

Abstract
Glucocorticoid hormones, via activation of their receptors, promote memory consolidation, but the exact underlying mechanisms remain elusive. We examined how corticosterone regulates AMPA receptors (AMPARs), which are crucial for synaptic plasticity and memory formation. Combining a live imaging fluorescent recovery after photobleaching approach with the use of the pH-sensitive GFP-AMPAR tagging revealed that corticosterone enhances the AMPAR mobile fraction and increases synaptic trapping of AMPARs in hippocampal cells. In parallel, corticosterone-enhanced AMPAR-mediated synaptic transmission. Blocking the mammalian target of rapamycin (mTOR) pathway prevented the effects of corticosterone on both AMPAR trapping—but not on the mobile fraction—and synaptic transmission. Blocking the mTOR pathway also prevented the memory enhancing effects of corticosterone in a contextual fear-conditioning paradigm. We conclude that activation of the mTOR pathway is essential for the effects of corticosterone on synaptic trapping of AMPARs and, possibly as a consequence, fearful memory formation.
Enhanced memory formation of emotionally arousing and stressful situations favors long-term behavioral adaptation to such conditions (De Kloet et al. 1999). Consolidation of emotionally arousing information is facilitated by corticosteroid hormones, which are released during and after exposure to stressful situations (Oitzl et al. 2001; Roozendaal et al. 2009). An important question is exactly how these hormones facilitate memory consolidation. Corticosterone, via activation of mineralocorticoid receptors and glucocorticoid receptors (GR), regulates AMPA receptor (AMPAR) function (Karst and Joëls 2005; Groc et al. 2008; Martin et al. 2009; Krugers et al. 2010), a critical end point for memory formation (Kessels and Malinow 2009; Mitsushima et al. 2011).
The intracellular mediators between steroid receptor activation and AMPAR function have not yet been resolved. One potential candidate is mammalian target of rapamycin (mTOR), a serine/threonine kinase critically involved in synaptic plasticity and memory formation (Tang et al. 2002; Glover et al. 2010) that controls initiation of protein translation through phosphorylation of several signaling targets including the p70-kDa ribosomal S6 kinase (p70S6K) and the eukaryotic initiation factor 4E-binding protein 1 (4EBP1). Activation of the mTOR pathway has also been implicated in the effects of stressful events and corticosteroid hormones on synaptic plasticity since stress exposure and GR activation suppress synaptic plasticity via activation of the mTOR pathway (Yang et al. 2008). These studies suggest that stress and GR activation, via activation of mTOR, enhance synaptic transmission and prevent subsequent synaptic plasticity, a mechanism to preserve stress-related information (Krugers et al. 2010). We tested the hypothesis that corticosterone action requires the mTOR signaling pathway to regulate AMPAR surface mobility, AMPAR function, and consequently memory formation.
Results
Imaging AMPA receptors
We first tested the involvement of the mTOR pathway on the surface expression of GluA1 and GluA2 AMPAR subunits in hippocampal cells. Corticosterone increased surface expression of both subunits, which was not affected by co-application of rapamycin (Fig. 1A–C). However, by combining a fluorescent recovery after photobleaching (FRAP) approach with the use of the pH-sensitive GFP-AMPAR tagging, we found that corticosterone alters the surface mobility of GluA2 containing AMPARs (Fig. 1D–I). More specifically, corticosterone increased the mobile fraction (Fig. 1E,G), the half time of fluorescence recovery T1/2 (Fig. 1E,H) and consequently, the diffusion coefficient of GluA2-containing AMPARs in dendritic spines is decreased (Fig. 1E,I). These effects could not be explained by altered surface diffusion since membrane-GFP diffusion remained unaffected by the corticosterone treatment (Fig. 2) indicating that the stress hormone selectively facilitates the mobility of GluA2, and promotes the synaptic trapping of AMPARs. Corticosterone effects on the mobile fraction were not affected by the mTOR antagonist rapamycin (Fig. 1F–H), but rapamycin incubation completely prevented the effect of corticosterone on the T1/2 and AMPAR diffusion coefficient (Fig. 1H, I).
Figure 1.

mTOR signaling is involved in the regulation of plasma membrane AMPAR lateral diffusion of corticosterone-treated rat hippocampal neurons. (A) Representative images of rat hippocampal neurons with labeling of GluA1 (in green) and GluA2 (in red) AMPAR subunits after treatment with vehicle (veh), corticosterone (cort, 100 nM), rapamycin (rapa, 50 nM), and rapamycin + corticosterone (rapa + cort). (B,C) Histograms showing the mean (±SEM) quantification of surface GluA1 (B) or GluA2 (C) AMPAR subunits. Data are expressed as ratio of control (vehicle condition). (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, one-way ANOVA, n > 10 cells in each group. (D) Sequential images from representative FRAP experiments performed on surface SEP-GluA2 from individual spine head (arrowheads) in control vehicle (veh) or in corticosterone (Cort, 100 nM) conditions, in the absence or presence of the potent mTOR inhibitor rapamycin (Rapa, 50 nM). (E) Normalized pooled and averaged FRAP curves from vehicle (n = 13 cells) and corticosterone (100 nM; n = 15 cells) treated hippocampal neurons. (F). Normalized pooled and averaged FRAP curves from rapamycin (50 nM; n = 13 cells) and rapamycin + corticosterone (Rapa, 50 nM, Cort, 100 nM; n = 16 cells) treated hippocampal neurons. (G–I). Histograms showing the means (±SEM) of synaptic SEP-GluA2 mobile fractions (G), the half time of fluorescence recovery (H), and the diffusion coefficient (I). One-way ANOVA was performed with a Bonferroni post-test for multiple comparison data sets. (*) P < 0.05, (**) P < 0.01.
Figure 2.

Blocking the mTOR signaling pathway does not impact the synaptic diffusion of membrane GFP. (A) Sequential confocal images of SEP-GluA2 in living rat hippocampal neurons. Bright SEP-GluA2 fluorescence is mainly due to surface expressed receptors and fluorescence is rapidly lost in pH 6.0 external solution. The fluorescence associated with SEP-GluA2 is almost totally abolished at low pH. (B) Representative trace showing the dynamic SEP-GluA2 fluorescence changes upon pH treatment described in A. (C) Sequential images from representative FRAP experiments performed on palmitoylated mGFP from individual spine head (arrowheads) in control vehicle (Ctrl; n = 28 cells) or in corticosterone (Cort, 100 nM; n = 28 cells) conditions, in the absence (Rapa, n = 22 cells) or in the presence of the potent mTOR inhibitor rapamycin (Rapa, 50 nM; n = 25 cells). (D–F) Histograms showing the means (±SEM) of synaptic mGFP mobile fractions (D) the half time of fluorescence recovery (E) and the diffusion coefficients (F) under the various conditions tested in (C). One-way ANOVA was performed with a Bonferroni post-test for multiple comparison data sets. (ns) Not significantly different.
Electrophysiology
We next examined the role of the mTOR pathway in hippocampal AMPAR function. Corticosterone increased the amplitude of miniature excitatory postsynaptic currents (mEPSCs) 3 h but not 1 h after treatment (Fig. 3A,B). These effects were long lasting, since the increase in amplitude of mEPSCs was still present when recorded 21 h after washing out of corticosterone (Fig. 3A,B). Both the GR-antagonist RU486 and the protein synthesis inhibitor cycloheximide (Fig. 3D,E) prevented the effects of corticosterone on the amplitude of mEPSCs, indicating that corticosterone-induced changes in AMPAR function start through genomic GR actions. The increase in the amplitude of mEPSCs in corticosterone was also fully blocked when rapamycin was co-incubated with corticosterone (Fig. 3G,H), pointing to mTOR as a necessary intracellular mediator of GR-dependent signaling. Frequencies of mEPSCs were not significantly altered by the pharmacological treatments (Fig. 3C,F,I).
Figure 3.

Corticosterone regulates AMPAR function via the mTOR pathway. (A) Representative traces of mEPSCs at 1, 3, or 24 h after vehicle (veh) or corticosterone (Cort, 100 nM) treatment on rat hippocampal neurons. (B). Amplitude of mEPSCs at 1, 3, or 24 h after vehicle (veh) or corticosterone (Cort, 100 nM) treatment. (*) P < 0.05, unpaired t-test, n > 10 in each group. (C) Frequency of mEPSCs at 1, 3, or 24 h after vehicle (veh) or corticosterone (Cort, 100 nM) treatment. (D) Representative traces of mEPSCs after treatment with corticosterone (Cort) and coapplication with vehicle (veh), the GR-antagonist RU486 (500 nM), and translation inhibitor cycloheximide (CX, 100 µM) for 3 h. (E) Amplitude of mEPSCs after treatment with corticosterone (Cort) and coapplication with vehicle (veh), the GR-antagonist RU486 (500 nM), and translation inhibitor cycloheximide (CX, 100 µM). Each condition (+control) was tested in a separate experimental series. (*) P < 0.05, unpaired t-test, n = 12 cells in each group. (F) Frequency of mEPSCs after treatment with corticosterone (Cort) and coapplication with vehicle (veh), the GR-antagonist RU486 (500 nM), and translation inhibitor cycloheximide (CX, 100 µM). (G) Traces of mEPSCs after treatment with vehicle (veh), corticosterone (Cort, 100 nM), rapamycin (rapa, 50 nM), or coapplication of rapamycin and corticosterone (rapa + cort). (H) Amplitude of mEPSCs after corticosterone treatment (Cort, 100 nM) and coapplication of vehicle (veh) or rapamycin (rapa, 50 nM). (*) P < 0.05, one-way ANOVA, veh (n = 8), cort (n = 7), rapa (n = 8), rapa + cort (n = 8). (I) Frequency of mEPSCs after treatment with vehicle (veh), corticosterone (Cort, 100 nM), rapamycin (rapa, 50 nM), or co-application of rapamycin and corticosterone (rapa + cort).
Fear conditioning
Finally, we tested whether hippocampus-dependent memory-enhancing effects of corticosterone are mediated via the mTOR pathway. Application of corticosterone (2 mg/kg) immediately after training in a weak contextual fear-conditioning paradigm (0.2 mA) enhanced the expression of contextual fear 24 h after training (Fig. 4). Post-training application of rapamycin by itself did not affect freezing behavior at 24 h after training. However, post-training administration of rapamycin prevented the corticosterone-induced increase in contextual fear memory at 24 h after training (Fig. 4).
Figure 4.
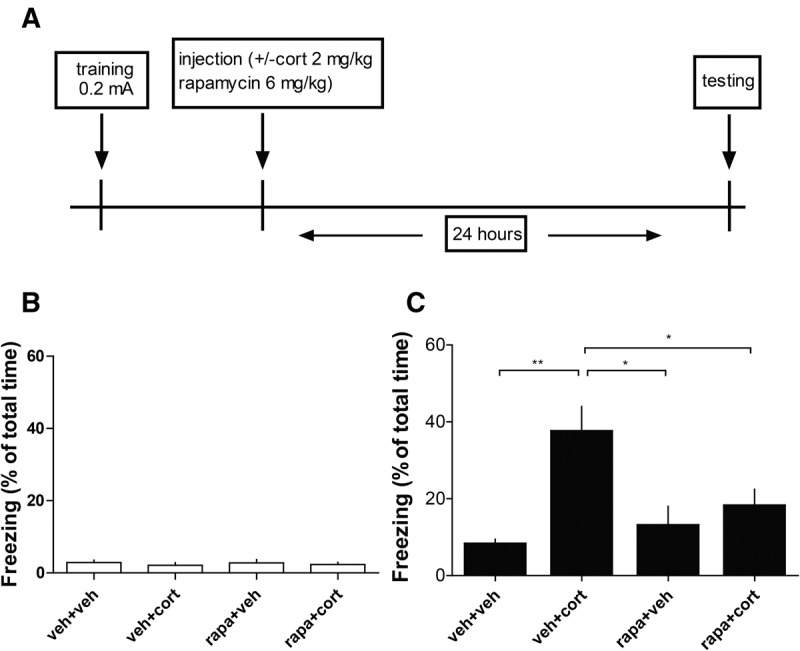
Corticosterone enhances memory consolidation via mTOR pathway. (A) Timeline of behavioral experiment. Animals received a footshock of 0.2 mA and were immediately injected with vehicle (veh), corticosterone (cort, 2 mg/kg), rapamycin (rapa, 6 mg/kg), or corticosterone (cort, 2 mg/kg) + rapamycin (rapa, 6 mg/kg intraperitoneally). Contextual fear was tested 24 h after training. (B) Freezing behavior (% of total time; mean ± SEM) of animals during free exploration, before footshock exposure and before being treated with vehicle (veh), corticosterone (cort, 2 mg/kg), or rapamycin (6 mg/kg) intraperitoneally immediately after training. (C) Freezing behavior (% of total time; mean ± SEM) of animals 24 h after being injected intraperitoneal immediately after training with vehicle (veh), corticosterone (cort, 2 mg/kg), rapamycin (6 mg/kg) intraperitoneally immediately after training. (*) P < 0.05, (**) P < 0.01. One-way ANOVA test, n = 7 animals (vehicle), seven animals (corticosterone), eight animals (rapamycin), eight animals (rapamycin + corticosterone).
Discussion
Various studies have shown that activation of GRs enhance hippocampus-dependent memory formation (Oitzl et al. 2001; Roozendaal et al. 2009; Zhou et al. 2010). Enhanced memory formation may involve BDNF-TrkB-MAPK-synapsin Ia/Ib signaling (Revest et al. 2005, 2010, 2014) and CaMKII-BDNF-CREB signaling (Chen et al. 2012). Yet, it remains to be determined how corticosteroid hormones regulate synaptic function, which is fundamental for memory formation (Rumpel et al. 2005; Kessels and Malinow 2009). We report that corticosterone via the mTOR pathway enhances synaptic retention of AMPARs, AMPAR function—which is a critical mechanism for memory formation (Kessels and Malinow 2009)—and improves contextual memory formation.
Corticosteroid hormones, via activation of GRs, have been reported to increase exocytosis of AMPARs (Yuen et al. 2011) and lateral diffusion of AMPARs (Groc et al. 2008). We report here that corticosterone not only enhances the mobile pool of AMPARs, but also enhances the synaptic retention of AMPARs and AMPAR-mediated synaptic function. This yields a picture that corticosterone acts on various pathways (exocytosis, lateral diffusion, and retention) to increase AMPAR function.
Previous studies have shown that serum- and glucocorticoid-inducible kinase and the activation of Rab4—which are involved in exocytotic processing—are involved in enhanced synaptic function by corticosteroid hormones (Yuen et al. 2011). Our results reveal that the corticosterone-induced increase in retention of AMPARs is regulated via the mTOR pathway. This effect is highly specific, since blocking the mTOR pathway did not prevent corticosterone effects on the mobile fraction of AMPARs. Exactly, how mTOR regulates synaptic retention and synaptic function of these receptors is not clear. Most likely, this effect involves translation of proteins, which regulate synaptic trapping of AMPARs. Importantly, these effects are highly relevant for behavior since preventing activation of the mTOR pathway prevented the effect of corticosterone on contextual memory consolidation. Taken together, a picture now emerges that corticosterone binds to GRs and increases AMPAR mobility via exocytosis (Yuen et al. 2011), lateral diffusion (Groc et al. 2008), but also by facilitating the synaptic retention of AMPARs via the mTOR pathway, contributing to enhanced memory consolidation.
Materials and Methods
Neuronal cultures
The experiments were carried out with permission of the local Animal Committee of the University of Amsterdam and the Centre National de la Recherche Scientifique, Institut de Pharmacologie Moléculaire et Cellulaire University of Nice, Sophia Antipolis. Primary hippocampal neurons were prepared from E18 pregnant Wistar rats as previously described (Loriol et al. 2013, 2014). Neurons were plated in Neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen), 0.5 mM glutamax, and penicillin/streptomycin on 12-mm glass coverslips precoated with 0.1 mg/mL poly-l-lysine. Neurons (40,000 cells per coverslip) were fed once a week for 3 wk in Neurobasal medium supplemented with 2% B27 and penicillin/streptomycin. For live-cell imaging, density of the cultures was 110,000 per 24-mm coverslip.
Immunocytochemistry
At DIV13-20 hippocampal neurons were incubated with GluR1 (Calbiochem (1:8) and GluR2 (Zymed (1:80) amino-terminal antibodies (10 µg/mL)) at 37°C for 15 min (Martin et al. 2008). Cells were preincubated at 37°C in 5% CO2 for 1 h in Neurobasal containing the potent mTOR inhibitor rapamycin (50 nM, Sigma) followed by corticosterone (100 nM, Sigma) or vehicle for 3 h in the presence of rapamycin. After washing in DMEM medium, the neurons were fixed for 5 min with 4% formaldehyde/4% sucrose in phosphate-buffered saline (PBS). Neurons were then washed three times in PBS for 30 min at room temperature and incubated with secondary antibody conjugated to Alexa488 (1:400) or Alexa568 (1:400) in staining buffer without TritonX-100 (0.2% BSA, 0.8 M NaCl, 30 mM phosphate buffer, pH 7.4) overnight at 4°C. Neurons were then washed three times in PBS for 30 min at room temperature and mounted. Confocal images were obtained with sequential acquisition settings at the maximal resolution of the microscope (1024 × 1024 pixels). Morphometric analysis and quantification were performed using MetaMorph software (Universal Imaging Corporation).
Live imaging
Neuronal transduction and transfection
Attenuated Sindbis virus expressing SEP-GluA2 was prepared and used as previously described (Martin et al. 2008, 2009). Neurons were transduced at a MOI of 1 between 18 and 20 DIV and incubated at 37°C under 5% CO2 for 24 h until use. For hippocampal neuron transfection, cells were incubated in a mix containing the Lipofectamin 2000 (Invitrogen) with 3 µg of plasmid DNA of palmitoylated membrane-anchored GFP (mGFP) and utilized 48 h post-transfection.
Imaging
Protocols were performed as previously described (Martin et al. 2008, 2009). Briefly, dendrites from live mGFP or SEP-GluA2 expressing neurons (19–21 DIV) were kept on a heated stage (set at 37°C) on a Nikon Ti inverted microscope and were continuously perfused at 1 mL/min with warm solution. GFP fluorescence was excited through a 100× oil-immersion lens (Numerical Aperture, 1.4) using a 488-nm laser light (50 mW, 1%–2%) and time series (1 image every 40 sec) were collected as a single image slice using a PerkinElmer Ultra-View spinning disk solution. For low pH external solution, equimolar MES (Sigma) was used instead of HEPES and pH adjusted to 6.0 and NH4Cl (50 mM) was used in place of equimolar NaCl to collapse pH gradient. All SEP-GluA2 experiments included a brief (10 sec) low pH wash at the beginning to ensure that the fluorescence from the area of interest comes from surface-expressed AMPARs.
Live SEP-GluA2-expressing neurons were preincubated at 37°C and 5% CO2 for 1 h in Neurobasal containing the potent mTOR inhibitor rapamycin (50 nM, Sigma) followed by corticosterone (100 nM, Sigma) or vehicle for 3 h in the presence of Rapamycin and finally live-imaged in Earle's buffer (50 mM HEPES–Tris pH 7.4, 140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, and 0.9 g/L glucose) containing the indicated drugs.
Electrophysiology
Coverslips were placed in a recording chamber mounted on an upright microscope (Nikon E600FN), continuously perfused with artificial cerebrospinal fluid (aCSF) (32°C, 2–3 mL/sec; containing in (mM): NaCl (120), KCl (3.5), MgSO4 (1.3), NaH2PO4 (1.25), CaCl2 (2.5), Glucose (10.0), and NaHCO3 (25.0), pH 7.4) and kept fully submerged. Whole-cell patch clamp recordings were made using an AXOPATCH 200B amplifier (Axon Instruments), with electrodes from borosilicate glass (1.5-mm outer diameter, Hilgerberg). The electrodes were pulled on a Sutter (USA) micropipette puller. The pipette solution contained (in mM): 120 Cs methane sulfonate; CsCl (17.5); HEPES (10); BAPTA (5); Mg-ATP (2); Na-GTP (0.5); QX-314 (10); pH 7.4, adjusted with CsOH; pipette resistance was between 3 and 6 MΩ. Under visual control (40× objective and 10× ocular magnification) the electrode was directed toward a neuron with positive pressure. Once sealed on the cell membrane (resistance ∼1 GΩ) the membrane patch under the electrode was ruptured by gentle suction and the cell was kept at a holding potential of −70 mV. The liquid junction potential caused a shift of no >10 mV, which was not compensated during mEPSCs recording. Recordings with an uncompensated series resistance of <15 MΩ and <2.5 times of the pipette resistance with a shift of <20% during the recording, were accepted for analysis. Data acquisition was performed with Pclamp 8.2 and analyzed off-line with Clampfit 9.0.
mEPSCs were recorded at a holding potential of −70 mV. Tetrodotoxin (0.25 µM, Latoxan) and bicuculline methobromide (20 µM, Biomol) were added to the buffer to block action potential-induced glutamate release and GABAA receptor-mediated miniature inhibitory postsynaptic currents (mIPSCs), respectively. During some recordings the non-NMDA-receptor blocker 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 µM, Tocris) was perfused to confirm that the mEPSCs were indeed mediated by AMPARs. The events were identified as mEPSCs when the rise time was faster than the decay time. mEPSCs were recorded for 5 min in each cell.
Corticosterone (100 nM, Sigma) or vehicle (<0.01% ethanol) was applied for 1 or 3 h. In one set of experiments, corticosterone was applied for 3 h, washed out and cultures were recorded 21 h after treatment. Cycloheximide (100 μM, Sigma), the GR antagonist RU486 (500 nM, Sigma), and rapamycin (50 nM, Sigma) were applied for 1 h before coapplication with corticosterone or vehicle.
Fear conditioning
Animals were housed individually 1 wk before the start of the experiment. Rats were trained in a fear-conditioning chamber (Context A; W × L × H: 30 × 24 × 26 cm) that contained a grid floor with 37 stainless steel rods and was connected to a shock generator and sound generator (Med-Farm LION-ELD) developed in-house. During training, one animal at a time was placed in the cage. After 3 min of free exploration, 1 footshock (2 sec, 0.2 mA; Cordero and Sandi 1998) was delivered and the animal was allowed to stay in the cage for 30 sec after the end of the footshock. Immediately after training, corticosterone (2 mg/kg, Sigma) or vehicle (ethanol, <0.01 %) was injected intraperitoneally (i.p.). At the same time rapamycin (6 mg/kg, Sigma) or DMSO (0.01%) was administered i.p. We have used a single dose of corticosterone in a range that mimics plasma corticosterone levels induced by stress and facilitates memory formation (Cordero and Sandi 1998; Miranda et al. 2008; Atsak et al. 2015). Then, the animals were transferred back into their home cage. Freezing behavior, defined as no body movements except those related to respiration, was determined every 2 sec throughout training (Zhou et al. 2010). Twenty-four hours later, one animal at a time was placed in context A for 3 min without receiving footshock and freezing behavior was scored.
Statistical analysis
Statistical analyses were calculated using Prism 4 (GraphPad software, Inc). Data are expressed as mean ± SEM Unpaired Student's t-tests and one-way ANOVA were performed with a Bonferroni post-test for multiple comparison data sets when required.
Acknowledgments
F.C. and S.M. designed and analyzed the live imaging experiments. F.C. performed the live imaging experiments. H.X., M.Z., and Y.Z. performed electrophysiological recordings. H.X. performed surface labeling and behavioral experiments. F.C., S.M., M.J., H.K., and H.X. wrote the manuscript. We thank Jeremy Henley (University of Bristol, UK) for the generous gift of pSinRep5 SEP-GluA2 and membrane-GFP plasmids. We gratefully acknowledge the “Agence Nationale de la Recherche” (ANR-2011-JSV4-003 1) and the French Government for the “Investments for the Future” LABEX “SIGNALIFE” (ANR-11-LABX-0028-01) to S.M. for financial support. We also thank the Royal Netherlands Academy for Arts and Sciences for support to H.X., M.Z., M.J., Z.X., and H.K. (Grants 05CDP013 and 11CDP017).
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.039420.115.
References
- Atsak P, Hauer D, Campolongo P, Schelling G, Fornari RV, Roozendaal B. 2015. Endocannabinoid signaling within the basolateral amygdala integrates multiple stress hormone effects on memory consolidation. Neuropsychopharmacology 40: 1485–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. 2012. Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci 15: 1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero MI, Sandi C. 1998. A role for brain glucocorticoid receptors in contextual fear conditioning: dependence upon training intensity. Brain Res 786: 11–17. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Oitzl MS, Joëls M. 1999. Stress and cognition: are corticosteroids good or Bad guys? Trends Neurosci 22: 422–426. [DOI] [PubMed] [Google Scholar]
- Glover EM, Ressler KJ, Davis M. 2010. Differing effects of systemically administered rapamycin on consolidation and reconsolidation of context vs. cued fear memories. Learn Mem 17: 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Choquet D, Chaouloff F. 2008. The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat Neurosci 11: 868–870. [DOI] [PubMed] [Google Scholar]
- Karst H, Joëls M. 2005. Corticosterone slowly enhances miniature excitatory postsynaptic current amplitude in mice CA1 hippocampal cells. J Neurophysiol 94: 3479–3486. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. 2009. Synaptic AMPA receptor plasticity and behavior. Neuron 61: 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugers HJ, Hoogenraad CC, Groc L. 2010. Stress hormones and AMPA receptor trafficking in synaptic plasticity and memory. Nat Rev Neurosci 11: 675–681. [DOI] [PubMed] [Google Scholar]
- Loriol C, Khayachi A, Poupon G, Gwizdek C, Martin S. 2013. Activity-dependent regulation of the sumoylation machinery in rat hippocampal neurons. Biol Cell 105: 30–45. [DOI] [PubMed] [Google Scholar]
- Loriol C, Cassé F, Khayachi A, Poupon G, Chafai M, Deval E, Gwizdek C, Martin S. 2014. mGlu5 receptors regulate synaptic sumoylation via a transient PKC-dependent diffusional trapping of Ubc9 into spines. Nat Commun 5: 5113. [DOI] [PubMed] [Google Scholar]
- Martin S, Bouschet T, Jenkins EL, Nishimune A, Henley JM. 2008. Bidirectional regulation of kainate receptor surface expression in hippocampal neurons. J Biol Chem 283: 36435–36440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Henley JM, Holman D, Zhou M, Wiegert O, van Spronsen M, Joëls M, Hoogenraad CC, Krugers HJ. 2009. Corticosterone increases AMPAR lateral diffusion and bidirectionally regulates synaptic AMPARs. PLoS One 4: e4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda MI, Quirarte GL, Rodriguez-Garcia G, McGaugh JL, Roozendaal B. 2008. Glucocorticoids enhance taste aversion memory via actions in the insular cortex and basolateral amygdala. Learn Mem 15: 468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsushima D, Ishihara K, Sano A, Kessels HW, Takahashi T. 2011. Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc Natl Acad Sci 108: 12503–12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oitzl MS, Reichardt HM, Joëls M, de Kloet ER. 2001. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc Natl Acad Sci 98: 12790–12795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revest JM, Di Blasi F, Kitchener P, Rougé-Pont F, Desmedt A, Turiault M, Tronche F, Piazza PV. 2005. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci 5: 664–672. [DOI] [PubMed] [Google Scholar]
- Revest JM, Kaouane N, Mondin M, Le Roux A, Rougé-Pont F, Vallée M, Barik J, Tronche F, Desmedt A, Piazza PV. 2010. The enhancement of stress-related memory by glucocorticoids depends on synapsin-Ia/Ib. Mol Psychiatry 12: 1140–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revest JM, Le Roux A, Roullot-Lacarrière V, Kaouane N, Vallée M, Kasanetz F, Rougé-Pont F, Tronche F, Desmedt A, Piazza PV. 2014. BDNF-TrkB signaling through Erk1/2MAPK phosphorylation mediates the enhancement of fear memory induced by glucocorticoids. Mol Psychiatry 19: 1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S. 2009. Stress, memory and the amygdala. Nat Rev Neurosci 10: 423–433. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. 2005. Postsynaptic receptor trafficking underlying a form of associative learning. Science 308: 83–88. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. 2002. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci 99: 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P-C, Yang C-H, Huang C-C, Hsu KS. 2008. Phosphatidylinositol 3-kinase activation is required for stress protocol-induced modification of hippocampal synaptic plasticity. J Biol Chem 283: 2631–2643. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. 2011. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry 16: 156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Bakker EH, Velzing EH, Berger S, Oitzl M, Joëls M, Krugers HJ. 2010. Both mineralocorticoid and glucocorticoid receptors regulate emotional memory in mice. Neurobiol Learn Mem 94: 530–537. [DOI] [PubMed] [Google Scholar]