

Summary
To investigate miRNA function in human acute myeloid leukemia (AML) stem cells (LSC), we generated a prognostic LSC-associated miRNA signature derived from functionally validated subpopulations of AML samples. For one signature miRNA, miR-126, high bioactivity aggregated all in vivo patient sample LSC activity into a single sorted population, tightly coupling miR-126 expression to LSC function. Through functional studies, miR-126 was found to restrain cell cycle progression, prevent differentiation, and increase self-renewal of primary LSC in vivo. Compared with prior results showing miR-126 regulation of normal hematopoietic stem cell (HSC) cycling, these functional stem effects are opposite between LSC and HSC. Combined transcriptome and proteome analysis demonstrates that miR-126 targets the PI3K/AKT/MTOR signaling pathway, preserving LSC quiescence and promoting chemotherapy resistance.
Graphical Abstract
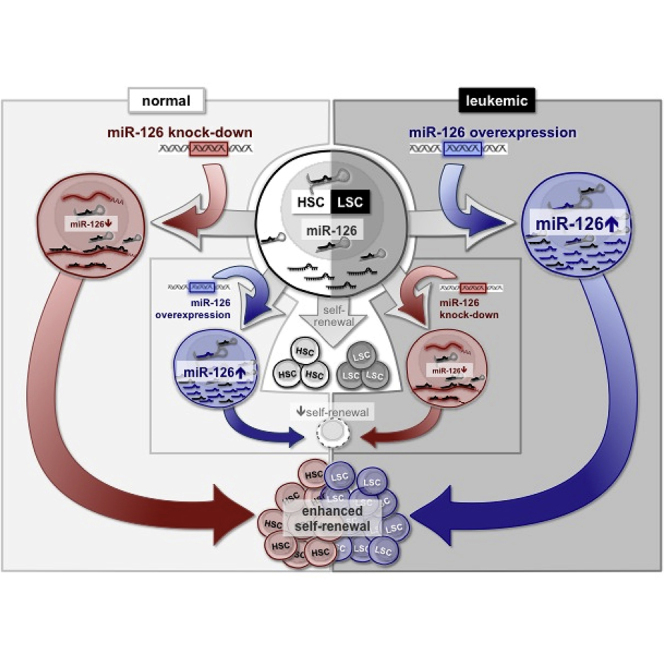
Highlights
-
•
Clinical outcome in AML correlates with LSC-associated miRNA expression
-
•
miR-126 targets multiple components of the PI3K/AKT/MTOR signaling pathway
-
•
miR-126 promotes chemotherapy resistance by preserving LSC in a quiescent state
-
•
miR-126 governs opposing self-renewal outcomes in normal and malignant stem cells
Lechman et al. show that miR-126 targets the PI3K/AKT/MTOR signaling pathway to preserve quiescence, increase self-renewal, and promote chemotherapy resistance of acute myeloid leukemia stem cells (LSC). Reducing the miR-126 level impairs LSC maintenance in contrast to expanding normal hematopoietic stem cells.
Significance
Leukemia stem cells play central roles in disease progression and recurrence due to their intrinsic capacity for self-renewal and chemotherapy resistance. However, few regulators of human LSC function are known. Our study establishes that miRNA plays a powerful role in governing the fundamental properties that define the stemness state of human LSC including quiescence, self-renewal, and chemotherapy response. Self-renewal regulators have remarkably parallel functions in malignant and normal stem cells, precluding their therapeutic targeting because of toxicity to normal stem cells. The opposing self-renewal outcomes governed by miR-126 within HSC and LSC indicate that despite shared stemness determinants, it may be possible to target therapeutically the networks that specifically control LSC through perturbation of miR-126 levels.
Introduction
Acute myeloid leukemia (AML) is organized as an aberrant developmental hierarchy maintained by functionally distinct leukemia stem cells (LSC) (Kreso and Dick, 2014). LSC are linked to therapy failure and disease recurrence, but they also share many biological properties with hematopoietic stem cells (HSC), including capacity for self-renewal and quiescence (Kreso and Dick, 2014). Several self-renewal regulators have been studied in both HSC and LSC contexts including PTEN, BMI1, GFI1, TEL1, STAT5, and JUNB; except for PTEN, loss of function typically impairs self-renewal of both LSC and HSC (Yilmaz and Morrison, 2008). LSC and HSC are both quiescent, although quiescence regulation is better understood in HSC. Several intrinsic and extrinsic signals converge upon cyclins and cyclin-dependent kinases (CDKs) that act upstream of Retinoblastoma (RB) family members to regulate early and late G1 progression in HSC (Viatour et al., 2008), while the G0 state is governed by MTORC1 and CDK6 (Laurenti et al., 2015, Rodgers et al., 2014). Quiescence and distinct G0 exit kinetics are essential HSC properties (Trumpp et al., 2010). Although LSC quiescence is less well defined, the known regulators appear to function similarly in LSC and HSC, with LSC quiescence often invoked as a mechanism of chemotherapy resistance (Holtz et al., 2007). Additional studies are required to determine if differences exist in self-renewal and quiescence regulation between LSC and HSC and whether it is possible to develop therapies that eradicate LSC while sparing HSC.
Transcriptional analysis of human HSC and functionally defined LSC have defined stemness signatures that are highly prognostic for patient survival, establishing that LSC-specific properties are clinically relevant (Eppert et al., 2011, Metzeler et al., 2013). However, little is known of how stemness programs are controlled. Several differentially expressed miRNAs were identified and found to control HSC (Hu et al., 2015, Lechman et al., 2012, Mehta et al., 2015, O'Connell et al., 2010) by coordinate repression of multiple targets (Ebert and Sharp, 2012). In hematopoiesis, most miRNAs affect progenitor lineage commitment and mature cell function (Undi et al., 2013), although HSC self-renewal can be governed by miR-125a/b, miR-29a, and miR-126 (Ooi et al., 2010, O'Connell et al., 2010, Guo et al., 2010, Lechman et al., 2012). miR-126 plays a role, conserved in both human and mouse, in maintaining HSC quiescence by attenuating the cellular response to extrinsic signals via targeting multiple components of the PI3K/AKT/GSK3B signaling pathway (Lechman et al., 2012). Thus, HSC expand without concomitant exhaustion upon miR-126 silencing.
Deregulation of miRNAs occurs in leukemia correlating with known risk categories and prognosis (Garzon et al., 2008, Li et al., 2008, Marcucci et al., 2009). Functionally, miRNA overexpression can induce murine leukemic transformation (Han et al., 2010, O'Connell et al., 2010, Song et al., 2013). Several LSC-associated miRNAs are functional: miR-17-92 polycistron maintained LSC in MLL models (Wong et al., 2010), whereas antagonizing miR-196 and miR-21 reduced LSC in an experimental human MLL model (Velu et al., 2014). Targeted miR-126 reduction in cell lines and primary AML samples reduced AML growth, although mechanisms were not reported (Dorrance et al., 2015, de Leeuw et al., 2014). These promising studies point to the importance of further understanding the role of miRNA in governing stemness in AML. Here, we investigated the role of miR-126 in governing LSC self-renewal, quiescence, and chemotherapy resistance.
Results
LSC miRNA Signature Is Prognostic for Patient Outcome
To determine whether miRNA are differentially expressed in LSC and HSC, we fractionated 16 AML patient samples and three lineage-depleted (Lin–) cord blood (CB) samples using CD34 and CD38 into four populations and subjected each to global miRNA profiling; the stem cell content of each fraction was functionally assayed by xenotransplantation (Figures 1A and S1A). Bioinformatic analysis of 25 LSC-enriched and 27 fractions devoid of LSC activity (Figure S1A) revealed a human LSC-associated miRNA signature derived from in vivo functionally validated AML patient samples (Figure 1B). In parallel, miRNAs enriched in HSC or committed progenitors were determined (Figure S1B). By comparing similar immunophenotypic AML and normal populations, several differentially expressed miRNAs were found (Figure S1C).
Figure 1.

Generation and Validation of an LSC-Enriched miRNA Signature
(A) Schematic depicting the strategy to fractionate human AML patient samples based on immuno-phenotypic staining for CD34 and CD38. Functional validation of sorted fractions was performed by xenotransplantation, the result of which was combined with miRNA expression profiling to generate stem cell-related miRNA expression profiles.
(B) Heatmap and summary of miRNAs enriched within the LSC and non-LSC populations.
(C) The optimized miRNA signature derived from regression analysis of the PMCC cohort and the weight each miRNA adds to the overall signature.
(D and E) Validation of the optimized LSC-associated miRNA signature shown in (C) in the TCGA cohort (n = 187) by (D) univariate analysis (hazard ratio [HR], 2.04; p < 0.0001) and (E) multivariate analysis (HR, 2.53; p = 0.003). See also Figure S1 and Table S1.
To determine if the LSC-associated miRNA signature was clinically relevant, a regression analysis was performed on 74 AML patients with normal cytogenetics (PMCC cohort, Table S1). An optimized LSC signature consisting of four miRNAs was identified, each with differential weights based on impact upon overall survival (OS) (Figure 1C). This signature was prognostic of OS in both univariate (Figure 1D) and multivariate analyses (Figure 1E) in an independent cohort. Together with prior studies showing that LSC-specific gene expression signatures are significantly prognostic (Eppert et al., 2011, Greaves, 2011), these data establish that LSC properties influence clinical outcomes and that miRNAs play a powerful role in regulating LSC stemness.
miR-126 Bioactivity Enriches for LSC Activity
Further functional studies on AML focused on miR-126 as it is a known HSC regulator (Lechman et al., 2012). qPCR independently confirmed that LSC-containing AML fractions generally expressed the highest miR-126 levels (Figure S2A). As miRNA expression does not uniformly equate with miRNA bioactivity, a miR-126 lentiviral reporter vector was used to investigate whether miR-126 is biologically active in LSC (Gentner et al., 2010); ΔNGFR levels indicate transduced cells, while EGFP levels are inversely correlated with miR-126 bioactivity (Figure S2B). Four primary AML samples (Table S2) were transduced with the reporter, transplanted into xenografts, and after 12 weeks the engrafting population was sorted solely on the basis of miR-126 bioactivity (Figure 2A). Each sorted population was transplanted into secondary mice and LSC activity scored after 8 weeks, based on whether the engrafting population recapitulated the same EGFP/ΔNGFR flow profile as the primary recipient (a cardinal property of cancer stem cells). Despite the presence of LSC activity in multiple subpopulations with CD34 and CD38 sorting (Table S2), miR-126 bioactivity aggregated all LSC activity into a single miR-126high population (Figure 2B). qPCR confirmed 40-fold higher mature miR-126 levels in LSC-engrafting fractions compared with non-engrafting fractions for three AML samples (Figure S2C). LSC-containing fractions also had the highest clonogenic (Figure S2D) and proliferative potential (Figure S2E). These data indicate that miR-126 bioactivity is directly linked to LSC function and that it is possible to exploit miRNA bioactivity for prospective LSC isolation, circumventing often unreliable and heterogeneously expressed cell surface markers (Kreso and Dick, 2014).
Figure 2.

miR-126 Bioactivity Marks the Functional LSC Compartment in Human AML
(A) Schematic describing the sorting scheme/scoring system for secondary mice. AML samples were transduced with an miR-126 reporter construct and transplanted into conditioned NSG mice for 12 weeks. Bone marrow was analyzed for engraftment using CD45+ΔNGFR+EGFP+ staining. Cells were sorted into four populations based on ΔNGFR (transduced cells) and EGFP expression (inverse of miR-126 bioactivity), counted, and injected into secondary mice for 8–10 weeks. When a ΔNGFR/EGFP profile is recapitulated in secondary mice, the mouse is scored as engrafted.
(B) Summary of the results of the miR-126 bio-reporter assays.
(C) Kaplan-Meier overall survival (OS) curves in the PMCC CN AML cohort (n = 74) according to the miR-126 expression level (HR, 2.23; p = 0.00901).
(D) Univariate Cox model analysis for miR-126 as prognostic for event-free survival in the PMCC cohort of CN AML patients (n = 74; p = 0.0207, log rank test, median split; HR, 1.8744; p = 0.0207, Wald test).
(E) Kaplan-Meier survival curves correlating miR-126 expression and relapse-free survival in the PMCC patient cohort. Univariate median split log rank test (HR, 1.7995; p = 0.033, Wald test).
(F) Univariate analysis for OS in the TGCA AML cohort that encompasses all levels of cytogenetic risk (n = 187) according to the miR-126 expression level (HR, 1.41; p = 0.0382). See also Figure S2.
Clinical Relevance of miR-126 Expression
To determine if miR-126 expression alone is prognostic, the PMCC cohort (Table S1) was investigated, and increased miR-126 expression was found to be associated with worse OS (median OS of 28.5 months [high expression] versus not reached [low expression]; Figure 2C), event-free survival (Figure 2D), and relapse-free survival (Figure 2E), a result in keeping with other studies (Dorrance et al., 2015, de Leeuw et al., 2014). Since miR-126 expression is high in patients with t(8; 21) and inv(16) (Li et al., 2008), we evaluated the prognostic value of miR-126 after excluding these patients from The Cancer Genome Atlas (TCGA) dataset. High miR-126 was associated with decreased survival in the TCGA dataset (median OS of 12.3 months [high expression] versus 18.5 months [low expression]; Figure 2F). The prognostic significance of miR-126 further strengthens the link between AML patient outcomes, stemness properties, and the regulatory of role of miRNA (Greaves, 2011).
Development of a Functionally Relevant Human AML Model for Mechanistic Studies
How miR-126 functions throughout the AML hierarchy is difficult to investigate since functional studies in primary AML cells are technically challenging and hitherto no human AML cell lines recapitulate the hierarchical organization of primary cells. Therefore, we developed an indefinitely growing AML culture system (8227) from a relapse sample that is organized as a functional hierarchy (Figure 3A) (E.L., unpublished data). Expression of CD34 and CD38 is tightly linked to the functional hierarchy; CD34+CD38− cells possess LSC activity and contain a quiescent population, by contrast CD34+CD38+ cells are enriched in clonogenic progenitors and the remaining 90% of CD34−CD38+ and CD34−CD38− cells are terminally differentiated CD15+CD14+ blasts (Figure 3A). We show through an integrated analysis of function, phenotype, miR-126 bioactivity, and promoter methylation status on all sorted fractions that high miR-126 levels correlate with the CD34+CD38− phenotype and LSC activity and are linked to EGFL7 expression and stem cell-specific promoter methylation patterns (Figure S3A and E.L., unpublished data). Thus, 8227 cells are a relevant model culture system for interrogating the functional effects of miR-126 activity within the context of a leukemic hierarchy.
Figure 3.

Enforced Expression and Knockdown of miR-126 Alters the Proliferation and Differentiation Status of Primitive 8227 AML Cells
(A) Illustration showing flow plots of CD34 and CD38 immunostained 8227 cultures. The red gated (CD34+CD38-) population is enriched in quiescent LSC and reinitiates the original hierarchy in vitro after flow sorting. The blue gated population (CD34+CD38+) is enriched in colony-forming unit (CFU) potential and represents the AML progenitor compartment. Both green and orange gated CD34− compartments are devoid of LSC and CFU activity, express CD15 and CD14 differentiation markers, and represent terminally differentiated mature AML blasts.
(B) Relative expression of mature miR-126-3p in 8227 cells 7 days after transduction with lentivectors expressing miR-126 (126OE) or an empty control vector (CTRL) measured by qPCR.
(C) The proportion of CD34+ cells over the time course of culture of 126OE and CTRL cells.
(D) Clonogenic potential of sorted subpopulations of 8227 cells after transduction with CTRL or 126OE vectors plated immediately post-sort.
(E) Percent BrdU incorporation into bulk cultures showing proliferation of CTRL and 126OE transduced 8227 cells over time.
(F) Ki67/Hoechst cell cycle staining of CD34+CD38− LSC-enriched 8227 cells.
(G and H) Percentage of total CD34+ (G) and primitive CD34+CD38– and CD34+CD38+ progenitor cells (H) at day 8 and day 15 post-sort in vitro in 8227 culture after sponge-mediated miR-126 knockdown.
(I) Day 0 post-sort colony-forming potential of sorted fractions of CTRL and 126KD 8227 cells.
(J) Proliferation measured by BrdU incorporation assay of CTRL or 126KD transduced 8227 cells in vitro.
(K) Cell cycle analysis of CD34+CD38− 8227 cells measured by Ki67/Hoechst staining.
Data are shown as means ± SEM of three biological replicate experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S3.
miR-126 Expression Induces Quiescence in Primitive AML Cells
To investigate the functional importance of miR-126 within the AML developmental hierarchy, 8227 cells were transduced with an mOrange (mO) lentivirus expressing miR-126 (126OE) or empty control vector (CTRL) (Figure S3B), and elevated miR-126 levels were confirmed (Figure 3B). Following in vitro propagation of transduced cells, the mO+CD34+CD38– (surrogate LSC) population was sorted and the proliferative, differentiation, and clonogenic capacity was evaluated over 28 days. By 7 days, primitive CD34+ cells increased (Figure 3C) and differentiated CD14+CD15+ cells decreased (Figure S3C) in the 126OE group. This proportional increase in CD34+ cells correlated to transient reductions in clonogenicity of day 0 bulk cultures (Figure S3D); a reduction primarily confined to CD34+CD38+ clonogenic fractions (Figure 3D). Bulk cultures of the 126OE group had significantly decreased (15%) bromodeoxyuridine (BrdU) incorporation at 3 hr (p = 0.002) and 16 hr (p = 0.001) compared with CTRL (Figure 3E). No differences in apoptosis were observed (data not shown). Cell cycle analysis of sorted 126OE populations at 7 days showed 2-fold increased proportions of quiescent (G0) CD34+CD38− cells and decreased S/G2/M cells (Figures 3F and S3E). By contrast, the G0 status of 126OE CD34+CD38+ and CD34− populations remained unaffected (data not shown). Thus, 126OE maintains 8227 cells in a more primitive state by increasing the proportion of quiescent CD34+CD38– cells, thereby decreasing the overall proliferative output and differentiation of AML blasts.
miR-126 Knockdown Provokes LSC Entry into Cycle
To determine the impact of miR-126 knockdown, 8227 cells were transduced with lentiviruses that were empty (CTRL) or expressing an miR-126 sponge (126KD) (Figure S3F) (Lechman et al., 2012). Following sorting and culture, 126KD of the EGFP+CD34+CD38– population resulted in increased output of CD34+ cells at all time points (Figure 3G), without increasing differentiation (Figure S3G). This effect was primarily localized to the CD34+CD38– compartment (Figure 3H). Clonogenic potential within the CD34+CD38− LSC-enriched compartment increased while no differences were observed in the CD34+CD38+ progenitor-enriched compartment (Figure 3I). 126KD increased BrdU incorporation by 20% at 3 hr (p = 0.0024) and 16 hr (p = 0.0093) (Figure 3J) without affecting apoptosis (data not shown). Upon 126KD, the proportion of cells in G0 was decreased (30%) and S/G2/M increased (3-fold) within EGFP+CD34+CD38− populations (Figures 3K and S3H). 126KD of CD34+CD38+ cells trended in the same direction (CTRL G0 16.07% versus 126KD G0 11.54%, p = 0.2); CD34− and non-transduced populations were unaffected (data not shown). Within bulk 126KD cultures, the increased cell cycle and clonogenicity (Figure S3I) was primarily due to effects on CD34+CD38– cells (Figure 3H). As LSC-enriched CD34+CD38– cells are less clonogenic than CD34+CD38+ cells, we interpret these data as 126KD driving CD34+CD38– cells out of their quiescent stem-like state and into a more committed population of proliferating clonogenic cells while retaining a primitive cell surface phenotype.
Enforced Expression of miR-126 Expands LSC In Vivo
To test the prediction that miR-126 maintains a primitive state by restraining entry into the cell cycle of LSC from patients, nine AML samples were transduced with 126OE and CTRL vectors and transplanted into NSG mice (Tables S2 and S3). Transduction efficiency and expression varied (Figures S4A and S4B), while leukemic engraftment was similar between CTRL and 126OE groups (Figure S4C). Although the initial transduction efficiency was ∼50% lower for 126OE than CTRL in six of nine AML samples, mOrange+ cells within the human CD45+ graft was higher for six of nine AML samples indicating a competitive advantage for 126OE groups (Figure S4D). Analysis of primitive cell engraftment used both CD34 and CD117, as CD117 is associated with AML clinical outcome and correlated with miR-126 expression (de Leeuw et al., 2014, Schneider et al., 2015). Phenotypic primitive cells were increased in 126OE groups for seven of nine samples (Figure 4A) with concomitant reduction of differentiated cells; four of nine samples showed a significant reduction for CD15+ cells (Figure 4B), and six of nine showed a trend for reduced CD14+ blasts (Figure S4E). 126OE caused an increase in CD15+ blasts for two samples (Figure 4B).
Figure 4.

Enforced Expression of miR-126 Expands Primary AML LSC
(A) Representative flow plots depicting changes in CD117+ and CD34+ levels upon 126OE and quantification of the percentage of CD117+ cells within the human CD45+mO++ graft.
(B) Representative flow plots depicting changes in the percentage of CD34+ and CD15+ cells within the human CD45+mO+ graft and quantification of changes in the percentage of AML cells expressing differentiation marker CD15. Data in (A) and (B) represent means ± SEM of 4–6 mice; ∗p < 0.05, ∗∗p < 0.01.
(C) CD45+mO+ AML cells were flow sorted from primary mice and transplanted in limiting doses into secondary recipients for 8–10 weeks. Human CD45+ marking of > 0.5% was considered positive for AML engraftment. Human grafts were confirmed to be CD33+CD19− AML. Limiting dilution analysis was performed using ELDA software. See also Figure S4 and Table S2.
To evaluate 126OE on LSC function within the xenografts, serial transplantation with limiting dilution analysis was used to quantify LSC numbers. In three samples, LSC frequency increased in the 126OE group (Figures 4C and S4F). Although individual patient samples exhibited variation, overall, 126OE increased LSC self-renewal and reduced differentiation leading to LSC expansion.
miR-126 Knockdown Targets LSC In Vivo
126KD was used to determine whether reducing miR-126 would impair AML engraftment or LSC function (Figure S5A). Total levels of human CD45+ (Figure S5B) or CD45+EGFP+ engraftment (Figure S5C) were unaffected in the 126KD group, although there was heterogeneity. By contrast, primitive CD117+ blasts were reduced in three of seven in the 126KD group, while two of seven had increased CD117+ blasts (Figure 5A); differentiated CD15+CD14+ cells were increased in four of seven samples (Figure 5B). The LSC frequency was reduced in two of three samples upon 126KD (Figures 5C and S5D). Together, these findings suggest that 126KD produces heterogeneous responses with LSC function and frequency reduced in a subset of AML patients.
Figure 5.

Diminished miR-126 Levels Reduce the Proportion of Primitive AML Cells
(A) Representative flow plots depicting changes in CD117+ and CD34+ levels upon 126KD and quantification of the percentage of CD117+ cells within the human CD45+EGFP+ graft.
(B) Representative flow plots depicting changes in the percentage of CD14+CD15+ cells within the human CD45+EGFP+ graft and quantification of changes in percentage of AML cells expressing differentiation markers CD14 and CD15. Data in (A) and (B) represent means ± SEM of 4–6 mice; ∗p < 0.05, ∗∗p < 0.01.
(C) CD45+EGFP+ AML cells were flow sorted from primary mice and transplanted at limiting doses into secondary recipients for 8–10 weeks. Human CD45+ marking of >0.5% was considered positive for AML engraftment. Human grafts were confirmed to be CD33+CD19– AML. Limiting dilution analysis was performed using ELDA software. See also Figure S5.
PI3K/AKT/MTOR Is Targeted by miR-126 in Primitive AML Cells
An integrated transcriptional and proteomic approach was employed to gain mechanistic insight into miR-126 functioning. Quantitative protein mass spectrometry (MS) was performed on bulk 126OE and CTRL 8227 cells resulting in the identification and quantification of 8,848 and 4,837 proteins, respectively. In parallel, gene expression profiling was undertaken on 126KD, 126OE or CTRL CD34+CD38–, and CD34+CD38+ 8227 cells. Gene set enrichment analysis (GSEA) of the proteomics dataset identified pathways and leading edge genes directly targeted by miR-126. In post-analysis, transcriptomic datasets were correlated with proteomic-modulated pathways (Figure 6A). The most significant pathways identified centered on PI3K/AKT/MTOR signaling, a miR-126 target pathway previously validated in primitive normal human CB cells (Lechman et al., 2012). In addition, the protein MS data revealed a strong quiescence signature (Figure S6A) substantiating the in vitro cell cycle effects (Figures 3 and S3). Additional BrdU labeling studies with miR-126OE and miR-126KD confirmed these cell cycle effects in vivo (Figures S6B and S6C). The proteomic analysis was validated and confirmed by western blot of 8227 cells showing that ADAM9, PIK3R2 (p85beta), and AKT levels are reduced in 126OE groups (Figure 6B). Although AKT is not a predicted miR-126 target, the protein MS data show that all three AKT isoforms are reduced by 126OE (Table S4). In addition, many predicted and validated miR-126 targets are signaling inputs for AKT activity (Martelli et al., 2010). To activate AKT, PDK1 is required to phosphorylate AKT on Thr308 in the activation loop. We found that pPDK1 Ser241 is reduced with 126OE, suggesting PDK1 activity is reduced by miR-126, further dampening AKT activation (Figure 6C). MTORC2 plays a critical role in AKT Ser473 phosphorylation, a prerequisite for full AKT activation. Our proteomics analysis found that MAPKAP1 (Sin1) was downregulated by 126OE (Figure 6A) and since MAPKAP1 is required for MTORC2 complex formation (Yang et al., 2006), its reduction is predicted to reduce MTORC2 activity. Finally, since PTEN antagonizes the PI3K/AKT signaling pathway by dephosphorylating phosphoinositides, and no change in total PTEN levels were observed by protein MS, we checked pPTEN Ser380 status and found increased pPTEN Ser380 phosphorylation; a modification thought to stabilize PTEN and maintain its function (Birle et al., 2002). Collectively, this integrated analysis provides strong data that miR-126 expression dampens many components of the PI3K/AKT/MTOR signaling pathway in primitive AML populations.
Figure 6.

PI3K/AKT/MTOR Is Targeted by miR-126 in Primitive AML Cells
(A) Functional enrichment map for protein MS-based expression revealing miR-126 modulated pathways. Blue nodes (circles) represent gene sets enriched in proteins downregulated in 8227 cells overexpressing miR-126. Green line (edge) width between nodes corresponds to the number of shared proteins. Predicted miR-126 targets (purple triangle) are connected to enriched pathways by gray edges and edge width is proportional to the overlap significance (Wilcoxon proteomics p < 0.05 and hypergeometric test p < 0.05). Downregulated genes from the transcriptomics data (red diamond) are connected to enriched pathways by orange edges (Wilcoxon proteomics p < 0.05, Wilcoxon transcriptomic p < 0.25, and hypergeometric p < 0.05). Thickest orange and gray edges have significant Wilcoxon and Fisher's exact test p < 0.05. Map includes only nodes (cyan border) that have significant overlap with miR-126 predicted targets and expression data and connected nodes belonging to same clusters (MCL cluster algorithm called from ClusterMaker2). Gene names in gray beside each cluster are the genes that are found in the specified cluster and overlap with predicted miR-126 targets repressed in 126OE.
(B) Western blot of ADAM9, PIK3R2, and AKT levels in 8227 cells transduced with miR-126OE or control lentivirus. GAPDH is the loading control.
(C) Western blot of phospho-PDK1 Ser241 and phospho-PTEN Ser280 levels in 8227 cells transduced with miR-126OE or control lentivirus. GAPDH is the loading control.
(D) Representative intracellular flow plots for the detection of CDK3 and pRB Ser807/811. Graph below represents three independent intracellular flow experiments for each condition where the mean fluorescence intensity was compared. Mean ± SEM; ∗p=<0.05 and ∗∗∗∗p=<0.0001.
(E) Graph depicting enhanced expansion of bulk 8227 cultures after enforced expression of CDK3 and mutCDK3. Fold expansion is normalized to mutCDK3 control culture day 7 after transduction. Data shown are the mean ± SEM of three replicate experiments; ∗p=<0.05.
(F) Graph showing clonogenic potential of primitive AML cells after enforced expression of CDK3 and mutCDK3. Colony counts are shown as the mean ± SEM of three replicate experiments.
(G) Graph depicting CDK3/OE rescue of CD34+ cell expansion upon 126OE. 8227 cells were transduced with miR-126, and CD34+CD38– cells were sorted and placed into culture. Cells were transduced with viral vectors expressing the mutCDK3 control vector or CDK3 vector. Flow cytometry was performed at day 3 and day 7. The percentage of CD34+ cells in double-transduced cultures is shown as the mean ± SEM of three replicate experiments, where ∗∗p < 0.01. See also Figure S6 and Table S3.
To characterize miR-126 targets not identified by proteomics or GSEA, all genes upregulated with 126KD and downregulated with 126OE (Figure S6D) were compared in collated lists of predicted miR-126 targets generated from four published prediction algorithms. Genes were ranked according to the level of perturbation by miR-126 (Figure S6E). Selected candidates including ADAM9, ILK, GOLPH3, CDK3, and TOM1 were confirmed as miR-126 targets using 3′ UTR luciferase reporter assays (Figure S6F) (Hamada et al., 2012, Oglesby et al., 2010).
PI3K/AKT signaling ultimately converges upon cyclins and CDK that promote RB1 phosphorylation and cell cycle entry. The uncovering of CDK3 as a potential miR-126 target was intriguing as miR-126 reduces cell cycle progression and CDK3 was previously identified as a gatekeeper of G0-G1 cell cycle control (Ren and Rollins, 2004). The PI3K/AKT/MTOR pathway regulates CDKN1B (p27kip) protein stability by controlling the levels of SKP2, a component of the SCFSKP2 ubiquitin ligase complex (Lin et al., 2009). Both chemical inhibition of PI3K or enforced expression of PTEN induces p27kip upregulation in quiescent cells (Collado et al., 2000, Lu et al., 1999) and CDK3 activity is downregulated with transient p27kip expression (Braun et al., 1998, Hsu et al., 2000). To test the hypothesis that miR-126 modulation of PI3K/AKT/MTOR signaling influences LSC function through CDK3, functional studies were undertaken. Intracellular flow cytometry of 8227 cells showed reduced CDK3 protein levels and pRB Ser807/811 levels upon 126OE (Figure 6D). CDK3/cyclin C phosphorylation of RB1 on Ser807/811 is required to induce cell cycle entry from a quiescent state (G0 exit) (Ren and Rollins, 2004) (Miyata et al., 2010) To verify that 126OE functions are dependent on CDK3 downregulation, lentiviruses expressing CDK3 or the CDK3 kinase mutant (CDK3mut) were generated (Figure S6G) (van den Heuvel and Harlow, 1993). Compared with CDK3mut, CDK3 significantly increased proliferation and clonogenicity of CD34+CD38– and CD34+CD38+ cells (Figures 6E and 6F), and partially reversed 126OE-induced expansion of CD34+ cells (Figures 6G and S6H). Collectively, these data suggest that miR-126 restricts LSC proliferation partly through targeting CDK3.
High miR-126 Bioactivity Endows LSC with Chemotherapy Resistance
To test whether the induction of LSC quiescence by 126OE is associated with chemotherapy resistance, 126OE or CTRL transduced 8227 cells were exposed to increasing concentrations of daunorubicin. 126OE increased the survival of CD34+ cells after 72 hr of treatment compared with CTRL (Figure 7A), an effect not seen in non-transduced cells (Figure S7A). Treatment of primary AML samples (Table S5) with daunorubicin plus cytarabine resulted in enrichment of primitive CD117+ cells (Figure S7B) and increased miR-126 levels in four of five samples (Figure 7B). Thus, primitive AML cells expressing the highest miR-126 levels are also the most resistant to anti-proliferative chemotherapy.
Figure 7.

High miR-126 Bioactivity Endows LSC with Chemotherapy Resistance
(A) Graphical representation of percent viable CD34+ 8227 cells with increasing doses of daunorubicin. CD34+ cell numbers were normalized to day 0 control transduced cells. Results are shown as the mean ± SEM of three biological replicate experiments; ∗∗∗∗p < 0.0001.
(B) Primary patient AML cells were plated onto MS5 stroma; after 24 hr cells were treated with vehicle or with daunorubicin (50 ng/ml)/AraC (500 ng/ml) for 72 hr. The miR-126 expression levels in daunorubicin/AraC-treated and control AML blasts were determined by qPCR. Results were normalized to RNU48 and are shown as the mean ± SD of four replicates; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(C) qPCR was performed on CD45dim sorted blasts from patient samples at diagnosis (n = 8, day 0) and at day 14 (n = 4) and day 30 (n = 5) after initiation of induction chemotherapy, as well as on day 30 after (unsuccessful) salvage chemotherapy (n = 3). Data shown are pooled from individual patients (see Figure S7C) and are shown as means ± SEM of combined individual patient samples. ∗p < 0.05.
(D and E) qPCR results of the relative levels of miR-126 in CD45dim (D, four AML patients, see Figure S7D) and CD45dimCD117+ (E, ten AML patients, see Figure S7E). AML blasts in paired diagnosis and relapse patient samples shown as the mean ± SEM of all patients combined; ∗p < 0.05.
(F) 8227 cells transduced with mutCDK3 and CDK3 lentiviruses were plated into a 96-well plate and treated with increasing doses of daunorubicin for 48 hr. Cells were stained for CD34 and live cells were identified by viability dye exclusion by flow cytometry. Results are shown as the mean ± SEM of four biological replicates; ∗p < 0.05 and ∗∗p < 0.01. See also Figure S7 and Table S4.
To determine if miR-126 expression could be linked to chemotherapy resistance in a clinical setting, biobanked samples were identified from eight AML patients who failed to achieve complete remission after induction therapy. CD45dim blasts were isolated from bone marrow at diagnosis (n = 8, day 0), day 14 (n = 4), and day 30 (n = 5) post-induction, and at day 30 after salvage chemotherapy (n = 3). In line with the in vitro findings, miR-126 expression was increased in six of eight samples (median, 3.4-fold; range, 0.3–9.4) after induction, and in two of three patients (including one in whom miR-126 expression was unchanged after induction) following salvage chemotherapy (median, 1.8-fold; range, 1.1–2.1) (Figures 7C and S7C). miR-126 expression was higher in relapse blasts compared with paired diagnostic samples in all four patients tested (Figures 7D and S7D). miR-126 expression in primitive CD45dimCD117+ cells was increased in eight of ten patients at relapse, with >100-fold enrichment in two patients (Figures 7E and S7E). Finally, enforced expression of CDK3 in 8227 cells rescued the 126OE effects by decreasing the proportion of CD34+ cells resistant to daunorubicin and cytarabine (Figure 7F). Overall, these data suggest that miR-126 confers resistance to chemotherapy, likely through the induction and maintenance of cellular quiescence by the targeting and repression of the PI3K/AKT/MTOR pathway.
Discussion
Our study establishes that miRNAs play a powerful role in governing the fundamental properties that define the stemness state of human LSC including quiescence, self-renewal, and chemotherapy response. miRNAs are differentially expressed within distinct cellular subsets that make up the AML hierarchy, with a restricted set expressed in an LSC-specific manner. The miRNA LSC signature was itself highly prognostic. This clinical association, together with the miR-126 functional data, establishes that miRNAs provide a layer of post-transcriptional control critical for maintaining the stemness state in AML. Although miR-126 governs the stemness and quiescence properties of both HSC and LSC, miR-126 perturbation results in divergent self-renewal outcomes. This discordance provides a novel avenue to therapeutically target LSC without attendant toxicity to HSC.
Our study provides a mechanistic link between quiescence control and the restraint of CDK3 expression by miR-126, thereby altering RB1 phosphorylation and delaying G0 exit in human primitive AML populations. Regulation of G0 exit kinetics is a fundamental HSC property, distinct from downstream progenitors, that is essential for maintaining HSC pool integrity (Laurenti et al., 2015, Nygren et al., 2006). CDK3 is poorly studied since all inbred mice carry a nonsense mutation in CDK3 (Ye et al., 2001). In quiescent human fibroblasts, CDK3 can complex with CCNC (cyclin C) and phosphorylate Rb1 (on residues S807 and S811) to directly initiate the cell cycle; when CDK3 levels are reduced, a 12-hr lag in G0 exit kinetics is induced but not a permanent block (Ren and Rollins, 2004). In murine LT-HSC, CCNC levels are highest during G0 exit (Passegué et al., 2005) and CCNC knockdown in human HSC increased quiescence, promoted HSPC expansion, and increased repopulation capacity (Miyata et al., 2010). In leukemia, CCNC deletion highly correlates with relapse (van Delft et al., 2011). Thus, it is likely that the miR-126/CDK3 regulatory axis also governs G0 exit kinetics in LSC, thereby providing new therapeutic opportunities for targeting quiescence control of LSC.
A model derived from our proteomic and transcriptomic data (Figure 8) depicts that upstream of CDK3, miR-126 represses multiple inputs converging on PI3K/AKT/MTOR signaling in LSC, paralleling miR-126 function in HSC (Lechman et al., 2012). Preclinical evidence indicates that activated PI3K/AKT/MTOR signaling plays a role in AML (Martelli et al., 2010) despite being rarely mutated and likely driven by upstream activation (Fransecky et al., 2015). Although inhibitors of AKT, MTOR, and PI3K are in clinical development, they have mostly failed for AML (Fransecky et al., 2015). While failure is attributed to feedback loops, our study provides an explanation that is embedded in the hierarchical organization of AML. PI3K/AKT/MTOR signaling is restricted to cycling leukemic progenitors; by contrast, quiescent LSC, a reservoir of leukemic relapse, have lower signaling and would then be spared following inhibitor treatment. In support of this prediction, AKT inhibition increased the fraction of G0 breast cancer cells, linking low AKT signaling to a G0-like state (Dey-Guha et al., 2011). In AKT knockout mice, HSC persisted in an enhanced G0 quiescent state, while AKT activation results in HSC hyper-proliferation and exhaustion (Juntilla et al., 2010, Kharas et al., 2010). Collectively, these reports suggest that the state of AKT activity plays a key role in governing quiescence of normal HSC and our data extend this concept to leukemia by showing that this pathway is tightly controlled at multiple points by miRNAs in order to maintain the human LSC state.
Figure 8.

miR-126 Represses Multiple AKT Inputs in LSC
LSC express high endogenous levels of miR-126 compared with more differentiated AML populations. High levels of endogenous or experimental miR-126 repress the level of several proteins regulating AKT (PI3K signaling, PI3CD, PIK3R2; integrin signaling, ADAM9, ITGA6, ILK, PARVB; RTK signaling, CRK, ABI1, CD97, CD84; MTOR signaling, MAPKAP1), reducing overall AKT levels and activity. Furthermore, high levels of miR-126 reduce pPDK1 Ser241, which phosphorylates AKT, and MAPKAP1, which is required for MTORC2 formation and full activation of AKT. Significantly diminished levels of AKT activity preferentially retain LSC in a quiescent state by increasing p27 levels, together with miR-126 targeted reduction of CDK3. Under high miR-126 levels, LSC that do enter the cycle are biased toward a self-renewal division. Reduction of LSC miR-126 levels through currently unspecified developmental cues (or lentiviral sponge-mediated) de-represses the expression and activity of multiple AKT signaling inputs. LSC now preferentially cycle and are biased toward differentiation divisions.
Although cell cycle regulation by miR-126 is similar between HSC and LSC, the functional consequence is the opposite: reduced miR-126 levels expand HSC in vivo, but impair LSC maintenance (see the model in Figure 8). With the exception of PTEN, known regulators of self-renewal have similar functions in normal and leukemic contexts (Yilmaz and Morrison, 2008). PTEN is rarely mutated in AML, yet experimental deletion results in HSC loss and LSC expansion, supporting our data on functional HSC–LSC divergence. Concordant with our findings of low PI3K/AKT/MTOR signaling in dormant LSC, rapamycin treatment only eliminates LSC during the early phases of leukemic initiation in PTEN mouse models when LSC are proliferating, but not when leukemia is fully developed and when some LSC are predicted to re-enter quiescence (Yilmaz and Morrison, 2008).
While targeting stemness represents a promising clinical direction, finding a selective therapeutic window might be challenging due to the shared determinants of stemness between HSC and LSC, and the likelihood of causing excessive toxicity (Kreso and Dick, 2014). The distinct function of miR-126 in HSC and LSC provides an opportunity to clinically target LSC while sparing HSC. Moreover, inhibiting miR-126 might overcome LSC chemo-resistance through cycle activation and increasing sensitivity to anti-proliferative drugs. Although targeting miRNA in vivo is still inefficient (Brown and Naldini, 2009), LNA miRNA decoy technology is effective clinically in hepatitis C (Janssen et al., 2013). Alternatively, targeting the LSC-specific pathways identified by miR-126 might also be an effective strategy.
Experimental Procedures
Patient-Derived Xenografts
NOD/Lt-scid/IL2Rɣnull (NSG) mice were bred at the University Health Network/Princess Margaret Cancer Center. Animal experiments were performed in accordance with national and institutional guidelines approved by the Canadian Counsel on Animal Care and approved by the University Health Network Animal Care Committee. Mouse xenografts were performed as described previously (Lechman et al., 2012). Briefly, NSG mice were sublethally irradiated (225 cGy) 1 day prior to injection. AML patient samples were thawed and plated in X-VIVO/20% BIT (Stem Cell Technologies) supplemented with Flt3-L (50 ng/ml), IL-6 (10 ng/ml), stem cell factor (50 ng/ml), thrombopoietin (125 ng/ml), IL-3 (10 ng/ml), granulocyte colony-stimulating factor (10 ng/ml) for 18 hr (Blair et al., 1998). Cells were transduced in 24-well culture plates at a multiplicity of infection of 30 with sensor lentivectors or for enforced expression and knockdown of miR-126. Transduced AML cells (5 × 105–1 × 106) were injected with 25 μl of PBS into the right femur of each recipient mouse. After 10–12 weeks, the mice were euthanized and bone marrow cells were flushed with 2 ml of PBS, 2% fetal calf serum, and 50 μl of cells were stained for surface markers.
Patient Samples and Treatment Protocols
Between 2003 and 2010, peripheral blood and bone marrow samples were collected from subjects with AML after obtaining informed consent according to procedures approved by the Research Ethics Board of the University Health Network (REB# 01-0573-C). Mononuclear cells were isolated and stored as previously described (Eppert et al., 2011). Cytogenetics were analyzed according to the revised MRC prognostic classification system (Grimwade et al., 2010). NPM1 and FLT3-ITD mutations were assessed as previously described (How et al., 2012).
The 74 patient samples used to optimize the miRNA prognostic signature (PMCC cohort) were diagnostic samples from individuals with de novo AML and normal cytogenetics. Although patients were not treated uniformly, all initially received induction chemotherapy followed by two cycles of consolidation in those who achieved complete remission (CR). First-line induction regimens included 3 + 7 (n = 69), NOVE-HIDAC (n = 1), and four patients were enrolled in clinical trials employing a 3 + 7 backbone with gemtuzumab ozogamicin (n = 2) or tipifarnib (n = 2). Treatment protocols were as previously described (Brandwein et al., 2008, Brandwein et al., 2009, How et al., 2012, Petersdorf et al., 2013). Allogeneic stem cell transplant (allo-SCT) was performed for high-risk patients in CR1 (n = 7), as well as for patients who achieved a second remission after relapse (n = 12) if they had an available donor, were younger than 70 years, lacked significant comorbidities, and had good performance status. Bio-informatic and clinical information for a second cohort of 187 de novo AML patients was obtained from TCGA and has been previously described (Cancer Genome Atlas Research Network, 2013).
See Supplemental Experimental Procedures for additional methods.
Author Contributions
Project Conceptualization, E.R.L, B.G., J.E.D., L.N, K.E., M.M., and J.C.Y.W.; Methodology, E.R.L. and K.E.; Investigation, E.R.L., E.M.S., P.V.G., N.T., S.M.D., A.T.G., G.K., J.E., A.M., W.C.C., K.G.H., K.E., R.M., B.L.E., J.L., and S.N.; Resources, M.M.; Data Curation, J.K.; Writing-Review and Editing, E.R.L. J.C.Y.W., and J.E.D.; Supervision, J.E.D., L.N., G.D.B., P.Z., and T.G.; Formal Analysis, S.W.K.N., J.K., B.N., R.I., V.V.; Visualization, K.K.; Funding Acquisition, T.G., J.E.D., and L.N.
Acknowledgments
We thank Dr. M Roehrl for mass spectrometer support, A Khandani and P. A. Penttilä for flow cytometry, and the Dick and Naldini laboratories for critical review. This work was supported by grants to L.N. from Telethon (TIGET grant), EU (FP7 GA 222878 PERSIST, ERC Advanced Grant 249845 TARGETING GENE THERAPY), and the Italian Ministry of Health and to J.E.D. from the Canadian Institutes for Health Research, Canadian Cancer Society, Terry Fox Foundation, Genome Canada through the Ontario Genomics Institute, Ontario Institute for Cancer Research with funds from the Province of Ontario, and a Canada Research Chair. E.M.S. is an EMBO Postdoctoral Fellow (ALTF 1595–2014) and is co-funded by the European Commission (LTFCOFUND2013, GA-2013-609409) and Marie Curie Actions. This research was funded in part by the Ontario Ministry of Health and Long Term Care (OMOHLTC). The views expressed do not necessarily reflect those of the OMOHLTC.
Published: January 28, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2015.12.011.
Accession Numbers
miRNA array, Illumina array, Nanostring data have been submitted to Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) with the following series accession numbers: miRNA, GEO: GSE55917; Illumina, GEO: GSE55814; and Nanostring, GEO: GSE55770. The MS data have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PRIDE: PXD001994.
Supplemental Information
References
- Birle D., Bottini N., Williams S., Huynh H., deBelle I., Adamson E., Mustelin T. Negative feedback regulation of the tumor suppressor PTEN by phosphoinositide-induced serine phosphorylation. J. Immunol. 2002;1950:286–291. doi: 10.4049/jimmunol.169.1.286. [DOI] [PubMed] [Google Scholar]
- Blair A., Hogge D.E., Sutherland H.J. Most acute myeloid leukemia progenitor cells with long-term proliferative ability in vitro and in vivo have the phenotype CD34+/CD71−/HLA-DR−. Blood. 1998;92:4325–4335. [PubMed] [Google Scholar]
- Brandwein J.M., Gupta V., Schuh A.C., Schimmer A.D., Yee K., Xu W., Messner H.A., Lipton J.H., Minden M.D. Predictors of response to reinduction chemotherapy for patients with acute myeloid leukemia who do not achieve complete remission with frontline induction chemotherapy. Am. J. Hematol. 2008;83:54–58. doi: 10.1002/ajh.21034. [DOI] [PubMed] [Google Scholar]
- Brandwein J.M., Leber B.F., Howson-Jan K., Schimmer A.D., Schuh A.C., Gupta V., Yee K.W.L., Wright J., Moore M., MacAlpine K. A phase I study of tipifarnib combined with conventional induction and consolidation therapy for previously untreated patients with acute myeloid leukemia aged 60 years and over. Leukemia. 2009;23:631–634. doi: 10.1038/leu.2008.341. [DOI] [PubMed] [Google Scholar]
- Braun K., Hölzl G., Soucek T., Geisen C., Möröy T., Hengstschläger M. Investigation of the cell cycle regulation of cdk3-associated kinase activity and the role of cdk3 in proliferation and transformation. Oncogene. 1998;17:2259–2269. doi: 10.1038/sj.onc.1202145. [DOI] [PubMed] [Google Scholar]
- Brown B.D., Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 2009;10:578–585. doi: 10.1038/nrg2628. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M., Medema R.H., Garcia-Cao I., Dubuisson M.L., Barradas M., Glassford J., Rivas C., Burgering B.M., Serrano M., Lam E.W. Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J. Biol. Chem. 2000;275:21960–21968. doi: 10.1074/jbc.M000759200. [DOI] [PubMed] [Google Scholar]
- Dey-Guha I., Wolfer A., Yeh A.C., G Albeck J., Darp R., Leon E., Wulfkuhle J., Petricoin E.F., Wittner B.S., Ramaswamy S. Asymmetric cancer cell division regulated by AKT. Proc. Natl. Acad. Sci. USA. 2011;108:12845–12850. doi: 10.1073/pnas.1109632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrance A.M., Neviani P., Ferenchak G.J., Huang X., Nicolet D., Maharry K.S., Ozer H.G., Hoellarbauer P., Khalife J., Hill E.B. Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia. Leukemia. 2015;29:2143–2153. doi: 10.1038/leu.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw D.C., Denkers F., Olthof M., Rutten A., Pouwels W., Schuurhuis G.J., Ossenkoppele G., Smit L. Attenuation of microRNA-126 expression that drives CD34+38- stem/progenitor cells in acute myeloid leukemia leads to tumor eradication. Cancer Res. 2014;74:2094–2105. doi: 10.1158/0008-5472.CAN-13-1733. [DOI] [PubMed] [Google Scholar]
- Ebert M.S., Sharp P.A. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149:515–524. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppert K., Takenaka K., Lechman E.R., Waldron L., Nilsson B., van Galen P., Metzeler K.H., Poeppl A., Ling V., Beyene J. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- Fransecky L., Mochmann L.H., Baldus C.D. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol. Cell. Ther. 2015;3:2. doi: 10.1186/s40591-015-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R., Volinia S., Liu C.-G., Fernandez-Cymering C., Palumbo T., Pichiorri F., Fabbri M., Coombes K., Alder H., Nakamura T. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008;111:3183–3189. doi: 10.1182/blood-2007-07-098749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner B., Visigalli I., Hiramatsu H., Lechman E., Ungari S., Giustacchini A., Schira G., Amendola M., Quattrini A., Martino S. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci. Transl. Med. 2010;2:58ra84. doi: 10.1126/scitranslmed.3001522. [DOI] [PubMed] [Google Scholar]
- Greaves M. Cancer stem cells renew their impact. Nat. Med. 2011;17:1046–1048. doi: 10.1038/nm.2458. [DOI] [PubMed] [Google Scholar]
- Grimwade D., Hills R.K., Moorman A.V., Walker H., Chatters S., Goldstone A.H., Wheatley K., Harrison C.J., Burnett A.K., National Cancer Research Institute Adult Leukaemia Working Group Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–365. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- Guo S., Lu J., Schlanger R., Zhang H., Wang J.Y., Fox M.C., Purton L.E., Fleming H.H., Cobb B., Merkenschlager M. MicroRNA miR-125a controls hematopoietic stem cell number. Proc. Natl. Acad. Sci. USA. 2010;107:14229–14234. doi: 10.1073/pnas.0913574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada S., Satoh K., Fujibuchi W., Hirota M., Kanno A., Unno J., Masamune A., Kikuta K., Kume K., Shimosegawa T. MiR-126 acts as a tumor suppressor in pancreatic cancer cells via the regulation of ADAM9. Mol. Cancer Res. 2012;10:3–10. doi: 10.1158/1541-7786.MCR-11-0272. [DOI] [PubMed] [Google Scholar]
- Han Y.-C., Park C.Y., Bhagat G., Zhang J., Wang Y., Fan J.-B., Liu M., Zou Y., Weissman I.L., Gu H. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J. Exp. Med. 2010;207:475–489. doi: 10.1084/jem.20090831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz M., Forman S.J., Bhatia R. Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer Res. 2007;67:1113–1120. doi: 10.1158/0008-5472.CAN-06-2014. [DOI] [PubMed] [Google Scholar]
- How J., Sykes J., Gupta V., Yee K.W.L., Schimmer A.D., Schuh A.C., Minden M.D., Kamel-Reid S., Brandwein J.M. Influence of FLT3-internal tandem duplication allele burden and white blood cell count on the outcome in patients with intermediate-risk karyotype acute myeloid leukemia. Cancer. 2012;118:6110–6117. doi: 10.1002/cncr.27683. [DOI] [PubMed] [Google Scholar]
- Hsu S.L., Hsu J.W., Liu M.C., Chen L.Y., Chang C.D. Retinoic acid-mediated G1 arrest is associated with induction of p27(Kip1) and inhibition of cyclin-dependent kinase 3 in human lung squamous carcinoma CH27 cells. Exp. Cell Res. 2000;258:322–331. doi: 10.1006/excr.2000.4933. [DOI] [PubMed] [Google Scholar]
- Hu W., Dooley J., Chung S.S., Chandramohan D., Cimmino L., Mukherjee S., Mason C.E., de Strooper B., Liston A., Park C.Y. miR-29a maintains mouse hematopoietic stem cell self-renewal by regulating Dnmt3a. Blood. 2015;125:2206–2216. doi: 10.1182/blood-2014-06-585273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen H.L.A., Reesink H.W., Lawitz E.J., Zeuzem S., Rodriguez-Torres M., Patel K., van der Meer A.J., Patick A.K., Chen A., Zhou Y. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- Juntilla M.M., Patil V.D., Calamito M., Joshi R.P., Birnbaum M.J., Koretzky G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115:4030–4038. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharas M.G., Okabe R., Ganis J.J., Gozo M., Khandan T., Paktinat M., Gilliland D.G., Gritsman K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115:1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A., Dick J.E. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Laurenti E., Frelin C., Xie S., Ferrari R., Dunant C.F., Zandi S., Neumann A., Plumb I., Doulatov S., Chen J. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. 2015;16:302–313. doi: 10.1016/j.stem.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechman E.R., Gentner B., van Galen P., Giustacchini A., Saini M., Boccalatte F.E., Hiramatsu H., Restuccia U., Bachi A., Voisin V. Attenuation of miR-126 activity expands HSC in vivo without exhaustion. Cell Stem Cell. 2012;11:799–811. doi: 10.1016/j.stem.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Lu J., Sun M., Mi S., Zhang H., Luo R.T., Chen P., Wang Y., Yan M., Qian Z. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.-K., Wang G., Chen Z., Teruya-Feldstein J., Liu Y., Chan C.-H., Yang W.-L., Erdjument-Bromage H., Nakayama K.I., Nimer S. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat. Cell Biol. 2009;11:420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Lin Y.Z., LaPushin R., Cuevas B., Fang X., Yu S.X., Davies M.A., Khan H., Furui T., Mao M. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- Marcucci G., Radmacher M.D., Mrózek K., Bloomfield C.D. MicroRNA expression in acute myeloid leukemia. Curr. Hematol. Malig. Rep. 2009;4:83–88. doi: 10.1007/s11899-009-0012-7. [DOI] [PubMed] [Google Scholar]
- Martelli A.M., Evangelisti C., Chiarini F., McCubrey J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1:89–103. doi: 10.18632/oncotarget.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A., Zhao J.L., Sinha N., Marinov G.K., Mann M., Kowalczyk M.S., Galimidi R.P., Du X., Erikci E., Regev A. The Microrna-132 and MicroRNA-212 cluster regulates hematopoietic stem cell maintenance and survival with age by buffering FOXO3 expression. Immunity. 2015;42:1021–1032. doi: 10.1016/j.immuni.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler K.H., Maharry K., Kohlschmidt J., Volinia S., Mrózek K., Becker H., Nicolet D., Whitman S.P., Mendler J.H., Schwind S. A stem cell-like gene expression signature associates with inferior outcomes and a distinct microRNA expression profile in adults with primary cytogenetically normal acute myeloid leukemia. Leukemia. 2013;27:2023–2031. doi: 10.1038/leu.2013.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y., Liu Y., Jankovic V., Sashida G., Lee J.M., Shieh J.-H., Naoe T., Moore M., Nimer S.D. Cyclin C regulates human hematopoietic stem/progenitor cell quiescence. Stem Cells. 2010;28:308–317. doi: 10.1002/stem.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren J.M., Bryder D., Jacobsen S.E.W. Prolonged cell cycle transit is a defining and developmentally conserved hemopoietic stem cell property. J. Immunol. 2006;177:201–208. doi: 10.4049/jimmunol.177.1.201. [DOI] [PubMed] [Google Scholar]
- Oglesby I.K., Bray I.M., Chotirmall S.H., Stallings R.L., O’Neill S.J., McElvaney N.G., Greene C.M. miR-126 is downregulated in cystic fibrosis airway epithelial cells and regulates TOM1 expression. J. Immunol. 2010;184:1702–1709. doi: 10.4049/jimmunol.0902669. [DOI] [PubMed] [Google Scholar]
- Ooi A.G.L., Sahoo D., Adorno M., Wang Y., Weissman I.L., Park C.Y. MicroRNA-125b expands hematopoietic stem cells and enriches for the lymphoid-balanced and lymphoid-biased subsets. Proc. Natl. Acad. Sci. USA. 2010;107:21505–21510. doi: 10.1073/pnas.1016218107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell R.M., Chaudhuri A.A., Rao D.S., Gibson W.S.J., Balazs A.B., Baltimore D. MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc. Natl. Acad. Sci. USA. 2010;107:14235–14240. doi: 10.1073/pnas.1009798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegué E., Wagers A.J., Giuriato S., Anderson W.C., Weissman I.L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersdorf S.H., Kopecky K.J., Slovak M., Willman C., Nevill T., Brandwein J., Larson R.A., Erba H.P., Stiff P.J., Stuart R.K. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121:4854–4860. doi: 10.1182/blood-2013-01-466706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S., Rollins B.J. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117:239–251. doi: 10.1016/s0092-8674(04)00300-9. [DOI] [PubMed] [Google Scholar]
- Rodgers J.T., King K.Y., Brett J.O., Cromie M.J., Charville G.W., Maguire K.K., Brunson C., Mastey N., Liu L., Tsai C.-R. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T., Flörcken A., Singh A., Türkmen S., Burmeister T., Anagnostopoulos I., Pezzutto A., Dörken B., Westermann J. Flow cytometric maturity score as a novel prognostic parameter in patients with acute myeloid leukemia. Ann. Hematol. 2015;94:1337–1345. doi: 10.1007/s00277-015-2400-5. [DOI] [PubMed] [Google Scholar]
- Song S.J., Ito K., Ala U., Kats L., Webster K., Sun S.M., Jongen-Lavrencic M., Manova-Todorova K., Teruya-Feldstein J., Avigan D.E. The oncogenic MicroRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013;13:87–101. doi: 10.1016/j.stem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A., Essers M., Wilson A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010;10:201–209. doi: 10.1038/nri2726. [DOI] [PubMed] [Google Scholar]
- Undi R.B., Kandi R., Gutti R.K. MicroRNAs as haematopoiesis regulators. Adv. Hematol. 2013;2013:1–20. doi: 10.1155/2013/695754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft F.W., Horsley S., Colman S., Anderson K., Bateman C., Kempski H., Zuna J., Eckert C., Saha V., Kearney L. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood. 2011;117:6247–6254. doi: 10.1182/blood-2010-10-314674. [DOI] [PubMed] [Google Scholar]
- van den Heuvel S., Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- Velu C.S., Chaubey A., Phelan J.D., Horman S.R., Wunderlich M., Guzman M.L., Jegga A.G., Zeleznik-Le N.J., Chen J., Mulloy J.C. Therapeutic antagonists of microRNAs deplete leukemia-initiating cell activity. J. Clin. Invest. 2014;124:222–236. doi: 10.1172/JCI66005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P., Somervaille T.C., Venkatasubrahmanyam S., Kogan S., McLaughlin M.E., Weissman I.L., Butte A.J., Passegué E., Sage J. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell. 2008;3:416–428. doi: 10.1016/j.stem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P., Iwasaki M., Somervaille T.C.P., Ficara F., Carico C., Arnold C., Chen C.-Z., Cleary M.L. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res. 2010;70:3833–3842. doi: 10.1158/0008-5472.CAN-09-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Inoki K., Ikenoue T., Guan K.-L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X., Zhu C., Harper J.W. A premature-termination mutation in the Mus musculus cyclin-dependent kinase 3 gene. Proc. Natl. Acad. Sci. USA. 2001;98:1682–1686. doi: 10.1073/pnas.041596198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O.H., Morrison S.J. The PI-3kinase pathway in hematopoietic stem cells and leukemia-initiating cells: a mechanistic difference between normal and cancer stem cells. Blood Cells. Mol. Dis. 2008;41:73–76. doi: 10.1016/j.bcmd.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.