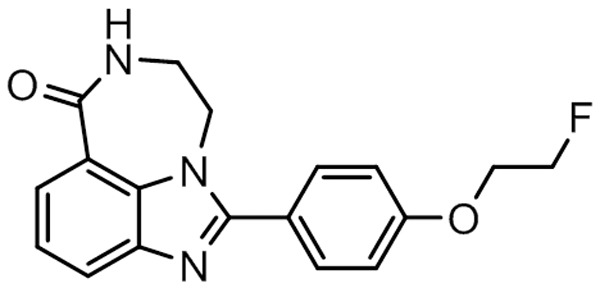
[18F]FTT structure.
In the current study, we measure the constitutive levels of PARP1 in multiple human breast cancer cell lines, including BRCA1 mutated, and correlate in vitro [18F]FTT uptake with constitutive PARP1 activity across cell lines. Furthermore, we demonstrate that constitutive PARP expression and activity is predictive of PET tracer uptake in breast cancer xenografts. Finally, cell uptake studies conducted in genetically engineered fibroblast cells confirm that [18F]FTT reflects PARP1 expression and not other PARP enzymes. These results confirm that [18F]FTT offers a noninvasive, in vivo means of evaluating tumor PARP1 expression and activity in breast tumors.
Materials and methods
Radiolabeling
Full automation of [18F]FTT synthesis was achieved successfully on an AllinOne module (TRASIS, Belgium) using a modified program from reported procedures [1]. In brief, 18F/F- was eluted from ion exchange QMA cartridge with eluent containing 7 mg of cryptand and 2 mg of potassium carbonate and azoetropically dried with acetonitrile (1 mL) at 100°C. A solution of tosylate precursor (0.8-1.0 mg) in DMF (0.8 mL) was added to the dried [18F]/F- and heated at 105°C for 10 minutes. After cooling, the reaction was quenched with 3 mL of HPLC mobile phase (17% acetonitrile in 20 mM ammonium bicarbonate aqueous solution). The quenched reaction mixture was passed through an Alumina N Plus cartridge (Waters, USA) to HPLC loop for purification using SB-C18 semi-preparative column (100 × 9.4 mm, Agilent, USA) with 5 mL/min flow rate. The retention time of [18F]FTT was about 20 min and collection time lasted 2 min. The collected product was diluted to 21 mL with water and enriched on C18 plus cartridge (Waters, USA). The cartridge was rinsed with water (10 mL), the product was eluted out and passed through 0.2 µm sterile nylon filter (Whatman, USA) into final production vial with ethanol (1.5 mL) and normal saline (14 mL). The entire synthesis required 55 min and gave 50-60% yield (decay corrected). The product radiochemical and chemical purity was >90% with a specific activity >2200 Ci/mmol. [18F]FTT was manufactured in accordance with cGMP guidelines for positron-emission tomography radiopharmaceuticals.
Cell culture
HCC1937 cell line (obtained from ATCC) was cultured in RPMI1640 (Gibco) supplemented with 10% FBS and 1% Penicillin Streptomycin. MDA-MB-231 cell line (ATCC) was cultured in MEM (Gibco) supplemented with 5% FBS, 1% L-Glutamine, 1% NEAA, 1% Sodium Pyruvate, 2% MEM vitamins, and 1% Penicillin Streptomycin. MCF7 (ATCC) cell line was cultured in DMEM (Gibco) with 10% FBS, 1% Penicillin Streptomycin, and 0.01 mg/mL human recombinant insulin.
Mouse embryonic fibroblasts (MEF) were cultured in DMEM with 10% FBS at 37°C. PARP1 or 2 double knockout MEFs originate from the cross-breeding of PARP1 or PARP2 null heterozygous mice to generate PARP1 or PARP2 homozygous double knockout models.
Western blot analysis
Whole cell extracts were prepared by lysing cells with RIPA buffer with protease (P8340, Sigma Life Sciences) and phosphatase inhibitors cocktail 2 and 3 (P5726, P0044, Sigma Life Sciences). Lysates were resolved on a polyacrylamide gel, transferred to a PVDF, and incubated with the primary antibodies BRCA1 anti-mouse (D-9: sc-6954; Santa Cruz Biotechnology), PAR (4335-MC-100-AC; Trevigen), PARP1 (9452; Cell Signaling Technologies) and β-actin anti-mouse (3700S, Cell Signaling and Technologies). Signal was detected and visualized using and LiCor Odyssey CLx Imager (Lincoln, NE).
PARP activity assay
Constitutive PARP1 activity of cell extracts from samples plated in quadruplicate was measured via a validated ELISA chemiluminescent assay of PAR (Trevigen, HT Chemiluminescent PARP/Apoptosis Assay, 4685-096-K). In brief, protein quantified cell lysates were deposited onto histone proteins in a 96-well plate and incubated with anti-PAR monoclonal antibody and then goat anti-mouse IgG-HRP conjugate. HRP substrate was used to generate a chemiluminescent signal, and light output detected by a Perkin Elmer Enspire Multimode Plate Reader (Waltham MA) and translated to relative PARP activity.
[18F]FluorThanatrace uptake assay
Each cell line was plated in quadruplicate. After approximately 24 hours of growth, cells were incubated with [18F]FTT diluted in PBS for a starting input of 150,000 cpm/well, or dual incubated with [18F]FTT and 10 μM olaparib, for various time points from 5 through 120 minutes. Activity was measured via a Perkin Elmer Wizard 2470 gamma counter. Co-incubation with competitive inhibitor olaparib was performed to determine specific [18F]FTT binding. The specific binding ratio was calculated as the following: (total binding - nonspecific binding)/nonspecific binding), where tracer uptake in the samples dual-incubated with olaparib was taken to represent non-specific binding.
For the uptake studies in engineered fibroblasts, cells were plated at 20,000 cells/well in 96 well format 24 hrs prior to the radioligand binding studies. Experiments were carried out in quadruplicate. On the day of experiment [18F]FTT was diluted in DMEM with and without 10 μM olaparib. The resulting solutions were added to the plate and allowed to incubate for 1 hr at 37°C. After 1 hr the solutions were aspirated and wells were washed with 200 μL of PBS three times. Next, radioactivity was assayed on a Perkin Elmer Wizard automatic gamma counter.
Xenograft models
Female Nude and SCID mice were purchased from Charles River Laboratories (Malvern, Pa) and acclimated for at least four days prior to tumor cell injection. Approximately 107 cells were suspended in a 1:1 mixture of FBS-free media and matrigel, and then subcutaneously injected into the subscapular region. At least 3 days prior to tumor cell injection, MCF7 mice were subcutaneously implanted with a 17-β-estradiol pellet (0.72 mg per pellet) 60-day release time (Innovative Research of America, Sarasota, FL). HCC1937 xenograft experiments were conducted in SCID mice, while MCF7 and MDA-MB-231 xenograft experiments were conducted in nude mice. Imaging studies were performed 10-14 days post implantation.
For imaging studies, anesthesia of tumor-bearing mice was maintained via a nose cone at 2-3% isoflurane, 1 L/min oxygen and body temperature was maintained by a heating pad placed under the animal. The mice were injected with 150-300 μCi of [18F]FTT and scanned for 60 minutes on a Philips Mosaic small animal PET scanner. Regions of interest were drawn manually over the tumors as well as a muscle region (background). The tumor to muscle ratio was calculated from the integrated counts/cc normalized to the injected dose from 40-60 min post intravenous injection of the radiotracer. For blocking studies, 1.25 mg of olaparib was dissolved in a 20% DMSO, Trappsol/PBS solution (0.25 mL) and injected (IP; 50 mg/kg) 30 min prior to ligand administration.
Results
Western blot analysis
The following three biologically contrasting human breast cancer cell lines were selected for all experiments: HCC1937, a triple negative breast cancer (TNBC) with a BRCA-/- mutation at codon 1755 within the BRCA1 C-terminal (BRCT) repeats that forms a truncated BRCA1 protein; MDA-MB-231, a TNBC with wild type BRCA1/2; and MCF7, a luminal A line with wild type BRCA1/2. Western blot for BRCA1 was performed and verified the absence of wild type BRCA1 protein in HCC1937, and confirmed the presence of BRCA1 in MDA-MB-231 and MCF7. In addition, HCC1937 showed higher levels of PAR and PARP1 by western blot analysis. See Figure 2. PARP1 quantification is presented in Figure 3A.
Figure 2.
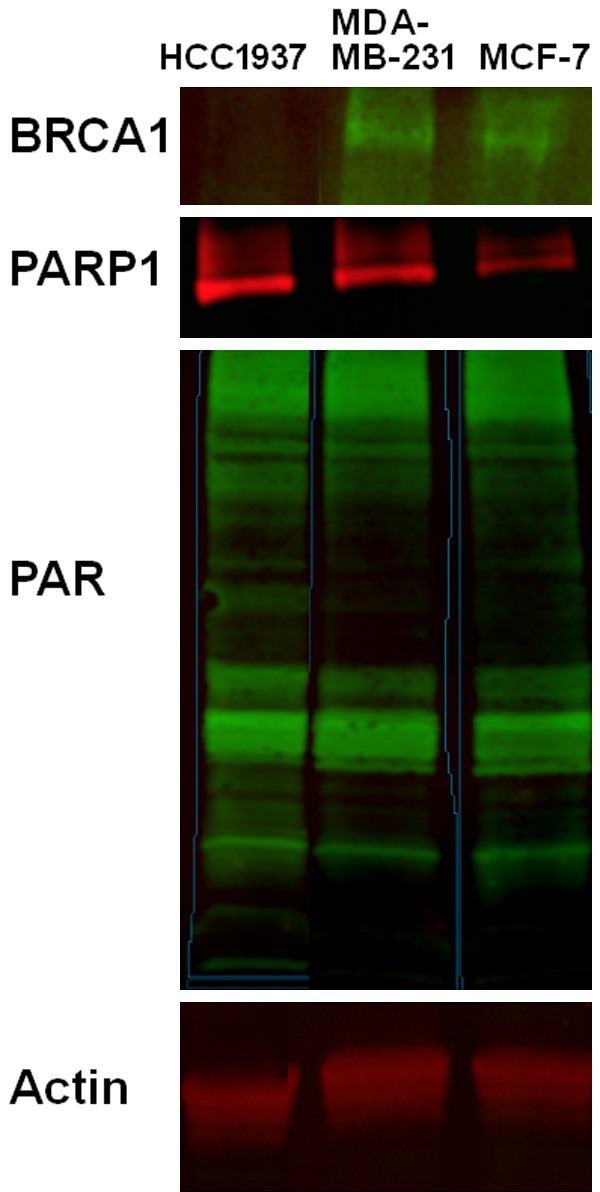
Western blot analysis of BRCA1, PAR, and PARP. Actin was used as a loading control. HCC1937 showed low expression of BRCA1 and confirmed the deleterious mutation in the BRCA1 gene. MDA-MB-231 and MCF-7 showed higher expression of BRCA1 compared to HCC1937. In addition, HCC1937 showed the highest expression of PAR and PARP compared with the other two cell lines.
Figure 3.
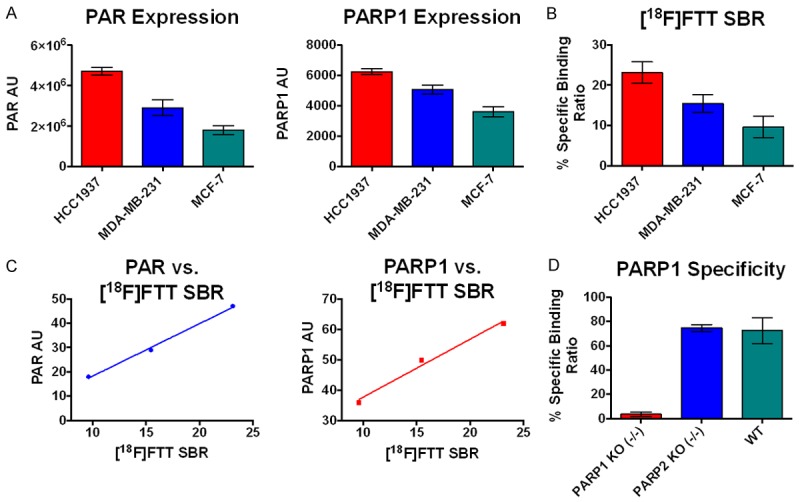
A. PAR expression measured via chemiluminescent ELISA assay, and PARP1 measured via western blot analysis. B. [18F]FTT specific binding ratio (SBR) radiotracer in vitro binding assay in cancer cell lines. C. Correlation of PAR and PARP vs. [18F]FTT SBR. D. [18F]FTT radiotracer in vitro binding assay in cancer cell lines.
PARP activity assay
Quantification of PAR has been verified as a marker of PARP1 activity [6,14], although in actuality it represents a balance between PARP1 (anabolism of PAR) and Poly-ADP-ribose glycohydrolase (PARG - catabolism of PAR) activity. We measured PAR via an established chemiluminescent ELISA assay across the three selected breast cancer cell lines. As predicted, HCC1937 demonstrated the highest levels of PAR (Figure 3A); this was expected given that BRCA1/2 deficient cells cannot undergo HR and are therefore dependent on PARP1 for DNA repair [6]. Both cell lines with preserved BRCA demonstrated lower levels of PAR, with MCF7 demonstrating the lowest level, and MDA-MB-231 an intermediate level (Figure 3A). It is not surprising that MDA-MB-231 has higher PARP activity than MCF7, as TNBC tumors frequently have HR defects in genes other than BRCA1/2 [15].
In vitro [18F]FTT uptake
An in vitro [18F]FTT cell uptake assay was performed to determine the binding of the tracer in each of the three cell lines, and to correlate the ratios with the PARP activity (PAR levels). Specific binding ratios were compared, rather than the total tracer binding, to eliminate the contribution of nonspecific tracer binding to the overall uptake measurements (see Methods section for specific binding ratio calculations). Specific binding ratios for the 120 minute incubation period are shown in Figure 3B. The relative specific binding ratios parallel the relative levels of PAR and PARP, implicating that either or both could be predictive of in vitro [18F]FTT uptake and both exhibited a linear correlation shown in Figure 3C.
Studies in the PARP1 k/o fibroblasts revealed a complete loss of specific tracer uptake (i.e., to the level of nonspecific binding). However, the specific uptake of radiotracer in the PARP2 k/o cells was identical to the wild type cells, suggesting that [18F]FTT is specific for PARP1 versus PARP2. See Figure 3D.
[18F]FluorThanatrace-PET imaging in tumor xenografts
HCC1937, MDA-MB-231, and MCF7 tumor xenografts underwent PET/CT scanning following injection with [18F]FTT. PET images, shown in Figure 4, demonstrated greatest [18F]FTT avidity within the HCC1937 tumors, while MCF7 tumors were the least avid. The tumor uptake ratios were calculated by normalizing tumor uptake to background muscle uptake. Relative tumor uptake ratios across the three cell lines correspond to in vivo tracer uptake. Furthermore, in vivo studies correspond to in vitro studies validating [18F]FTT as a marker of PARP expression and activity. As expected from our previous studies [1], pretreatment with olaparib (50 mg/kg, IP) blocked the uptake of [18F]FTT in the HCC1937 tumors (Figure 4).
Figure 4.
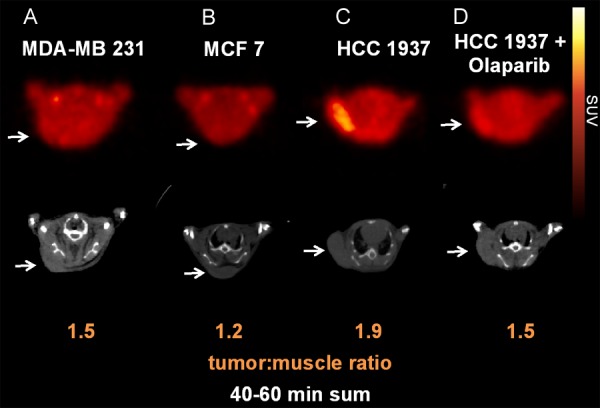
[18F]FluorThanatrace-PET-CT images of tumor xenografts, with tumors implanted in the subcutaneous posterior back/shoulder region, marked by arrows. Coronal slices through the tumor bed for: A. MDA-MB 231; B. MCF7; and C. HCC 1937 xenografts; D. HCC 1937 xenograft pretreated 30 min prior to imaging with 50 mg/kg olaparib, IP.
Discussion
For the past three decades, PARP1 has been actively investigated as a therapeutic target due to its role in DNA repair [7]. More recently, the unique selectivity of PARPi against homologous recombination deficient tumors has been exploited to target breast tumors in BRCA1/2 mutation carriers. While clinical trials of PARPi in BRCA1/2 mutation breast cancer patients are promising, not all patients benefit from therapy. Both initial and acquired resistance is observed in a significant proportion of patients. Multiple clinical trials of PARP inhibitors as a single agent in patients with BRCA1/2 mutation tumors demonstrate a substantial proportion of cases without treatment efficacy [6,15,16]. For this reason, much work has gone into identifying biomarkers of PARPi sensitivity and resistance. Resistant tumors may reflect underlying tumor heterogeneity as well as resistance mechanisms acquired from prior treatment with conventional chemotherapy, particularly platinum agents [6,17,18]. Research implicates a host of genetic factors that may contribute to insensitivity to PARPi, including reversion of the initial BRCA1/2 truncation [6,19,20], loss of p53-binding protein 53BP1 [6,21,22] up-regulation of P-glycoprotein efflux pumps [6,23,24], and up-regulation of various proteins involved in mediating DNA repair, such as RAD51 [25]. Importantly, loss of PARP1 expression also appears to be a mechanism of resistance to PARPi [8,10,11]. PTEN loss may confer selective sensitivity to PARPi [6,26]. However, many of these proposed sensitivity and resistance mechanisms have not been directly confirmed in tumor specimens from PARPi-treated patients and therefore remain speculative [6].
PARPi likely offers therapeutic utility beyond breast and ovarian tumors with BRCA1/2 mutations. Indeed, PARPi has shown efficacy in sporadic ovarian cancer, although it is unclear if these tumors had somatic BRCA mutations [27]. Many sporadic breast tumors lack BRCA1/2 mutation but possess defects in other key HR-related genes and are biologically similar to BRCA1/2 mutation cancers; such tumors are said to possess “BRCA-ness” [28]. There is significant overlap in the genetic profiles of BRCA1/2 breast cancers and triple negative (estrogen receptor negative, progesterone negative, and HER2/neu negative) breast cancer; 75-80% of breast cancers arising in BRCA1 carriers and approximately 50% percent of those in BCRA2 carriers are TNBC [17,29]. Furthermore, over 90% of BRCA1 mutation-related breast cancers exhibit molecular and histologic features similar to TNBC [6,30,31]. Genomic instability is characteristic of both BRCA-mutated and sporadic TNBC, and provides the basis for interest in TNBC as an additional target for PARPi [17].
TNBC accounts for approximately 15-20% of breast cancer diagnoses, and have a higher likelihood of recurrence and death compared to other breast tumors [17,32]. Evidence suggests that PARP1 is up-regulated in TNBCs as compared to receptor-positive cancers [33], and PARPi therapy could potentially be extended to TNBCs harboring defects in HR other than hereditary BRCA1/2 mutations. However, within the TNBC cohort, there is extensive biologic diversity, resulting in variable clinical outcomes to chemotherapy [17,34,35]. To date, the greatest obstacle to identifying sporadic breast cancer patients who may benefit from PARPi is the lack of definitive biomarker for HR deficiency, as a multitude of genes appear to contribute. Just as for BRCA1/2 mutation-related breast cancers, successful application of PARPi to sporadic breast cancers will require a biomarker assay for HR deficiency.
We have demonstrated higher constitutive PARP expression and activity in a BRCA1 mutant human breast cancer line compared to BRCA1/2 preserved lines. The differences in baseline PARP expression and activity across the three breast cancer cell lines are predictive of [18F]FTT microPET uptake. Thus, [18F]FTT-PET provides a noninvasive assay of PARP expression and activity. Further studies are needed to evaluate [18F]FTT-PET as a predictive assay of PARPi response in BRCA1/2 cancer patients as well as in cohorts likely to have other deficiencies in HR, namely TNBC patients. Future clinical trials are warranted to evaluate [18F]FTT-PET as a marker of breast cancer PARP activity in humans, and to assess the tracer as a predictor of PARPi response. [18F]FTT-PET may bypass the need for multiple laboratory biomarkers, some of which have likely not yet even been identified, and may offer a single direct assessment of sensitivity to PARPi.
Acknowledgements
This work was supported by grants from the Department of Energy (DE SC0012476) and the Basser Center for BRCA at Penn Medicine’s Abramson Cancer Center.
References
- 1.Zhou D, Chu W, Xu J, Jones LA, Peng X, Li S, Chen DL, Mach RH. Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography. Bioorg Med Chem. 2014;22:1700–1707. doi: 10.1016/j.bmc.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 3.Thomas C, Tulin AV. Poly-ADP-ribose polymerase: machinery for nuclear processes. Mol Aspects Med. 2013;34:1124–1137. doi: 10.1016/j.mam.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J Biol Chem. 1984;259:4770–4776. [PubMed] [Google Scholar]
- 5.Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: A new era of targeted therapy. Maturitas. 2015;81:5–9. doi: 10.1016/j.maturitas.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Basu B, Sandhu SK, de Bono JS. PARP inhibitors: mechanism of action and their potential role in the prevention and treatment of cancer. Drugs. 2012;72:1579–1590. doi: 10.2165/11635510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 7.Ferraris DV. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem. 2010;53:4561–4584. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- 8.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- 9.Saffhill R, Ockey CH. Strand breaks arising from the repair of the 5-bromodeoxyuridinesubstituted template and methyl methanesulphonate-induced lesions can explain the formation of sister chromatid exchanges. Chromosoma. 1985;92:218–224. doi: 10.1007/BF00348697. [DOI] [PubMed] [Google Scholar]
- 10.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pettitt SJ, Rehman FL, Bajrami I, Brough R, Wallberg F, Kozarewa I, Fenwick K, Assiotis I, Chen L, Campbell J, Lord CJ, Ashworth A. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One. 2013;8:e61520. doi: 10.1371/journal.pone.0061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiner T, Lacy J, Keliher EJ, Yang KS, Ullal A, Kohler RH, Vinegoni C, Weissleder R. Imaging therapeutic PARP inhibition in vivo through bioorthogonally developed companion imaging agents. Neoplasia. 2012;14:169–177. doi: 10.1593/neo.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlucci G, Carney B, Brand C, Kossatz S, Irwin CP, Carlin SD, Keliher EJ, Weber W, Reiner T. Dual-Modality Optical/PET Imaging of PARP1 in Glioblastoma. Mol Imaging Biol. 2015;17:848–55. doi: 10.1007/s11307-015-0858-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, Issaeva N, Sleeth K, Sharma RA, Helleday T. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70:5389–5398. doi: 10.1158/0008-5472.CAN-09-4716. [DOI] [PubMed] [Google Scholar]
- 15.Audeh MW. Novel treatment strategies in triple-negative breast cancer: specific role of poly (adenosine diphosphate-ribose) polymerase inhibition. Pharmgenomics Pers Med. 2014;7:307–316. doi: 10.2147/PGPM.S39765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livraghi L, Garber JE. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med. 2015;13:188. doi: 10.1186/s12916-015-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 18.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A’Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JH, de Bono JS, Kaye SB. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010;28:2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 19.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 20.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NM, Borst P, Jonkers J. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiarugi A. A snapshot of chemoresistance to PARP inhibitors. Trends Pharmacol Sci. 2012;33:42–48. doi: 10.1016/j.tips.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Schild D, Wiese C. Overexpression of RAD51 suppresses recombination defects: a possible mechanism to reverse genomic instability. Nucleic Acids Res. 2010;38:1061–1070. doi: 10.1093/nar/gkp1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–322. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–861. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 28.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 29.Bayraktar S, Gluck S. Systemic therapy options in BRCA mutation-associated breast cancer. Breast Cancer Res Treat. 2012;135:355–366. doi: 10.1007/s10549-012-2158-6. [DOI] [PubMed] [Google Scholar]
- 30.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, Gilks CB, van de Rijn M, Perou CM. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 32.Pal SK, Mortimer J. Triple-negative breast cancer: novel therapies and new directions. Maturitas. 2009;63:269–274. doi: 10.1016/j.maturitas.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 33.Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of Poly (ADPRibose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer. 2010;1:812–821. doi: 10.1177/1947601910383418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232:142–150. doi: 10.1002/path.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oakman C, Moretti E, Pacini G, Santarpia L, Di Leo A. Triple negative breast cancer: a heterogeneous subgroup defined by what it is not. Eur J Cancer. 2011;47(Suppl 3):S370–372. doi: 10.1016/S0959-8049(11)70207-6. [DOI] [PubMed] [Google Scholar]