

Abstract
Because extracellular signal-regulated kinase 5 (ERK5) inhibits endothelial inflammation and dysfunction, activating ERK5 might be a novel approach to protecting vascular endothelial cells (ECs) against various pathological conditions of the blood vessel. We have identified small molecules that protect ECs via ERK5 activation and determined their contribution to preventing cardiac allograft rejection. Using high throughput screening (HTS), we identified certain statins and anti-malarial agents including chloroquine (CQ), hydroxychloroquine (HCQ), and quinacrine (QC) as strong ERK5 “activators”. Pitavastatin enhanced ERK5 transcriptional activity and Kruppel-like factor-2 (KLF2) expression in cultured human and bovine ECs, but these effects were abolished by the depletion of ERK5. CQ and HCQ up-regulated ERK5 kinase activity and inhibited VCAM-1 expression in an ERK5-dependent but MEK5- and KLF2/4-independent manner. Leukocyte rolling and vascular reactivity were used to evaluate endothelial function in vivo, and we found that EC-specific ERK5 knockout (ERK5-EKO) mice exhibited increased leukocyte rolling and impaired vascular reactivity, which could not be corrected by pitavastatin. The role of endothelial ERK5 in acute cardiac allograft rejection was also examined by heterotopic grafting of the heart obtained from either wild type (WT) or ERK5-EKO mice into allomismatched recipient mice. A robust increase in both inflammatory gene expression and CD45-positive cell infiltration into the graft was observed. These tissue rejection responses were inhibited by pitavastatin in WT but not ERK5-EKO hearts. Our study has identified statins and anti-malarial drugs as strong ERK5 activators and shown that ERK5 activation is preventive of endothelial inflammation and dysfunction and acute allograft rejection.
Introduction
Extracellular signal-regulated kinase 5 (ERK5), an atypical mitogen activated protein kinase with transcriptional activity, has not only a kinase domain but also a transcriptional activity domain, the latter being regulated by an intra-molecular interaction independent of its kinase activity (1). Using tamoxifen-inducible endothelial specific ERK5 knockout (ERK5-EKO) mice, we have demonstrated that ERK5 possesses endothelial protective effects (1). We and others have shown that the laminar shear stress-mediated endothelial protection is due to ERK5 activation (2, 3), which inhibits leukocyte-endothelial interaction and adhesion molecule expression (1, 4–7). ERK5-KLF2 signaling is involved in the laminar-flow-induced eNOS expression and anti-inflammatory effects (3). These results collectively suggest that activating ERK5 is a novel approach to protecting ECs. To translate this idea into therapy, we performed high throughput screening (HTS) of a large number of molecules to identify novel activators of ERK5 transcriptional activity. Of the most effective compounds screened were pitavastatin and quinacrine (QC, an old, but well known anti-malarial drug).
Statins (or HMG-CoA reductase inhibitors) are principal therapeutic agents used in the treatment of hypercholesterolemia (8). However, they also exert cholesterol-independent effects such as improving endothelial function (9–12), reducing the progression of cardiac allograft vasculopathy (13), and improving 8-year survival of heart transplant recipients (13). Although some in vitro studies suggest that the cholesterol-independent statin effects may be achieved via Rho/ROCK (9), PI3-K-Akt (10), ERK5-KLF2 (11, 12, 14), and KLF4 (15) signaling, molecular mechanisms through which statins improve vascular function and inhibit cardiac allograft vasculopathy remain unclear.
Due to their anti-inflammatory effects, anti-malarial drugs quinacrine (QC), chloroquine (CQ), and hydrochloroquine (HCQ) are being used for the treatment of not only malaria but also for autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (16). Recently, the Hopkins Lupus Cohort and LUMINA (Lupus in Minorities: Nature Versus Nurture) nested case-control study showed that HCQ treatment was associated with a long-term protective effect on cardiovascular diseases (17), probably by inhibiting inflammation (16). The possible role of antimalarial agents as prostaglandin antagonists via inhibiting phospholipase A2 was suggested (18), but recent publications challenged this concept (19–21). Therefore, the target molecule(s) of anti-malarial agents in regulating inflammation remains unknown. More importantly, to our knowledge there has never been a study on the role of anti-malarial agents in cardiac allograft rejection.
The combination of HTS and studies on molecular mechanisms of ERK5 activators has allowed us to uncover the crucial role of ERK5, which is activated by pitavastatin and anti-malarial drugs, in inhibiting endothelial inflammation and dysfunction. Furthermore, we found the crucial role of endothelial ERK5 in preventing cardiac allograft rejection. To our knowledge this is the first report to define the role of endothelium during the course of cardiac allograft rejection, and will suggest a new therapeutic application of known drugs including anti-malaria drugs against endothelial inflammation and dysfunction.
Methods
Reagents and antibodies
Pitavastatin was kindly provided by Kowa Company Ltd (Tokyo, Japan). Rosuvastain was kindly supplied by Astra Zeneca, London, UK). 6-Mecaptopurine monohydrate (6-MP) was purchased from Alfa Aesar (A Johnson Mathey Company, Cat. # A12197), and quinacrine dihydrochloride (QC) from Sigma-Aldrich (Cat. # Q3251). Chloroquine phosphate (CQ, CAS 50-63-5) and hydroxychloroquine sulfate (HCQ, CAS 747-36-4) were from Santa Cruz Biotechnology (Dallas, Texas). Anti-phospho-ERK5 (Thr218/Tyr220, #3371) and anti-ERK5 (#3372) were from Cell Signaling (Cell Signaling Technology Inc, Danvers, MA). Anti-tubulin (T-5168) was from Sigma (St. Louis, MO), anti-MEK5 (# KAP-MA003E) from StressGen (San Diego, CA), anti-CD45 (Sc-103012) from Santa Cruz, and anti-CD3 (# ab5690) from Abcam (Cambridge, MA).
Cell culture and transfection
HUVECs were cultured as described previously (22). BAECs were cultured in M199 medium (Invitrogen, Cat. #11150-059, Grand Island. NY) supplemented with 10% fetal bovine serum Clone III (HyClone, Cat. # SH 30109.03, Logan, UT), 1% NEM-amino acid (Invitrogen, Cat. #11130-051), and 1% Antibiotic-Antimycotic (Cellgro, Cat. #30-004Cl, Mediatech Inc., Manassas, VA). HPAECs were cultured in M200 medium (Cascade Biologics, Cat. # M-200-500, Life Technologies Coporation, Grand Island, NY) supplemented with 2% fetal bovine serum (FBS, Atlanta Biologicals, Cat. # S11050, Lawren ceville, GA), and low serum growth supplement (LSGS, Cascade Biologicals, Cat. # S-003-10). The HeLa cell line stably expressing ERK5 was cultured in DMEM containing 10% FBS (Atlanta Biologicals, Cat. # S11050, Lawrenceville, GA) and 100 μg/ml G418 (Calbiochem, Cat. #345810, San Diego, CA). All cells were maintained at 37 °C in the humidified atmosphere of 5% CO2 and 95% air.
High Throughput Screening (HTS) for activators of ERK5 transcriptional activity
We generated a stable HeLa cell line co-expressing pG5-Luc, which contains five Gal4 binding sites upstream of a minimal TATA box, and pBIND-ERK5 fused to Gal4 (pBIND-ERK5). Using these reporter cells, the MicroSource SPECTRUM Collection (http://www.msdiscovery.com/spectrum.html), which consists of about 2000 structurally diverse compounds selected by medicinal chemists and biologists for testing a wide range of biological activities, was screened at the University of Rochester HTS Core facility. Included in this collection are the following three classes of compounds: i) Known drugs that have reached clinical trial stages within the USA, have been assigned the USAN or USP status, and are found in the USP Dictionary of USAN and International Drug Names (2005, US Pharmacopeia); ii) known drugs from Europe and Asia; and iii) natural compounds with already known pharmacologic effects. A major advantage of screening this group of compounds is that most of them have known biological activities that can be investigated in more detail if a specific hit is found.
Stable cells plated on a 384-well plate (2500 cells/well) were treated with test compounds at the concentration of 5 μM for 18 hrs. The level of luciferase activity was assayed using a Luciferase kit (Promega corporation, Madison, WI) and a series of positive and negative control compounds were used as references.
ERK5 transcriptional activity assay (not used in HTS study)
Cells were transfected with pG5-Luc and Gal4-ERK5 constructs using Opti-MEM (Invitrogen, Carlsbad, CA) containing Plus-Lipofectamine mixture. After transfection, cells were treated with various concentrations of pitavastatin, CQ, and HCQ for 24 hrs. Cells were then harvested, and the level of luciferase activity was assayed using a dual-luciferase reporter system (Promega Corporation, Madison, WI) and measured using a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA). Transfections were performed in triplicates, and each experiment was repeated at least three times. Since Gal4-ERK5 also contains the Renilla luciferase gene, the expression and transfection efficiencies can be normalized by Renilla luciferase activity. We verified our HTS data by detecting both luciferase and Renilla activity, which also controlled for toxic effects of the compounds.
Mammalian two-hybrid assay to study the effect of CQ on SMRT1-PPARγ binding
Cells were transfected with the pG5-luc vector and various pBIND (Gal4) and pACT (VP16) plasmids (Promega, Madison, WI), as indicated, for 4 hrs. Cells were then washed, and complete M200 medium supplemented with LSGS and 2% FBS was added. In CQ treated samples, cells were then incubated five more hours prior to CQ treatment. The pG5-luc vector contains five Gal4 binding sites upstream of a minimal TATA box, which in turn, is upstream of the firefly luciferase gene. pBIND containing Gal4 was fused with SMRT1, and pACT containing VP16 was fused with PPARγ. Cells were harvested 24 hrs after transfection unless otherwise indicated, and luciferase activity was assayed as described previously(1).
Real-time RT-PCR
Total RNA from cells was isolated using an RNeasy Plus Mini Kit (Cat. # 74136, QIAGEN, Valencia, CA) according to the manufacturer’s instruction. Total RNA from cardiac allografts harvested 5 days after transplantation was isolated using an RNeasy Fibrous Tissue Mini Kit (Cat. #74704, QIAGEN, Valencia, CA). At the time of harvest, cardiac allografts were stored in RNAlater solution (Cat. # AM7020, Ambion, Grand Island, NY) until use. To isolate total RNA, stored hearts were taken out of the storage solution and minced using a razor blade and transferred to a 1.5 mL tube with 600 μL of buffer RLT Plus (β-ME added). The lysate was loaded onto a QIAshredder spin column placed in a 2 ml collection tube, centrifuged for 2 min at 15,000 rpm, and purification was continued according to the manufacturer’s instruction. First strand cDNA was synthesized, and target cDNA levels were quantified by real-time reverse transcriptase polymerase chain reaction (RT-PCR) using a MyiQ™2 Optics Module (Bio-Rad Laboratories, Inc.) as described previously (22).
Following primers were used: human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward: 5′-GGT GGT CTC CTC TGA CTT CAA CA-3′ and reverse: 5′-GTT GCT GTA GCC AAA TTC GTT GT-3′; human KLF2 forward: 5′-GCA CGC ACA CAG GTG AGA AG-3′ and reverse: 5′-ACC AGT CAC AGT TTG GGA GGG-3′; human endothelial nitric oxide synthase (eNOS)-forward: 5′-GTG GCT GTC TGC ATG GAC CT-3′ and reverse: 5′-CCA CGA TGG TGA CTT TGG CT-3′; human KLF4-forward: 5′-ACC AGG CAC TAC CGT AAA CAC A-3′ and reverse: 5′-GGT CCG ACC TGG AAA ATG CT-3′; human VCAM1 forward: 5′-GGT GGG ACA CAA ATA AGG GTT TTG G-3′ and reverse: 5′-CTT GCA ATT CTT TTA CAG CCT GCC-3′; mouse IL-1 forward: 5′-CAA CCA ACA AGT GAT ATT CTC CAT G-3′ and reverse: GAT CCA CAC TCT CCA GCT GCA-3′; mouse IL-6 forward: 5′-CAG AAT TGC CAT CGT ACA ACT CTT TTC TCA-3′ and reverse: 5′-AAG TGC ATC ATC GTT GTT CAT ACA-3′; mouse IL-10 forward: 5′-GGT TGC CAA GCC TTA TCG GA-3′ and reverse: 5′-ACC TGC TCC ACT GCC TTG CT-3′; mouse IFN-γ forward: 5′-TCA AGT GGC ATA GAT GTG GAA GAA-3′ and reverse: 5′-TGG CTC TGC AGG ATT TTC ATG-3′; mouse TNFα forward: 5′-CAT CTT CTC AAA ATT CGA GTG ACA A-3′ and reverse: 5′-TGG GAG TAG ACA AGG TAC AAC CC-3′; mouse β-actin forward: 5′-AGA GGG AAA TCG TGC GTG AC-3′ and reverse: 5′-CAA TAG TGA TGA CCT GGC CGT-3′ (23); mouse MCP1 forward: 5′-CCA CTC ACC TGC TGC TAC TCA T-3′ and reverse: 5′-TGG TGA TCC TCT TGT AGC TCT CC-3′ (24).
KLF2 and eNOS promoter activity
Cells were transfected with a reporter gene encoding KLF2 and eNOS promoter as described previously (22, 25). Transfected cells were then treated with various concentrations of pitavastatin for 24 hrs. KLF2 and eNOS promoter luciferase activity was assayed using a dual-luciferase reporter assay system. Experiments were repeated three times.
In vitro ERK5 kinase assay
In vitro ERK5 kinase activity was measured as we described previously (22). For each reaction, 1 μg of purified GST-MEF2C-GST fusion protein on beads and 0.5 μg of GST-ERK5 fusion protein were added to 30 μL of reaction buffer (10 mM MgCl2, 10 mM MnCl2 and 25 mM HEPES, pH7.5). Different doses of pitavastatin were added to the mixture, and reactions were allowed to take place for 30 min at 30 °C in the dark with vigorous shaking. Cold ATP (final concentration 1 mM) and 1 μCi of γ-32P-ATP were added to the reaction mixture, which was then gently mixed and further incubated as before for 30 min. Finally, 6X sample buffer was added to terminate the reaction. The samples were boiled, electrophoresed on SDS-polyacrylamide gels and transferred to polyvinilidene difluoride membranes. The film exposed to the membrane was developed. MEF2C phosphorylation was quantified using Fujifilm Image Gauge software (Version 4.0). Amounts of GST-MEF2C fusion protein used in each sample were visualized by Poncreau S staining. Amounts of recombinant GST-ERK5 fusion protein used in each sample were detected by Western blotting using anti-ERK5 antibody.
Western blot analysis
Western blotting was performed as described previously (22).
Animal model
Tamoxifen (4-OHT: 4-hydroxytamoxifen)-inducible EC-specific ERK5 KO mice (VeCad-CreER-ERK5/flox/flox, or inducible ERK5-EKO mice) were generated as we described previously (22). To induce deletion of the endothelial Erk5 gene, ERK5-EKO mice were injected with peanut oil with or without 2 mg of 4-OHT for 5 consecutive days. Sustained reduction of endothelial ERK5 was confirmed by Western blotting using mouse endothelial cells isolated from ERK5-EKO mice two weeks after 4-OHT injection as described previously (22). At age 8–10 weeks, mice (C57BL/6) made diabetic by IP injection of a single dose of freshly prepared STZ solution (150 mg/kg body wt in citrate saline, pH 4.5) using 26.5 Gauge needle. Diabetic status was confirmed by blood glucose measurements. Animals were maintained according to the protocol approved by the University of Rochester Institutional Animal Care and Use Committee.
Analysis of metabolic parameters
Mice were anesthetized and blood was collected from the abdominal artery. Plasma was prepared and total cholesterol, HDL, and non-HDL (LDL and VLDL) concentrations were measured enzymatically using commercially available kits (Cat. # ab65390, Abcam, Kendall Square, Cambridge, MA) according to the manufacturer’s instructions. Plasma glucose was measured using blood glucose meter (Advocate, Model: TD-4223F, Diabetic Supply of Suncoast, Inc., Baton Rouge, LA).
Leukocyte rolling assay and vessel diameter measurements
Leukocyte rolling on the endothelium, leukocyte rolling flux (number of rolling leukocytes passing a perpendicular line placed across the target vessel in one minute), and vascular reactivity were measured and analyzed as described previously (22).
Abdominal cardiac transplantation
To determine the effect of ERK5 activation by statin on allograft rejection and immune responses, heterotopic cardiac transplantation was performed. Two weeks before transplantation, VeCad-CreER-ERK5/flox/flox mice (inducible ERK5-EKO) and VeCad-CreER-WT mice (being used as a control), both with C57BL/6 genetic background, were injected with 4-OHT. Balb/c male mice (8–10 week-old) were used as recipients. Cardiac transplantation was performed as described previously (26). Briefly, donor’s ascending aorta and pulmonary artery were anastomosed to recipient’s abdominal aorta and inferior vena cava, respectively. All anastomoses were done in an end-to-side fashion. Following cardiac transplantation, manual palpation was performed twice a day to gauge allograft rejection, and cessation of beating was interpreted as rejection. This step was done in a blind fashion.
To assess the progress of graft rejection and immune responses, the transplanted heart was harvested 5 days after the surgery. The recipient mice were anesthetized with intraperitoneal injection of ketamine (50 mg/kg) and xylazine (10 mg/kg) in saline, sacrificed, and the grafted hearts harvested. The vascular anastomoses were cut off and discarded, and the heart was divided into two segments: the apex segment was stored in RNAlater solution (Cat No.: AM7020, Ambion, Grand Island, NY) for RNA isolation, the remaining heart was fixed in acidic methanol (Methanol: Acetic acid: ddH2O = 6:3:1), embedded in paraffin, sectioned (5 μm), and stained by H&E or specific antibodies.
Histomorphometric assessment of allograft rejection
Parenchymal rejection (PR) was assessed in allografts 5 days after transplantation using the modified grading system of the Working Formulation of the International Society for Heart and Lung Transplantation and was scored on a scale of 0 to 4 (0, no rejection; 1, focal mononuclear cell infiltrates; 2, focal mononuclear infiltrates with necrosis; 3, multifocal infiltrates with necrosis; and 4, widespread infiltrates with hemorrhage and/or vasculitis). Occlusion (GAD) was scored on a scale of 0 to 4 on the extent of luminal occlusion averaged over 10 arteries (0, <10 %; 1, 10–25%; 2, 25–50%; 3, 50–75%; and 4, >75% occlusion of the vascular lumen) as described previously (27).
Statistical Analysis
Data are expressed as mean ± SD. Nonparametric methods were used to analyze differences between independent groups. Comparisons between two groups for graft survival were performed by using Kaplan-Meier and log-rank tests. Comparisons among more than two groups were analyzed by Kruskal-Wallis test, followed by Bonferroni multiple-comparison test. Statistic analysis was performed using GraphPad Prism software for Macintosh version 5.00 (GraphPad Software, SanDiego, CA). The sample size, the time intervals, and where the correlation data come from, were listed in each figure and figure legend. A p value less than 0.05 was accepted as being statistically significant and indicated by one asterisk (*), and a value less than 0.01 is indicated by two asterisks (**).
Results
High throughput screening for activators of ERK5 transcriptional activity
To perform a cell-based assay for ERK5 transcriptional activity, we generated a HeLa cell line stably expressing pG5-Luc and pBIND-ERK5 vectors. The pG5-luc vector contains five Gal4 binding sites upstream of a minimal TATA box, which in turn, is upstream of the firefly luciferase gene. The pBIND-ERK5 vector contains Gal4 fused with ERK5. When ERK5 transcription activity is activated, Gal4 of pBIND-ERK5 bind the Gal4 binding site of pG5-luc, which results in increased firefly luciferase expression in cells. For ERK5 activator screening, cells were treated with compounds in the the MicroSource SPECTRUM Collection for 18 hrs and luciferase activity determined. A scoring system was generated based on the intensity of luciferase activity ranging from 4000 (highest) to 0 (lowest) in an arbitrary unit, and statistical assessment of this assay gave a Z’ factor = 0.57, which is suitable for HTS. From the screening, three compounds that had scores >500 were selected and re-assayed in duplicates. Among the three compounds identified as novel ERK5 activators, two were pitavastatin and 6-mercaptopurine (6-MP, an activator of Nur77, which is an ERK5 downstream target) (12, 28), and the third was QC (Fig. 1A). Pitavastatin, 6-MP, and QC were then confirmed to increase ERK5 transcriptional activity in a dose-dependent manner using the same HeLa cell line (Fig. 1B–D).
Figure 1. HTS identified small molecules as ERK5 transactivators.

(A) Structures of compounds with high scores and rosuvastatin obtained from PubChem database: 6-Mercaptopurine (CID 667490), Quinacrine (CID237), Pitavastatin (CID5282452), Chloroquine (CID2719), Hydroxychloroquine (CID3652), and Rosuvastatin (CID5282455). (B–D) HeLa cells stably expressing pG5-Luc and pBIND-ERK5 were treated with pitavastatin (B), 6-MP (C), or QC (D) at various doses for 24 hrs, and ERK transcriptional activity measured. (E) HUVECs were transfected with pG5-Luc and pBIND-ERK5 constructs and treated with pitavastatin, CQ, or HCQ at various doses for 24 hrs. For B–E, luciferase activity was assayed using the Dual-Luciferase Reporter Assay system; (Promega, Madison, WI) and a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA). Values are mean±SD (n=3) and represent the firefly luciferase:Renilla luciferase ratio. **p< 0.01 versus untreated control.
To examine whether pitavastatin, 6-MP, and QC could also activate ERK5 transcriptional activity in ECs, we used human pulmonary aortic endothelial cells (HPAECs) and bovine aortic endothelial cells (BAECs). Although 6-MP increased ERK5 transcriptional activity in the genetically engineered HeLa cells, we could not detect any significant activation in ECs (Fig. S1A, C). In contrast, both pitavastatin and QC increased ERK5 transcriptional activity in ECs we tested (Fig. S1B–D).
Among the 2,000 compounds screened, several different statins were present. Interestingly, however, not all increased ERK5 transcriptional activity. Rosuvastatin showed no activation (score < 50) while simvastatin marginally increased ERK5 transcriptional activity (score =220). Pitavastatin, simvastatin, and atorvastatin increased ERK5 transcriptional activity in human umbilical vein endothelial cells (HUVECs) (Fig. S1E and G). Since KLF2 promoter activity is positively affected by certain statins only (12), it is likely that the similar statin selectivity exists for ERK5 activation. We also determined the effect of CQ and HCQ on ERK5 transcriptional activity in HUVECs. As shown in Fig. 1E, they dose-dependently increased ERK5 transcriptional activity. It was reported that as much as 100 μM CQ or HCQ was necessary in in vitro experiments to generate an intracellular drug level similar to that in patients receiving 400 mg/day HCQ for antirheumatic therapy (29). Therefore, the 5 μM dose we used should be regarded as highly selective for the drug effect. To further characterize pitavastatin-induced ERK5 activation, we examined if this statin was able to induce ERK5-TEY motif phosphorylation in HUVECs and found that it could do so at a level similar to that achieved by steady laminar flow (Fig. 2A). Since KLF2 and eNOS are regulated by ERK5 in ECs (2, 3), we then examined the effect of pitavastatin on KLF2 and eNOS promoter activity in HUVECs and found that both were dose-dependently increased by pitavastatin treatment (Fig. 2B–C). ERK5 siRNA was then used to determine the role of ERK5 in pitavastatin-induced eNOS and KLF2/4 mRNA expression in HUVECs. Indeed, ERK5 depletion inhibited the pitavastatin effect on eNOS and KLF2/4 mRNA expression (Fig. 2D–G).
Figure 2. Pitavastatin activates ERK5 and its downstream targets.

(A) Effects of pitavastatin treatment on ERK5 phosphorylation in HUVECs. Steady laminar flow (s-flow), a well-known activator of EC ERK5 in vitro (46), was used as a positive control. Immunoblots with anti-phospho-ERK5 (top) and anti-ERK5 (bottom) are shown on the left and the graph on the right show quantified data of phospho-ERK5 immunoblots. Levels of ERK5 phosphorylation are expressed relative to those of untreated control cells (0 min). Values are mean±SD, n=3 for each condition. (B–C) HUVECs were co-transfected with CA-MEK5α (positive control) or an empty vector and the luciferase reporter gene driven by either the KLF2 promoter (B) or the eNOS promoter (C). Twenty-four hrs after transfection, cells were treated with various doses of pitavastatin for 24 hrs. Luciferase activities were assayed as described in Fig. 1. Values represent firefly luciferase:Renilla luciferase ratios. Values are mean±SD (n=3) for each condition. *p<0.05, **p<0.01 versus untreated cells (white bar). (D–G) After transfection with ERK5 siRNA or control siRNA (48 hrs), HUVECs were treated with various concentrations of pitavastatin for 24 hrs. ERK5 depletion was confirmed by Western blotting using anti-ERK5 (D). Relative mRNA expression levels of eNOS (E), KLF2 (F), and KLF4 (G) were measured by qPCR and expressed relative to the GAPDH mRNA expression level. Values represent the ratio of mean value of pitavastatin-treated group to mean value of untreated control group. Values are mean±SD (n=3). **p<0.01 versus untreated control.
Pitavastatin directly activates ERK5 kinase activity in a cell-free system and increases KLF2 expression in a MEK5 independent manner
To determine the role of MEK5, an upstream kinase of ERK5 (Fig. 3A), in the action of pitavastatin, we depleted MEK5 by siRNA in HUVECs. In these cells, we also expressed constitutively active form of MEKK3 (mitogen-activated protein kinase kinase kinase 3, CA-MEKK3), which is an upstream kinase of MEK5, and detected ERK5 transcriptional activity. As shown in Fig. 3B and C, MEK5 depletion abolished CA-MEKK3-mediated ERK5 transcriptional activity. In contrast, we detected no inhibition by MEK5 siRNA on pitavastatin-induced ERK5 transcriptional activity (Fig. 3D), suggesting that pitavastatin exerts its effect directly on ERK5. This possibility was tested using a cell-free system, in which recombinant ERK5 kinase activity was assayed using GST-MEF2C (myocyte enhancer factor-2C) as a substrate in the presence of pitavastatin. Pitavastatin dose-dependently activated ERK5 kinase activity with an IC50 of ≈ 0.43 μM (Fig. 3E). We then examined the involvement of MEK5 in pitavastatin-induced KLF2/4 and eNOS expression in ECs and found that MEK5 depletion had no effect on the basal KLF2 induction (Fig. 3F) and only ≈ 40–50% inhibition in pitavastatin-induced KLF4 and eNOS induction (Fig. 3G, H). Of note, since we observed complete inhibition of steady laminar flow-induced eNOS mRNA expression by MEK5 siRNA (Fig. 3I), the level of MEK5 depletion by siRNA was biologically effective.
Figure 3. Pitavastatin directly activates ERK5 kinase activity and induces KLF2/4 and eNOS expression.

(A) Scheme of ERK5 signaling pathway (3, 47, 48), (B–D) HUVECs were transfected with MEK5 siRNA or control siRNA for 48 hrs. MEK5 depletion was confirmed by Western blotting with anti-MEK5 (B). Cells were then co-transfected with a pG5-Luc and a pBIND-ERK5 constructs with pcDNA3.1 (control) or the CA-MEKK3 vector (C), or treated with pitavastatin (24 hrs) (D). Luciferase activity was assayed as described in Fig. 1. Values represent firefly luciferase:Renilla luciferase ratios. Values are mean±SD (n=3) **p< 0.01 versus control siRNA, N.S: not significant. (E) Kinase activity of GST-ERK5 recombinant protein was activated by pitavastatin in an in vitro kinase assay using GST-MEF2C as a substrate. (F–I) Following MEK5 siRNA transfection, HUVECs were treated with 1 μM pitavastatin (F–H) or subjected to s-flow (I) for 24 hrs. Relative mRNA expression levels of KLF2 (F), KLF4 (G), and eNOS (H, I) were measured by qPCR and expressed relative to the GAPDH mRNA expression level and normalized against the mean level of expression in control siRNA transfected cells. Values are mean±SD (n=3) **p<0.01 N.S: not significant.
To determine the involvement of MEK5 kinase activation on pitavastatin-induced ERK5 TEY motif phosphorylation, we transfected ECs with MEK5 siRNA. As shown in Fig. S2A, we found that the deletion of MEK5 inhibited this phosphorylation, suggesting that MEK5 is involved in ERK5 TEF motif phosphorylation. In contrast, MEK5 depletion did not inhibit ERK5 transcriptional activity (Fig. 3D and S2B). These data are consistent with our earlier suggestion (2) that the regulation of ERK5 transcriptional activity can be independent of ERK5 kinase activity or ERK5 TEF motif phosphorylation.
The crucial role of ERK5, but not KLF2/4 or MEK5, in the anti-malarial agent-induced anti-inflammatory effect on ECs
Of the three anti-malarial drugs (CQ, HCQ, and CQ) found as ERK5 activators, CQ and HCQ but not QC are commonly used to treat SLE and rheumatoid arthritis (16) as they reduce the severity of renal dysfunction through their immune-modulatory, anti-inflammatory, and anti-thrombotic effects. Thus, we investigated effects of CQ and HCQ on ERK5 signaling. Although pitavastatin increased KLF2/4 expression (Fig. 4A) and CQ increased ERK5 phosphorylation (Fig. 4B), neither CQ nor HCQ was able to up-regulate KLF2/4 expression (Fig. 4A). However, they inhibited tumor necrosis factors-α (TNFα)-induced vascular cell adhesion molecule 1 (VCAM-1) expression (Fig. 4C), suggesting that both CQ and HCQ had an anti-inflammatory effect via KLF2/4-independent mechanisms. Interestingly, their inhibitory effect on TNF-α-induced VCAM-1 mRNA expression was abolished by the depletion of ERK5 (Fig. 4C) but not MEK5 (Fig. 4D). These data show a crucial role of ERK5 but not KLF2/4 or MEK5 in certain anti-inflammatory effects of these anti-malarial agents on ECs.
Figure 4. CQ and HCQ inhibit TNFα-induced VCAM-1 expression via ERK5 but not MEK5.

(A) HUVECs were treated for 24 hrs with pitavastatin (Pita, 1 μM), CQ (5 μM) or HCQ (5 μM). Relative mRNA expression levels of KLF2, KLF4 and eNOS were measured by qPCR and expressed relative to the GAPDH mRNA expression level. (B) Effects of CQ (5 μM) and s-flow treatment on ERK5 phosphorylation in HUVECs. Immunoblots with anti-phospho-ERK5 (top) and anti-ERK5 (bottom) are shown on the left and the graph on the right show quantified data of phospho-ERK5 immunoblots. Levels of ERK5 phosphorylation are expressed relative to those of untreated control cells (0 min). Values are mean±SD, n=3 for each condition. (C–D) After 48 hrs transfection with ERK5 siRNA (C), MEK5 siRNA (D) or control siRNA, HUVECs were treated for 1 hr with Pita (1 μM), CQ (10 μM) or HCQ (10 μM), followed by further incubation with TNF-α (10 ng/mL, 3hrs). Relative VCAM-1 mRNA expression levels were measured by qPCR and expressed relative to the GAPDH mRNA expression level. (A–D) Values represent the ratio of mean value of drug-treated group to mean value of untreated control group. Values are mean±SD, n=3 each **p<0.01 versus untreated control. (E) HUVECs were transfected with various plasmids as indicated. Two hours later, transfection medium was removed, and complete M200 medium was replaced. Five hours later, cells were treated with CQ or vehicle, and after 24 hrs luciferase activity was assayed. Values are mean ± S.D (n=6) for each condition. (F) HUVECs were transfected with ERK5 (siERK5) or control siRNA (si-Ctrl) for 48 hrs. Cells were then transfected with various plasmid, followed by CQ stimulation as described in (E), and after 24 hrs luciferase activity was assayed. Values are mean ± S.D (n=3) for each condition.
ERK5 is not only functional as a kinase or a co-activator, but also can release a co-repressor, SMRT, from PPARs by direct binding with PPARs as we have reported previously (1). It has been reported that PPARs can inhibit NF-κB transcriptional activity by releasing a co-repressor such as Bcl6, SMRT1 and NCoR1(30–33). Therefore, we examined whether CQ could release SMRT1 from PPARγ as we had previously shown by steady laminar flow(1). We found that CQ treatment indeed disrupted binding between PPARγ and SMRT (Fig. 4E). Interestingly, the deletion of ERK5 not only inhibited this disruption, but also accelerated binding between PPARγ and SMRT1 after CQ treatment (Fig. 4E, F). These results can provide a mechanistic insight how CQ can inhibit TNF-induced NF-κB activation without increasing KLF2 expression and also provides a new functional mechanism of ERK5 in regulating inflammation.
EC-specific ERK5 depletion counters the protective effect of pitavastatin against endothelial dysfunction
Leukocyte rolling and adhesion are characteristics of vascular inflammation, and vascular reactivity to acetylcholine (ACh) is an indicator of normal vascular function. Since increased leukocyte rolling (34) and decreased ACh-induced vasodilation (35) have been reported in diabetes (DM) (22), we used this model to study vaso-protective effects of pitavastatin. In a streptozotocin (STZ)-treated mouse diabetic model, leukocyte rolling was increased and ACh-induced vasodilation was dampened compared with those of the vehicle-treated mice (Fig. 5A–D and supplemental videos 1–3). When DM mice were treated with pitavastatin, we observed a clear recovery from the DM-induced vascular inflammation and dysfunction (Fig. 5A–D and supplemental video 3). Unlike the effect of lowering blood glucose levels by insulin on leukocyte rolling/adhesion (22), pitavastatin affected neither blood glucose nor total cholesterol levels (Fig. 5E), demonstrating that pitavastatin’s action on leukocyte rolling/adhesion is independent of both blood glucose and cholesterol lowering effects.
Figure 5. Effects of pitavastatin in diabetic (A–E) and inducible ERK5-EKO mice (F–J).

Wild type mice were intraperitoneally injected STZ (one dose, 150 mg/kg) or vehicle (0.1 M citrate buffer pH 4.5). Four and half days later, blood glucose was measured to confirm STZ-induced hyperglycemia. Five days later, mice were orally administrated pitavastatin (0.3 mg/kg body weight) or vehicle (PBS: phosphate buffered saline, pH 7.4) twice a day for 3 days. (A) On day 8, leukocyte rolling was recorded with a digital video camera for 2 min, as previously described (22). (B) Leukocyte rolling velocity, (C) leukocyte rolling flux (number of rolling leukocytes passing a perpendicular line placed across the observed vessel in one minute) and (D) vascular dilation in response to ACh stimulation were quantified. To analyze these parameters, image analysis software (NIS elements, Nikon) was used. See also videos 1, 2, and 3. (E) Total cholesterol, HDL, non-HDL (LDL and VLDL), and glucose levels in the plasma were measured as described in Methods. (F) Inducible ERK5-EKO mice were injected with 4-OHT or peanut oil (vehicle) for 5 consecutive days. Two weeks later, mice were orally administrated pitavastatin or vehicle (PBS) twice a day for 3 days, and leukocyte rolling was recorded. (H) Leukocyte rolling velocity, (I) leukocyte rolling flux, and (J) vascular dilation in response to ACh stimulation were examined. See also videos 4, 5, 6 and 7. In each group, 5 venules/animal were used for analysis. (G) Total cholesterol, HDL, and non-HDL were measured as described in Methods. In peanut oil treated ERK5-EKO mice, normal vasodilation was observed. However, in 4-OHT-treated ERK5-EKO mice, it was significantly dampened, and pitavastatin failed to improve the vessel reactivity (J). Values are mean±SD (n=3–5). *p<0.05, **p<0.01 versus vehicle.
To test the role of pitavastain-mediated endothelial ERK5 activation in these vaso-protective effects, we assayed leukocyte rolling and ACh-induced vasodilation in inducible ERK5-EKO mice. Leukocyte rolling was increased and ACh-induced vasodilation was decreased in 4-hydroxytamoxifen (4-OHT) treated ERK5-EKO, but not in VE-Cad-CreER/wild type (WT) mice (Fig. 5F–J and supplemental videos 4–6). Pitavastatin and the depletion of endothelial ERK5 affected neither total cholesterol nor blood glucose levels (Fig. 5G). Interestingly, pitavastatin had no effect on leukocyte rolling and vessel reactivity in ERK5-EKO mice (Fig. 5F, H–J and supplemental videos 6–7). These results show a critical role for endothelial ERK5 in mediating the vaso-protective effects of pitavastatin.
Endothelial ERK5 mediates pitavastatin’s effects on increased cardiac allograft survival and attenuated graft vasculopathy
Inflammation plays a major role in graft rejection. Since pitavastatin activates ERK5 and reduces inflammatory responses, we examined if this statin could have a protective effect against acute cardiac allograft rejection. We performed a total allomismatched heterotopic cardiac transplantation, in which hearts from either VE-Cad-CreER/WT or ERK5-EKO mice (C57BL/6 background) were transplanted to the abdominal aorta of BALB/c recipient mice. Pitavastatin treatment significantly prolonged the survival of VE-Cad-CreER/WT allografts (Fig. 6A left) but was unable to prolong the survival of ERK5-EKO allografts (Fig. 6A middle and right).
Figure 6. Pitavastatin inhibits allograft rejection via activation of endothelial ERK5.

After 2 weeks of 4-OHT injection, VE-Cad-CreER/WT or ERK5-EKO hearts were transplanted to the recipient abdominal aorta. The recipients were then orally administrated pitavastatin (3 mg/kg) or vehicle (sterilized distilled water) twice a day. To gauge the graft rejection, manual palpation was performed twice a day until the donor heart stops beating. (A) Kaplan-Meier plots indicate that pitavastatin prolongs VE-Cad-CreER/WT (left), but not ERK5-EKO (middle, right), allograft survival. (B) Five days after transplantation, relative mRNA expression levels of TNFα, IFNγ, MCP-1, IL-1, IL-6, and IL-10 in allografts were measured by qPCR and expressed relative to the GAPDH mRNA expression level. In each allograft group, values represent the ratio of mean value of pitavastatin-treated samples to mean value of vehicle treated samples. Values are mean±SD, n value as indicated in graphs. n.s: not significant.
Since cytokines and MCP-1 are involved in allograft rejection (36, 37), we investigated pitavastatin’s effects on their expression in harvested allografts. In VE-Cad-CreER/WT allografts after 5 days of transplantation, pitavastatin treatment increased IL-10 expression while decreasing the expression of TNF-α, IFNγ, MCP-1, and IL-1. IL-6 mRNA expression had a tendency to be also reduced by pitavastatin treatment in VE-Cad-CreER/WT allografts after 5 days of transplantation, but the difference did not reach statistical significance. However, these changes were not found in the ERK5-EKO allografts (Fig. 6B). Taken together, these data suggest that pitavastatin’s effect on allograft survival is mediated by endothelial ERK5 that regulates expression of inflammatory molecules.
Five days after transplantation, vehicle-treated VE-Cad-CreER/WT allografts showed severe coronary arteriolitis with perivascular edema and myocardial necrosis. Inflammatory cells and thrombi were present in the vessel lumen of vehicle-treated VE-Cad-CreER/WT allografts. Although these lesions were morphologically different from chronic graft arterial disease lesions, we examined parenchymal rejection (PR) and graft arterial disease (GAD) using the GAD scoring system and quantified the extent of early luminal occlusions as reported previously (27, 36). PR and GAD scores were significantly lower in pitavastatin-treated VE-Cad-CreER/WT allografts than in vehicle treated VE-Cad-CreER/WT allografts, but there was no difference between pitavastatin- and vehicle-treated ERK5-EKO allografts (Fig. 7A–B). However, we found similar luminal occlusions in both VE-Cad-CreER/WT and ERK5-EKO allografts (Fig. 7A), suggesting that pitavastatin prevents vascular occlusion due to accumulated inflammatory cells and thrombosis in allografts and that this graft survival effect is mediated by endothelial ERK5.
Figure 7. Pitavastatin inhibits inflammatory cell infiltration into allograft vessels, which is abolished in ERK5-EKO allografts.

Histological analyses of allografts 5 days after transplantation (A) Upper: representative cross section images of H&E stained allografts; lower: high power views of infiltrated inflammatory cells into the myocardium space, where damage to myocytes and distorted tissue architecture can be noted. Arrows indicate accumulation of infiltrated inflammatory cells. Scale bar: 50 μm. (B) PR scores (left) and GAD scores (right) as described in the Methods are shown. Values are mean±SD, WT+vehicle (n=8), WT+Pita (n=10), ERK5-EKO+vehicle (n=7), and ERK5-EKO+Pita (n=7). **p < 0.01, *p < 0.05. N.S: not significant.
Since cardiac allograft rejection is associated with accumulation of inflammatory cells and increased expression of pro-inflammatory molecules (36), we examined CD45- and CD3-positive cell infiltration in allografts. Such cells were detected on 5 days after transplantation in vehicle-treated allografts (Fig. 8A–B). The number of CD45-positive, but not CD3-positive cells, was decreased in pitavastatin-treated VE-Cad-CreER/WT allografts. However, there was no difference in the extent of infiltration of CD45-positive cells between vehicle- and pitavastatin-treated ERK5-EKO allografts (Fig. 8A–B).
Figure 8. Pitavastatin inhibits CD45+, but not CD3+, cell infiltration into allografts, which is abolished by specific deletion of EC ERK5.
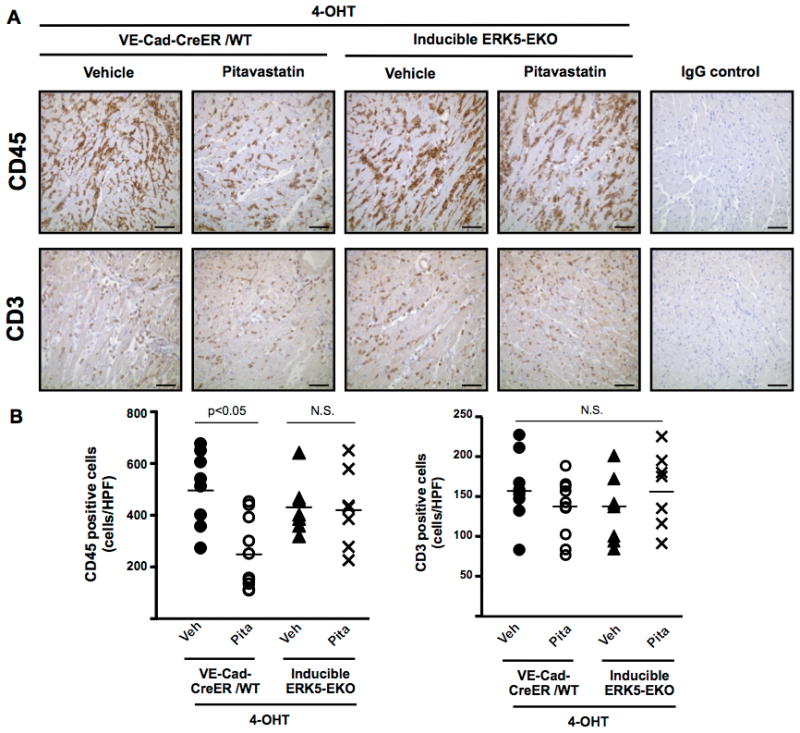
(A) CD45-positive (anti-CD45 staining) and CD3-positive (anti-CD3 staining) cells in the myocardium of allografts 5 days after transplantation. Scale bar =50 μm. (B) Twenty fields per graft were quantified for CD45- and CD3-positive cell number. Values are mean±SD, WT+vehicle (n=8), WT+Pita (n=10), ERK5-EKO+vehicle (n=7), and ERK5-EKO+Pita (n=7). For IgG control, WT+vehicle and ERK5-EKO+Pita samples were used. *p < 0.05.
Discussion
We identified certain statins and anti-malarial agents as potent ERK5 activators by HTS, which are shown to induce anti-inflammatory effects via activating endothelial ERK5. Pitavastatin is one of the statins identified as an ERK5 activator. Interestingly, depletion by siRNA of ERK5 but not MEK5 inhibited pitavastatin-mediated KLF2 induction, strongly suggesting that there is a direct interaction between ERK5 and pitavastatin. Indeed, our in vitro kinase assay showed activation of recombinant ERK5 by pitavastatin in a cell-free system. Importantly, our study shows that pitavastatin reduced the extent of vessel occlusion by inflammatory cells and thrombi in allografts but this beneficial effect was completely abolished by ERK5 depletion in ECs. In a clinical study (38), the contribution of coronary endothelial dysfunction after heart transplantation has been suggested, but the mechanism for this remains largely unclear. To our knowledge this is the first report that defines the crucial role of endothelial ERK5 in the process of cardiac allograft rejection, especially under statins treatment. Although the anti-inflammatory role of ERK5 (22) and statins (12) was reported, the role of ERK5 in statin-induced protection against endothelial dysfunction and cardiac allograft rejection has not been explored in vivo. In this study, we have established using both in vitro and in vivo approaches that via endothelial ERK5, certain statins and anti-malaria drugs provide protection against endothelial dysfunction. Our comprehensive approaches to detect small molecules by HTS and to determine not only their molecular mechanism in vitro but also pathological consequence in vivo have established the critical role of ERK5 in endothelial protection and identified endothelial ERK5 as a novel drug target for heart transplantation. Lastly, we have also established a novel screening assay for detecting ERK5 activators, which provides a technical contribution for further search of compounds that possibly prevent endothelial dysfunction and subsequent cardiac allograft rejection.
In a total allomismatched cardiac transplantation model, pitavastatin significantly prolonged allograft survival, reduced the expression of inflammatory gene expression while increasing anti-inflammatory IL-10 gene expression. A decrease in CD45-positive cells was observed in pitavastatin-treated VE-Cad-CreER/WT allografts, but such a decrease was not found in pitavastatin-treated ERK5-EKO allografts. In contrast to a previous report (39), infiltration of CD3-positive cells was not affected by pitavastatin. Since ERK5 is not essential for T-cell development and survival (40), the lack of pitavastatin’s effects on T-cell infiltration may be reasonable.
Anti-malarial therapy for patients with autoimmune diseases has been an effective treatment since 1955 as this class of drugs improves the survival and remission rates with a good safety profile (16, 17, 41). It is worth noting that anti-malarial agents are recommended for patients with lupus nephritis as they reduce the severity of renal dysfunction through their immune-modulatory, anti-inflammatory, and anti-thrombotic effects, which subsequently prevent endothelial dysfunction and reduce renal inflammation (16). In our study, we found that the anti-inflammatory effect of CQ and HCQ was mediated by an ERK5-dependent but MEK5- or KLF2/4-independent mechanism. To our knowledge, this is the first report defining endothelial ERK5 as a primary action site and proposing a novel application of ERK5 activators such as anti-malarial agents in cardiac allograft transplantation. Since ERK5 also increases the expression of other anti-inflammatory genes and the activity of transcription factors including peroxisome proliferator-activated receptors and Nur77(1, 42, 43), these molecules may be also involved in the CQ- and HCQ-induced anti-inflammatory effects, which need further investigation. Taken together, our results have revealed a novel role for ERK5 as a direct target of certain statins and anti-malarial agents, and as such they prevent endothelial inflammation and dysfunction and suppress acute rejection of cardiac allografts.
As shown in Fig. S2C, we could not find ERK1/2 activation by pitavastatin in ECs. However, it has been reported that pitavastatin can inhibit (not activate) lysophosphatidylcholine-induced ERK1/2 activation in vascular smooth muscle cells (44). Since we did not observe inhibition of the basal level of ERK1/2 activity by pitavastatin, ERK1/2 may not be a major player in regulating NF-κB activation in ECs.
Although we detected a significant increase in ERK5 TEY motif phosphorylation by pitavastatin and CQ, we could not detect visible band-shift of ERK5 by pitavastatin and CQ compared with steady laminar flow stimulation. As we reported previously, ERK5 band shift is not only caused by phosphorylation, but also by other post-translational modification such as SUMOylation and ubiquitination, and possibly by other molecules interactions (2). As we showed in the study, we found that both pitavastatin and CQ increased ERK5 transactivation in a MEK5 independent manner. In addition, ERK5 has multiple phosphorylation sites besides the TEY motif (22, 45). These data suggest that both pitavastatin and CQ increase TEY motif phosphorylation but may not induce phosphorylation of other sites or other molecular association with ERK5 including MEK5 (1). Therefore, it is possible that the cause of band shift induced by CQ and pitavastatin is different from that induced by steady laminar flow (1).
When we overexpressed ERK5 wild type and ERK5 TEF motif phosphorylation site mutant (ERK5-AEF), ERK5-AEF inhibited pitavastatin and CQ-induced ERK5 transcriptional activity (Fig. S2D, E). The deletion of MEK5 inhibited pitavastatin-induced ERK5 TEY motif phosphorylation, but not ERK5 transcriptional activity (Fig. S2A, B). Taken together, although the induction of ERK5 TEY motif phosphorylation by pitavastatin is not necessary for the pitavastatin-induced increase of ERK5 transcriptional activity, the basal level of ERK5 TEY motif phosphorylation is necessary for full activation of ERK5 transcriptional activity induced by pitavastatin.
Supplementary Material
Acknowledgments
The authors thank Drs. Cathy Tournier and Xin Wang for providing ERK5flox/flox mice, Carolyn J Giancursio, Hannah Cushman, Shane A Cieri for technical support, and Yingqian Xu for preparation of HUVECs.
Sources of Funding
This work is supported by grants from the National Institute of Health (NIH) to Drs. Abe (HL-108551, HL-064839, and HL-102746), Fujiwara (HL-064839), Smrcka (GM-81772), Morrell (HL-093179), from the American Heart Association (AHA) to Drs. Fujiwara (11GRNT5850001), Morrell (13EIA14250023), Le (13SDG14500033), and Heo (12SDG11690003), and from Japan Society for the Promotion of Science (JSPS) to Dr. Izawa-Ishizawa.
Nonstandard Abbreviations and Acronyms
- 4-OHT
4-hydroxytamoxifen
- 6-MP
6-mercaptopurine
- ACh
acetylcholine
- BAECs
bovine aortic endothelial cells
- CA-MEKK3
constitutively active form of MEKK3 (mitogen-activated protein kinase kinase kinase 3)
- CQ
chloroquine
- DM
diabetes mellitus
- EC
endothelial cells
- ERK5
extracellular signal-regulated kinase 5
- ERK5-EKO
inducible endothelial ERK5 knock out
- GAD
graft arterial disease
- HCQ
hydroxychloroquine
- HPAECs
human pulmonary aortic endothelial cells
- HTS
high throughput screening
- HUVECs
human umbilical vein endothelial cells
- KLF
Kruppel-like factor
- MEK5
mitogen-activated protein kinase/ERK kinase 5
- PR
parenchymal rejection
- QC
quinacrine
- SLE
systemic lupus erythematosus
- STZ
streptozotocin
- TNFα
Tumor necrosis factor-α
- VCAM1
vascular cell adhesion molecule 1
- WT
wild type
Footnotes
Disclosures
None
References
- 1.Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woo CH, Shishido T, McClain C, Lim JH, Li JD, Yang J, Yan C, Abe J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res. 2008;102:538–545. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- 3.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci. 2000;902:230–239. doi: 10.1111/j.1749-6632.2000.tb06318.x. discussion 239–240. [DOI] [PubMed] [Google Scholar]
- 6.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamawaki H, Lehoux S, Berk BC. Chronic physiological shear stress inhibits tumor necrosis factor-induced proinflammatory responses in rabbit aorta perfused ex vivo. Circulation. 2003;108:1619–1625. doi: 10.1161/01.CIR.0000089373.49941.C4. [DOI] [PubMed] [Google Scholar]
- 8.Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–1357. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 9.Sawada N, Liao JK. Rho/Rho-Associated Coiled-Coil Forming Kinase Pathway as Therapeutic Targets for Statins in Atherosclerosis. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu K, Tian S, Zhou H, Wu Y. Statins protect human endothelial cells from TNF-induced inflammation via ERK5 activation. Biochem Pharmacol. 2013;85:1753–1760. doi: 10.1016/j.bcp.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–726. doi: 10.1161/CIRCULATIONAHA.104.525774. [DOI] [PubMed] [Google Scholar]
- 13.Wenke K, Meiser B, Thiery J, Nagel D, von Scheidt W, Krobot K, Steinbeck G, Seidel D, Reichart B. Simvastatin initiated early after heart transplantation: 8-year prospective experience. Circulation. 2003;107:93–97. doi: 10.1161/01.cir.0000043241.32523.ee. [DOI] [PubMed] [Google Scholar]
- 14.Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr, Garcia-Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–26719. doi: 10.1074/jbc.C500144200. [DOI] [PubMed] [Google Scholar]
- 15.Ohnesorge N, Viemann D, Schmidt N, Czymai T, Spiering D, Schmolke M, Ludwig S, Roth J, Goebeler M, Schmidt M. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4) J Biol Chem. 2010;285:26199–26210. doi: 10.1074/jbc.M110.103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SJ, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nature reviews. Nephrology. 2011;7:718–729. doi: 10.1038/nrneph.2011.150. [DOI] [PubMed] [Google Scholar]
- 17.Alarcon GS, McGwin G, Bertoli AM, Fessler BJ, Calvo-Alen J, Bastian HM, Vila LM, Reveille JD, Group LS. Effect of hydroxychloroquine on the survival of patients with systemic lupus erythematosus: data from LUMINA, a multiethnic US cohort (LUMINA L) Annals of the rheumatic diseases. 2007;66:1168–1172. doi: 10.1136/ard.2006.068676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loffler BM, Bohn E, Hesse B, Kunze H. Effects of antimalarial drugs on phospholipase A and lysophospholipase activities in plasma membrane, mitochondrial, microsomal and cytosolic subcellular fractions of rat liver. Biochim Biophys Acta. 1985;835:448–455. doi: 10.1016/0005-2760(85)90114-6. [DOI] [PubMed] [Google Scholar]
- 19.Bugge E, Gamst TM, Hegstad AC, Andreasen T, Ytrehus K. Mepacrine protects the isolated rat heart during hypoxia and reoxygenation--but not by inhibition of phospholipase A2. Basic Res Cardiol. 1997;92:17–24. doi: 10.1007/BF00803753. [DOI] [PubMed] [Google Scholar]
- 20.Gorbachev AV, Gasparian AV, Gurova KV, Gudkov AV, Fairchild RL. Quinacrine inhibits the epidermal dendritic cell migration initiating T cell-mediated skin inflammation. Eur J Immunol. 2007;37:2257–2267. doi: 10.1002/eji.200636708. [DOI] [PubMed] [Google Scholar]
- 21.Holscher C. Quinacrine acts like an acetylcholine receptor antagonist rather than like a phospholipase A2 inhibitor in a passive avoidance task in the chick. Neurobiology of learning and memory. 1995;63:206–208. doi: 10.1006/nlme.1995.1022. [DOI] [PubMed] [Google Scholar]
- 22.Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, Xu H, Morrell C, Sullivan MA, Cohen MS, Serafimova IM, Taunton J, Fujiwara K, Abe J. A Crucial Role for p90RSK-Mediated Reduction of ERK5 Transcriptional Activity in Endothelial Dysfunction and Atherosclerosis. Circulation. 2013;127:486–499. doi: 10.1161/CIRCULATIONAHA.112.116988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Overbergh L, Valckx D, Waer M, Mathieu C. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine. 1999;11:305–312. doi: 10.1006/cyto.1998.0426. [DOI] [PubMed] [Google Scholar]
- 24.Kagari T, Doi H, Shimozato T. The importance of IL-1 beta and TNF-alpha, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J Immunol. 2002;169:1459–1466. doi: 10.4049/jimmunol.169.3.1459. [DOI] [PubMed] [Google Scholar]
- 25.Nigro P, Satoh K, O’Dell MR, Soe NN, Cui Z, Mohan A, Abe J, Alexis JD, Sparks JD, Berk BC. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2011;208:53–66. doi: 10.1084/jem.20101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosuge H, Suzuki J, Gotoh R, Koga N, Ito H, Isobe M, Inobe M, Uede T. Induction of immunologic tolerance to cardiac allograft by simultaneous blockade of inducible co-stimulator and cytotoxic T-lymphocyte antigen 4 pathway. Transplantation. 2003;75:1374–1379. doi: 10.1097/01.TP.0000061601.26325.82. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu K, Libby P, Shubiki R, Sakuma M, Wang Y, Asano K, Mitchell RN, Simon DI. Leukocyte integrin Mac-1 promotes acute cardiac allograft rejection. Circulation. 2008;117:1997–2008. doi: 10.1161/CIRCULATIONAHA.107.724310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pires NM, Pols TW, de Vries MR, van Tiel CM, Bonta PI, Vos M, Arkenbout EK, Pannekoek H, Jukema JW, Quax PH, de Vries CJ. Activation of nuclear receptor Nur77 by 6-mercaptopurine protects against neointima formation. Circulation. 2007;115:493–500. doi: 10.1161/CIRCULATIONAHA.106.626838. [DOI] [PubMed] [Google Scholar]
- 29.French JK, Hurst NP, O’Donnell ML, Betts WH. Uptake of chloroquine and hydroxychloroquine by human blood leucocytes in vitro: relation to cellular concentrations during antirheumatic therapy. Annals of the rheumatic diseases. 1987;46:42–45. doi: 10.1136/ard.46.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM. Transcriptional Repression of Atherogenic Inflammation: Modulation by PPAR{delta} Science. 2003 doi: 10.1126/science.1087344. [DOI] [PubMed] [Google Scholar]
- 31.Takata Y, Liu J, Yin F, Collins AR, Lyon CJ, Lee CH, Atkins AR, Downes M, Barish GD, Evans RM, Hsueh WA, Tangirala RK. PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci U S A. 2008;105:4277–4282. doi: 10.1073/pnas.0708647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plutzky J. The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Circ Res. 2011;108:1002–1016. doi: 10.1161/CIRCRESAHA.110.226860. [DOI] [PubMed] [Google Scholar]
- 33.Barish GD, Yu RT, Karunasiri MS, Becerra D, Kim J, Tseng TW, Tai LJ, Leblanc M, Diehl C, Cerchietti L, Miller YI, Witztum JL, Melnick AM, Dent AL, Tangirala RK, Evans RM. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 2012;15:554–562. doi: 10.1016/j.cmet.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Booth G, Stalker TJ, Lefer AM, Scalia R. Mechanisms of amelioration of glucose-induced endothelial dysfunction following inhibition of protein kinase C in vivo. Diabetes. 2002;51:1556–1564. doi: 10.2337/diabetes.51.5.1556. [DOI] [PubMed] [Google Scholar]
- 35.Taylor PD, Wickenden AD, Mirrlees DJ, Poston L. Endothelial function in the isolated perfused mesentery and aortae of rats with streptozotocin-induced diabetes: effect of treatment with the aldose reductase inhibitor, ponalrestat. Br J Pharmacol. 1994;111:42–48. doi: 10.1111/j.1476-5381.1994.tb14021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosuge H, Haraguchi G, Koga N, Maejima Y, Suzuki J, Isobe M. Pioglitazone prevents acute and chronic cardiac allograft rejection. Circulation. 2006;113:2613–2622. doi: 10.1161/CIRCULATIONAHA.105.594101. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki J, Koga N, Kosuge H, Ogawa M, Haraguchi G, Maejima Y, Saiki H, Isobe M. Pitavastatin suppresses acute and chronic rejection in murine cardiac allografts. Transplantation. 2007;83:1093–1097. doi: 10.1097/01.tp.0000259650.67061.16. [DOI] [PubMed] [Google Scholar]
- 38.Hollenberg SM, Klein LW, Parrillo JE, Scherer M, Burns D, Tamburro P, Oberoi M, Johnson MR, Costanzo MR. Coronary endothelial dysfunction after heart transplantation predicts allograft vasculopathy and cardiac death. Circulation. 2001;104:3091–3096. doi: 10.1161/hc5001.100796. [DOI] [PubMed] [Google Scholar]
- 39.Khatri P, Roedder S, Kimura N, De Vusser K, Morgan AA, Gong Y, Fischbein MP, Robbins RC, Naesens M, Butte AJ, Sarwal MM. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J Exp Med. 2013;210:2205–2221. doi: 10.1084/jem.20122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ananieva O, Macdonald A, Wang X, McCoy CE, McIlrath J, Tournier C, Arthur JS. ERK5 regulation in naive T-cell activation and survival. Eur J Immunol. 2008;38:2534–2547. doi: 10.1002/eji.200737867. [DOI] [PubMed] [Google Scholar]
- 41.Fessler BJ, Alarcon GS, McGwin G, Jr, Roseman J, Bastian HM, Friedman AW, Baethge BA, Vila L, Reveille JD, Group LS. Systemic lupus erythematosus in three ethnic groups: XVI. Association of hydroxychloroquine use with reduced risk of damage accrual. Arthritis Rheum. 2005;52:1473–1480. doi: 10.1002/art.21039. [DOI] [PubMed] [Google Scholar]
- 42.Woo CH, Massett MP, Shishido T, Itoh S, Ding B, McClain C, Che W, Vulapalli SR, Yan C, Abe J. ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor delta (PPARdelta) stimulation. J Biol Chem. 2006;281:32164–32174. doi: 10.1074/jbc.M602369200. [DOI] [PubMed] [Google Scholar]
- 43.Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol. 2000;20:8382–8389. doi: 10.1128/mcb.20.22.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamakawa T, Tanaka S, Kamei J, Kadonosono K, Okuda K. Pitavastatin inhibits vascular smooth muscle cell proliferation by inactivating extracellular signal-regulated kinases 1/2. J Atheroscler Thromb. 2003;10:37–42. doi: 10.5551/jat.10.37. [DOI] [PubMed] [Google Scholar]
- 45.Morimoto H, Kondoh K, Nishimoto S, Terasawa K, Nishida E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J Biol Chem. 2007;282:35449–35456. doi: 10.1074/jbc.M704079200. [DOI] [PubMed] [Google Scholar]
- 46.Yan C, Takahashi M, Okuda M, Lee JD, Berk BC. Fluid shear stress stimulates big mitogen-activated protein kinase 1 (BMK1) activity in endothelial cells. Dependence on tyrosine kinases and intracellular calcium. J Biol Chem. 1999;274:143–150. doi: 10.1074/jbc.274.1.143. [DOI] [PubMed] [Google Scholar]
- 47.Le NT, Corsetti JP, Dehoff-Sparks JL, Sparks CE, Fujiwara K, Abe J. Reactive Oxygen Species, SUMOylation, and Endothelial Inflammation. International journal of inflammation. 2012;2012:678190. doi: 10.1155/2012/678190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chao TH, Hayashi M, Tapping RI, Kato Y, Lee JD. MEKK3 directly regulates MEK5 activity as part of the big mitogen- activated protein kinase 1 (BMK1) signaling pathway. J Biol Chem. 1999;274:36035–36038. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.