

Abstract
Emerging resistance to existing antibiotics is an inevitable matter of concern in the treatment of bacterial infection. Naturally occurring unique class of natural antibiotic, platensimycin, a secondary metabolite from Streptomyces platensis, is an excellent breakthrough in recent antibiotic research with unique structural pattern and significant antibacterial activity. β-Ketoacyl-(acyl-carrier-protein (ACP)) synthase (FabF) whose Gram-positive bacteria need to biosynthesize cell membranes is the target of inhibition of platensimycin. So, isolation, retrosynthetic analysis, synthesis of platensimycin, and analogues of platensimycin synthesized till today are the objectives of this review which may be helpful to further investigate and to reveal untouched area on this molecule and to obtain a potential antibacterial lead with enhanced significant antibacterial activity.
1. Introduction
The majority of the “illness” lies in the fact that when immune system is defeated, it is in war with pathogens. Development starting from ethnic to modern synthetic approaches in drug discovery mainly provides better weapons to combat and survive against pathogens. In contrast to medical development, pathogens also have acquired protection called “resistance.” More or less all classes of antibiotics are resistant to bacteria; hence a novel process to the discovery of antibiotic with new mechanism of action is essential [1, 2]. Microbial and chemical groups at Merck in the year 2006 have found three novel chemical classes, using antisense technology, from their older microbial screening library [3]. Two compounds, platensimycin and platencin, were found as potent inhibitors of fatty acid biosynthesis. Platensimycin selectively inhibits fatty acid acyl carrier protein synthase II (FabF). Platensin is a balanced dual inhibitor of both FabF and FabH (fatty acid acyl carrier protein synthase III) [4]. The third compound lucensimycin A was found to inhibit ribosomal protein synthesis. FabF is one of the enzymes which catalyze the biosynthesis of fatty acids in bacteria. It makes FabF an essential target for inhibiting bacterial growth in resistant bacteria. Previously two classes of inhibitor, cerulenin [5] and thiolactomycin [6–8], were reported, but inhibitory activity was poor (IC50 ranges within 1.3–13 μg/mL) with poor antimicrobial activity (Streptococcus aurous, MIC—64 μg/mL) [9]. On the other hand, platensimycin had shown selective FabF inhibitory activity on S. aurous and E. coli with IC50 value of 48 nM and 68 nM [3]. The ability of this class of compounds to bind and inhibit FabF enzyme has given a new class of antibiotics. Among them platensimycin and platencin are most promising and need further investigation. There might be an argument that if fatty acid synthesis is an attractive target for antibacterials, why there have not many drugs or natural inhibitors targeting this pathway been isolated? One of the reasons may be that the Streptomyces and related Actinomyces, the organisms that have delivered most of the existing antibiotics, are constrained in their ability to produce fatty acid synthesis inhibitors by the adjacent relationship between the synthetic pathways of fatty acids and polyketides. These pathways share many chemical, mechanistic, and structural features, and, thus, fatty acid synthesis inhibitors recurrently inhibit polyketide synthesis (e.g., cerulenin is a powerful inhibitor of polyketide synthesis). An organism producing a fatty acid synthesis inhibitor must retain not only a resistant form of the fatty acid synthetic enzyme but also a resistant form of the antibiotic-producing polyketide synthase. Furthermore, if several polyketides are needed for survival of the organism in its ecological niche, then resistant forms of each of these pathways would be prerequisite. Such precincts seem likely to severely narrow the opportunities to evolve fatty acid synthesis inhibitors and may account for the scarceness of such antibiotics. However, various Streptomyces produce analogues of thiolactomycin, which has not been reported to block polyketide synthesis, thus demonstrating that development of fatty acid synthesis inhibitors by this organism is conceivable [10].
2. Isolation of Platensimycin
Platensimycin was isolated by Wang et al. in 2006 at concentration of 2 to 4 mg/L from fermentation broth of S. platensis (MA7327 and MA7331) using SephadexLH20 liquid chromatography medium by reversed-phase HPLC chromatography [3]. In a subsequent study, three-step isolation was modified by Singh et al. and they established two-step method eliminating the SephadexLH20 step [11]. It was also isolated from Streptomyces platensis (MA7327), recovered from soil samples collected in Eastern Cape, South Africa [12].
3. Structure of Platensimycin
Platensimycin (Figure 1) consists of a benzoic acid moiety substituted at ortho and para with hydroxyl group and in meta position is conjugated with a unique pentacyclic ketolide by an amide linkage [13]. The structure was established by combination of DQ-COSY and TOCSY correlations (2D NMR), UV, and IR spectroscopy and confirmed by X-ray crystallography [14].
Figure 1.
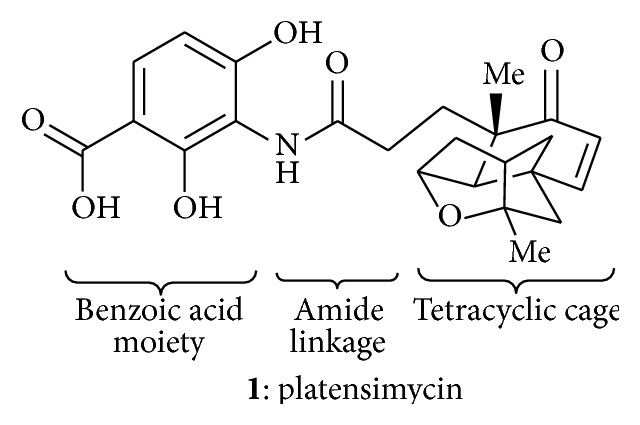
Structure of platensimycin.
4. Synthesis of Platensimycin
Platensimycin consists of an aromatic acid conjugated with aliphatic moiety by an amide linkage. The effective synthetic strategy is to synthesize the aromatic and aliphatic parts separately and then combine them by amide linkage. Many synthetic methods are available for the synthesis of those two building blocks.
4.1. First Total Synthesis of Platensimycin
Nicolaou et al. in the year 2006 first reported the total synthesis of platensimycin [26].
4.1.1. Retrosynthetic Analysis of Platensimycin
Using retrosynthetic analysis (Scheme 1) they separated the aromatic amine 2 and the carboxylic acid 3 by disconnection of amide linkage. The carboxylic acid was further simplified to a simplified enyne in successive three retrosynthetic steps. This resulted in two target molecules to synthesize the tetracyclic carboxylic acid and the aromatic amine from simplified starting material.
Scheme 1.

Retrosynthetic analysis of platensimycin.
4.1.2. Synthesis of Tetracyclic Cage
The simplified enone generated from 3-ethoxycyclohex-2-enone which was used as a starting material (Scheme 2). Allylic bromide 9 [27] (LDA, 92%) and propargyl bromide (LDA, 97%) were used as reagents of choice to generate the bis-alkylated enone 10 from 8. Reduction followed by acidic hydrolysis and reintroduction of the TBS ether produced enone 11 from enone 10 (yield 84%). Spirocycle 12 was generated by cycloisomerization of 11 [28, 29]. Oxidation of 12 produced bis-enone 13 [30] which upon acid hydrolysis gave desired aldehyde 6. Secondary alcohol 14 was prepared by addition of samarium(II) iodide in a dilute solution of aldehyde 6 HFIP in THF/HMPA followed by NH4Cl solution. Esterification of 14 with TEA resulted in the formation of cage-like structure 4 which on treatment with KHMDS and MeI followed by KHMDS and allyl iodide produced olefin 16. Vinyl pinacol boronate and 16 reacted in presence of the Grubbs second generation catalyst to produce vinyl boronate 19 which on reacting with trimethylamine N-oxide gave 20. Following Pinnick protocol 20 was converted to desired carboxylic acid 3.
Scheme 2.

Synthesis of tetracyclic cage. (i) LDA, (ii) LDA, (iii) DIBAL-H, then HCl, (iv) TBSCL, (v) [CpRu(MeCN)3]PF6, (vi) LiHMDS, TMSCl, (vii) Pd(OAc)2, (viii) HCl aq., (ix) Sml2, HFlP, (x) TFA, (xi) KHMDS, MeI, (xii) KHMDS, (xiii) Me3NO, and (xiv) NaClO2.
4.1.3. Synthesis of Aromatic Amine
The synthesis of the aromatic amine 2 was started from 2-nitroresorcinol 21 by protecting with MOM ether, followed by catalytic hydrogenation, 24 was formed (Scheme 3). Again protecting the amino group and followed by silylation, lithiation, and quenching with methyl cyanoformate 24 was carboxylated and by unprotecting amino group using thermolysis, desired aniline 2 was synthesized.
Scheme 3.

Synthesis of aromatic amine and platensimycin. (i) NaH, MOMCL, (ii) H2, Pd/C cat., (iii) Boc2O, (iv) nBuLi, and (v) 205°C.
4.1.4. Synthesis of Platensimycin Core
The total synthesis of platensimycin was completed by the coupling of carboxylic acid 3 with aniline 2 which was achieved by treatment with HATU followed by hydrolysis with LiOH (Scheme 3).
4.2. Another Approach for Synthesizing Tetracyclic Cage by Nicolaou et al.
After successfully reporting the first total synthetic strategy of platensimycin, Nicolaou et al. [31] report a new synthetic strategy that starts from the readily available and inexpensive (R)-(−)-carvone to the tetracyclic enone 2.
4.2.1. Retrosynthetic Strategy of Tetracyclic Enone
A five-step retrosynthetic disconnection approach was shown to generate commercially available (R)-(−)-carvone 31 from 26 (Scheme 4).
Scheme 4.

Retrosynthetic analysis of platensimycin.
4.2.2. Synthesis of Tetracyclic Enone
(R)-(−)-carvone 31 on reaction with Grignard's reagent 32 using CeCl3 [32] and by oxidizing with PCC gave the corresponding enone 33. Enone 33 was treated with Hg(OAc)2 and later on with NaBH4 to generate approximately 1 : 1 mixture of exo- and endotertiary alcohol, which on dehydration using Martin's sulfurane reagent produced exocyclic alkene 35 which was treated with TMSCl and HMDS and followed by an electrophilic quench with PhSeCl and subsequent oxidative elimination (H2O2) to give enone 36. Enone 37 was treated with SmI2 for radical cyclization, and by adding mentioned reagent (Scheme 5) in subsequent 8 steps of tetracyclic cage 26 was synthesized.
Scheme 5.

Synthesis of tetracyclic cage. (i) CeCl3, (ii) PCC, and (iii) Hg(OAc)2, then NaBH4, (iv) Martin's sulfurane, (v) TMSCl, Lil HMDS, and then PhSeCl, (vi) H2O2, Py, (vii) AcOH, MW, (ix) Sml2, (x) DIAD, ArCO2H, PPh3, Ar=p-NO2C6H4, (xi) KOH, (xii) L-selectride (H3O+), (xiii) PCC, (xiv) TMSCl, Lil HMDS, and (xv) IBX or Pd(OAc)2.
4.3. Matsuo's Synthesis of Tetracyclic Cage
4.3.1. Retrosynthetic Analysis of Tetracyclic Cage
Matsuo et al. [33] offered another route for stereocontrolled synthesis of tetracyclic enone 4. They expected that the transannular radical cyclization of 44 will produce enone 4. A four-step retrosynthetic analysis is producing enone 47 and siloxydiene 48. The key feature of plan is that all stereocenters in 4 are controlled by the stereochemistry presented in 47 (Scheme 6).
Scheme 6.

Retrosynthetic analysis of platensimycin.
4.3.2. Synthesis of Tetracyclic Cage
O-TBS and O-benzoyl-protected enones 47a and 47b were prepared from 1,3-cyclohexadiene 49 through allylboration, selective oxidation, and utilizing the Dess-Martin oxidation, with the yield of (97%). Diels-Alder reaction in between 47a and 47b and siloxydienes produced two stereogenic mixtures 46a, 46b and 46c, 46d. The mixture of two inseparable diastereomers (46b and 46d) was employed in the next step. Following Noyori's procedure [26] followed by hydrolysis of the benzoyl group and separation of diastereomers 53 was given which upon catalytic oxidation using palladium(II) chloride and copper(II) acetate produced 54a and 54b in 10 : 1 ratio. Vinyl triflate 55 was produced from 54 and reduced to 56 which was transformed to 57. Transannular radical cyclization of monothioacetal 57 gave desired 4 (Scheme 7).
Scheme 7.

Synthesis of tetracyclic cage. (i) allylBBr2, then HOOH NaOH, (ii) t-BuOOH, VO(sese)2, (iii) Dess-Martin, periodinane, (iv) Silica gel, (v) TBSCl imidazole/BzCl, Py, (vi) PMSOTf (cat.) (TMSOCH2)2, (vii) aqNaOH separation of diastereomers, (viii) PdCl2 Cu(OAc)2 DMA, O2, (ix) KHMDS, (x) Pd(OAc)2(PPh3)2 HCOOH, Bu3N, (xi) PhSH, (xii) Bu3SnH AIBN toluene, reflux, and (xiii) 1N aq HCL.
4.4. Kaliappan's Synthesis of Tetracyclic Cage
4.4.1. Retrosynthetic Analysis of Tetracyclic Cage
Kaliappan and Ravikumar [34] showed seven-step retrosynthetic analysis using a 5-exo-trig radical cyclization strategy [35, 36] to generate Wieland-Miescher ketone 64 (Scheme 8).
Scheme 8.

Retrosynthetic analysis of platensimycin.
4.4.2. Synthesis of Tetracyclic Cage
Synthesis was started from Wieland-Miescher ketone 64 [37], a chiral starting material, which on two-step reduction yielded an inseparable diastereomeric mixture of alcohols 66 [38]. Corresponding aldehyde 68 containing oxoethylene group was synthesized from alcohols 66 by Mandai protocol [39]. Using Ohira-Bestmann reagent 74 [40, 41], enyne 69 was formed which was treated with AIBN in tert-BuOH and PPTS to initiate the radical cyclization resulting formation of tetracyclic cage 72. L-Selectride, THF, and TFA/CH2Cl2 were consecutively added with 72 to produce the desired enone 58 (Scheme 9).
Scheme 9.

Synthesis of tetracyclic cage. (i) NaBH4, ETOH, 00C, 2 h, (ii) TBSCI, Im, DMF, r.t., 2 h, (iii) DIBAL-H CH2Cl2, (iv) NaH, cat. KHTHF, r.t., 12 h, (v) Decalin, 180°C, 5d, (vi) K2CO3, MeOH, (vii) TBAF, THF, reflux, (viii) IBX PhF/DMSO 65°C, (ix) TBTH, AIBN t-BuOH, reflux, 12 h, (x) PPTS, CH2Cl2, r.t., 6 h, (xi) L-selectride, THF, −78°C to r.t., and (xii) TFA/CH2Cl2, 0°C, 2 h.
4.5. Corey's Synthetic Strategy
4.5.1. Synthesis of Tetracyclic Cage
Lalic and Corey [42] used methoxy α-naphthol 75 as the starting material which upon reaction with bistrifluoroacetoxyiodobenzene and ethylene glycol in acetonitrile at 0°C produced 6-methoxy-1,4-naphthoquinone-4-ethylene ketal 76. An enantioselective conjugation of 2-propenyl group to ketal 76 using 2-propenyl trifluoroborate, Rh-BINAP, BF4 catalyst, and triethylamine [43–45] produced the chiral ketone 77. cis-tetralin 78 was generated from 77 by reduction of the carbonyl group, hydroxyl protection, and reductive cleavage of the ethylene ketal subunit. Demethylation of 78 produced phenol 79 which was converted to tricyclic 80 by etherification followed by reaction with Br2 in CH2Cl2. Heating with tetra-n-butylammonium fluoride in THF at 130°C, tetracyclic cage 81 was produced which on catalytic diastereoselective hydrogenation produced tetrahydro derivative 82. The saturated ketone 82 was transformed into the corresponding α,β-enone 4 using the 2-iodoxybenzoic acid sequence (Scheme 10) [46, 47].
Scheme 10.

Synthesis of tetracyclic cage. (i) PhI(O2CCF3)2, OHCH2CH2OH, MeCN, 0°C, 2 h. (ii) (S)-BINAP, [Rh(cod)2]BF4, C7H8, H2O, Et3N, CH3-CH(BF3K)=CH2. (iii) (1) NaBH4, MeOH, (2) MEMCl, i-Pr2NEt, CH2Cl2, (3) TsOH, Me2CO, 0°C, (4) i-Bu2AIH, CH2Cl2, 0°C, (5) Et3SiH, (CF3CO)2O, CH2Cl2, −20°C. (iv) PhSH, Cs2CO3, DMF, D 170°. (v) (1) i-Pr3SiCl, imidazole, CH2Cl2, 23°C, 12 h and (2) Br2, CH2Cl2, −78°C. (vi) n-Bu4NF, THF, D 130°C. (vii) [Rh(cod)2]BF4, (R,R)-DIOP, H2 600 psi, CH2Cl2, 16 h. (viii) (1) TMSOTf, Me3N, CH2Cl2 and (2) IBX, MPO, DMSO.
4.6. Yamamoto's Synthetic Strategy
4.6.1. Retrosynthetic Analysis of Tetracyclic Core
Li et al. [47] used an intramolecular Robinson annulation approach [48] in the retrosynthetic analysis presented in (Scheme 11). The bicyclic compound 84 to give the tetracyclic core structure 4 is key step in the retrosynthesis by Robinson annulation event.
Scheme 11.

Retrosynthetic analysis of platensimycin.
4.6.2. Synthesis of Tetracyclic Core
The bicyclic ketone 84 can be synthesized from known lactone 85 which could be generated from ketone 86, through a Baeyer-Villiger oxidation/rearrangement sequence [49, 50] and by utilizing Bronsted acid assisted chiral Lewis acid catalyzed Diels-Alder reaction, and subsequent N-nitrosoaldol addition/decarboxylation, ketone 86 could be easily prepared from inexpensive, commercially available starting materials (Scheme 12).
Scheme 12.

Synthesis of tetracyclic cage. (i) CH2Cl2, −78°C, 14 h, (ii) LDA, THF, −78°C, then PhNO, −78°C, 2 h, and then LiOH dioxane/H2O 30°C, 20 h, (iii) H2O2/NaOH Et2O/H2O 0°C to r.t., 45 min, (iv) CuBr·Me2S, CH2CMgBr THF/Me2S, −40°C to r.t., (v) 4.5 mol% HNTf2, CH2ClCH2Cl, 70°C, 45 min, (vi) DIBAL-H, toluene, −78°C, 30 min, then Et2AlCN BF3·OET2, 20 min, (vii) DIBAL-H/n-BuLi, −78°C to 0°C, 25 min, (viii) NaH, CH3COCH2P(O)(OET)2, THF, 0°C, 20 min, (ix) 2.02 eq NaIO4, 3.5 mol% RuCl3 6 : 1 CH3CN/H2O r.t., 3 h, and (x) 1 eq pyrrolidine-2-carboxylic acid, DMF, r.t., 5 days, then 2N NaOH(aq) 0°C to r.t., 40 min.
Diels-Alder reaction between methyl acrylate 87 and methyl cyclopentadiene 88 produced adduct 89. The reaction was catalyzed by BLA and carbon-based Bronsted acid [51]. Ketone 90 was obtained in one pot reaction from adduct 89 using lithium enolate and lithium hydroxide in dioxane. Baeyer-Villiger oxidation in basic condition [52] of ketone 90 gave lactone 85. Vinyl lactone 91 was obtained from lactone 85 using cuprate reagent [53] and trifluoromethanesulfonimide [54, 55]. DIBAL-H reduction followed by cyanation produced cyanide 92a, 92b, which was reduced and subjected to Wadsworth-Emmons conditions [56] to give enone 93; ruthenium-catalyzed oxidation [57] produced aldehyde 84. Using L-proline as the chiral control element followed by sodium hydroxide treatment gave the desired tetracyclic core structure 4.
5. Pharmacology of Platensimycin
Bacterial cell wall synthesis, protein synthesis, and DNA replication are the predominant targets for widely used antibiotics. But the emergence of resistance to antibiotics demands new antibacterial targets. Fatty acid synthase (FAS) pathway is now an attractive target for antibacterial agents because, as a new target, FAS inhibition will not suffer from bacterial resistance immediately and biosynthesis pathway of bacteria, plants, and parasites (FAS II in which component proteins are dissociated) is different from mammals (FAS I in which component proteins are generally single-chain, multidomain homodimers or two-chain heterodimers carrying all proteins of the pathway) in subcellular organization of components which demonstrate a target specificity for the FAS II inhibitors. A general scheme for type II fatty acid biosynthesis is shown in Figure 2[9]. A recent development in finding inhibitors of fatty acid biosynthesis is the discovery of platensimycin which shows broad-spectrum Gram-positive antibacterial activity (Staphylococcus aureus (MRSA) and Enterococci (VRE), MIC < 1.0 μg mL−1) by selectively inhibiting cellular lipid biosynthesis [58]. The mechanism of action is the selective inhibition of elongation of FabF (a condensing enzyme) in the bacterial fatty acid synthetic pathway by intercalating with the malonyl binding site of the catalytic triad of FabF acyl enzyme intermediate. Inverse correlation of FabF expression levels with the sensitivity of S. aureus to the drug platensimycin confirms that FabF is the useful target for antibacterial action. Platensimycin exhibited an IC50 of 48 nM and 160 nM against FabF in S. aureus and E. coli, respectively. But the drug shows weak inhibition against FabH with an IC50 value of 67 mM. Further studies have shown that in vitro binding of platensimycin with FabF is relatively weak, which leads to the discovery of its binding with acyl-thioester intermediate of the FabF pathway. From a crystal structure of a Cys-163-Gln FabF mutant, which simulates acyl-thioester intermediate, it was found that platensimycin bind with the active site of FabF with the carboxylic acid group lying in the malonate-binding site coplanar with the amide side chain of Gln163 [59].
Figure 2.

Fatty acid biosynthesis pathway of bacteria (FAS II).
Though Brinster et al. [60] explained an alternative hypothesis that FAS II inhibition is not a suitable target for Streptococcus agalactiae (lactobacillales). The need for synthesized fatty acid by their own (streptococci, pneumococci, enterococci, and staphylococci) reduces in the presence of exogenous fatty acid in both in vitro and in vivo conditions. Human serum has a high composition of fatty acids, so FAS II inhibitor may not affect at all Gram-positive pathogens in vivo. But the findings with S. agalactiae can be reasonably extended to all Gram-positive bacteria, which has started a vigorous debate. Also there is no clear explanation how bacteria incorporates exogenous fatty acids into their cell [61, 62]. Platensimycin exhibited antibacterial activity against efflux-negative Escherichia coli (tolC), but not against wild-type E. coli, specifying that efflux mechanisms, and not compound specificity, limit the effectiveness of platensimycin in E. coli and possibly other Gram-negative bacteria [3].
6. Platensimycin Analogues
The major drawback to most natural products, including platensimycin, is poor pharmacokinetic properties and negligible oral bioavailability. Only the continuous infusion of a high platensimycin dose showed effectiveness in mice infected with S. aureus [3]. For the improvement of pharmacokinetic profile, many researchers have developed platensimycin analogues (Table 1); however potent antimicrobial activity than that of platensimycin is yet to get.
Table 1.
Antimicrobial activity of synthesized platensimycin analogue.
| Sl. number | Analogues name | Analogues structure | Antimicrobial activity | Reference |
|---|---|---|---|---|
| 1 | (−)-Platencin |
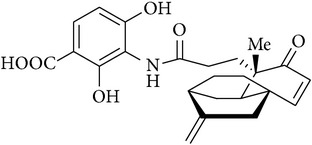
|
Inhibits both FabF and FabH with similar potency | [12] |
|
| ||||
| 2 | 7-Phenylplatensimycin |
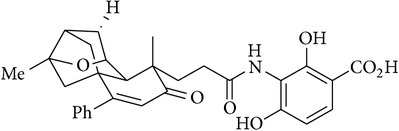
|
MIC against S. aureus (MSSA), 0.25 μg/mL | [15] |
|
| ||||
| 3 | 11-Methyl-7-phenylplatensimycin |
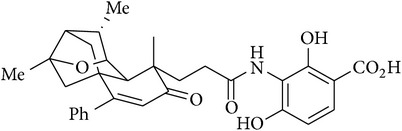
|
MIC against S. aureus (MSSA), <0.25 μg/mL | [15] |
|
| ||||
| 4 | Sulfonamide analogues of platensimycin |
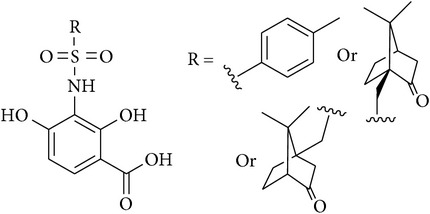
|
Not tested | [16] |
|
| ||||
| 5 | Oxazinidinyl platensimycin |
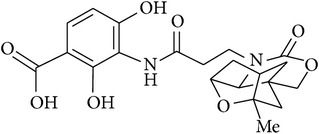
|
MIC of 90 μg/mL against S. aureus, S. agalactiae, and B. subtilis | [17] |
|
| ||||
| 6 | Isoplatencin |
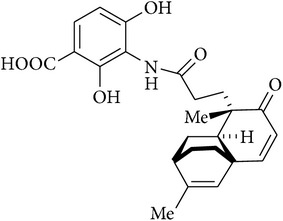
|
MIC against S. aureus (MSSA), 0.4 μg/mL | [18] |
|
| ||||
| 7 | Cl-iso-platencin |
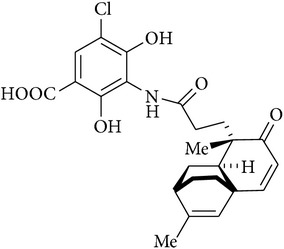
|
MIC against S. aureus (MSSA), >25.6 μg/mL | [18] |
|
| ||||
| 8 | Cl-platencin |
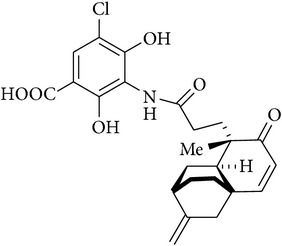
|
MIC against S. aureus (MSSA), >25.6 μg/mL | [18] |
|
| ||||
| 9 | Dehydrohomoplatencin |
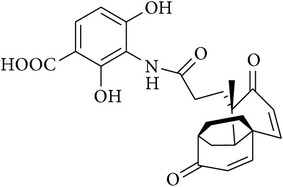
|
MIC against S. aureus (MSSA), 0.4 μg/mL | [19] |
|
| ||||
| 10 | Isoplatensimycin |
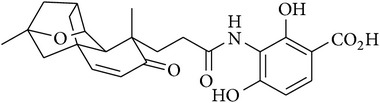
|
MIC against S. aureus (MSSA), 128 μg/mL | [20] |
|
| ||||
| 11 | Carbaplatensimycin |
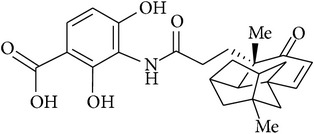
|
MIC against S. aureus, 0.4–1.1 μg/mL | [21] |
|
| ||||
| 12 | Platensimycin B1 and B3 |
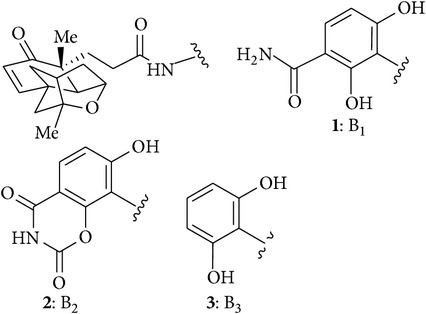
|
Not tested | [22] |
|
| ||||
| 13 | Platensimide A |
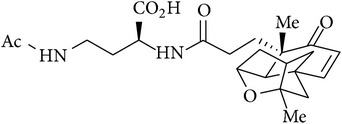
|
Not tested | [22] |
|
| ||||
| 14 | Homoplatensimide A and homoplatensimide A methyl ester |
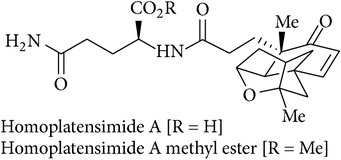
|
Not tested | [22] |
|
| ||||
| 15 | Dialkylamino-2,4-dihydroxybenzoic acids analogues of Platensimycin |
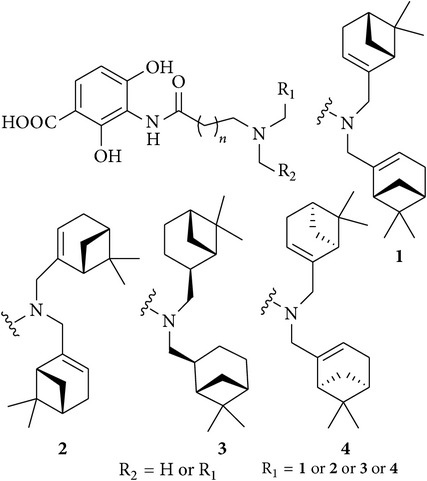
|
Out of 18 Synthesized derivatives, four derivatives which have the substitution 1, 2, 3, and 4, respectively, have shown potential activity against B. subtilis (MIC 2–8 μg/mL) | [23] |
|
| ||||
| 16 | (−)-nor-platencin |
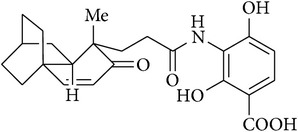
|
MIC against S. aureus (MSSA), 5 μg/mL | [24] |
|
| ||||
| 17 | Adamantaplatensimycin |
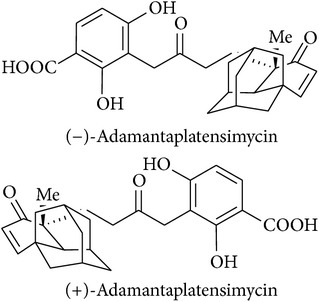
|
MIC of (−)-adamantaplatensimycin against S. aureus (MSSA), 1.3–1.8 μg/mL; MIC of (+)-adamantaplatensimycin against S. aureus (MSSA), >88 μg/mL | [25] |
7. Conclusion
Development of bacterial resistance to the existing antibiotics is an alarming situation in the 20th century. Finding new target for bacterial demolition is essential. Platensimycin serving as a potential antibiotic interacting with FabF may bypass bacterial resistance. It should be noted that starting from 2006 till now a huge number of scientists provided many synthetic strategies and derivatives which are very encouraging. Though some evidence like incorporation of exogenous fatty acid inside bacteria when supplied makes the hope regarding platensimycin is uncertain, overall, isolation of platensimycin as a selective FabF inhibitor, complex synthesis of tetracyclic cage, and enhancement of its pharmacokinetic properties by its derivative synthesis are the excellent works in the era of medicinal chemistry. It is expected that this review might be helpful for the medicinal chemist.
Acknowledgment
The authors are thankful to the Department of Pharmacy, Tripura University (A Central University).
Abbreviations
- Ac:
Acetyl
- AIBN:
2,2′-Azobis(isobutyronitrile)
- BINAP:
2,2′-Bis(diphenylphosphino)-1,10-binaphthalene
- Boc:
tert-Butoxycarbonyl
- Bz:
Benzoyl
- DIBALH:
Diisobutylaluminium hydride
- DMF:
N,N,-Dimethylformamide
- DMSO:
Dimethyl sulfoxide
- HATU:
O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HMPA:
Hexamethyl phosphoramide
- KHMDS:
Potassium hexamethyldisilazide
- LDA:
Lithium diisopropylamide
- PCC:
Pyridinium chlorochromate Piv pivaloyl
- PPTS:
Pyridinium-p-toluenesulfonate
- Py:
Pyridine
- TEA:
Triethylamine
- Tf:
Trifluoromethanesulfonyl
- TFA:
Trifluoroacetic acid
- TFE:
2,2,2-Trifluoroethanol
- THF:
Tetrahydrofuran
- TMS:
Trimethylsilyl.
Conflict of Interests
There is no conflict of interests.
References
- 1.Walsh C. T. Where will new antibiotics come from? Nature Reviews Microbiology. 2003;1(1):65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 2.Singh S. B., Barrett J. F. Empirical antibacterial drug discovery—foundation in natural products. Biochemical Pharmacology. 2006;71(7):1006–1015. doi: 10.1016/j.bcp.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 3.Wang J., Soisson S. M., Young K., et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441(7091):358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 4.Wang J., Kodali S., Lee S. H., et al. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(18):7612–7616. doi: 10.1073/pnas.0700746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsumae A., Nomura S., Hata T. Studies on cerulenin. IV. Biological characteristics of cerulenin. The Journal of Antibiotics. 1964;17:1–7. [PubMed] [Google Scholar]
- 6.Miyakawa S., Suzuki K., Noto T., Harada Y., Okazaki H. Thiolactomycin, a new antibiotic. IV. Biological properties and chemotherapeutic activity in mice. The Journal of Antibiotics. 1982;35(4):411–419. doi: 10.7164/antibiotics.35.411. [DOI] [PubMed] [Google Scholar]
- 7.Noto T., Miyakawa S., Oishi H., Endo H., Okazaki H. Thiolactomycin, a new antibiotic. III. In vitro antibacterial activity. The Journal of Antibiotics. 1982;35(4):401–410. doi: 10.7164/antibiotics.35.401. [DOI] [PubMed] [Google Scholar]
- 8.Oishi H., Noto T., Sasaki H., et al. Thiolactomycin, a new antibiotic. I. Taxonomy of the producing organism, fermentation and biological properties. The Journal of Antibiotics. 1982;35(4):391–395. doi: 10.7164/antibiotics.35.391. [DOI] [PubMed] [Google Scholar]
- 9.Kodali S., Galgoci A., Young K., et al. Determination of selectivity and efficacy of fatty acid synthesis inhibitors. The Journal of Biological Chemistry. 2005;280(2):1669–1677. doi: 10.1074/jbc.m406848200. [DOI] [PubMed] [Google Scholar]
- 10.Campbell J. W., Cronan J. E., Jr. Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annual Review of Microbiology. 2001;55:305–332. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]
- 11.Singh S. B., Jayasuriya H., Ondeyka J. G., et al. Isolation, structure, and absolute stereochemistry of platensimycin, a broad spectrum antibiotic discovered using an antisense differential sensitivity strategy. Journal of the American Chemical Society. 2006;128(36):11916–11920. doi: 10.1021/ja062232p. [DOI] [PubMed] [Google Scholar]
- 12.Nicolaou C. K., Tria S. G., Edmonds J. D., Kar M. Total syntheses of (±)-platencin and (−)-platencin. Journal of the American Chemical Society. 2009;131(43):15909–15917. doi: 10.1021/ja906801g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Häbich D., Von Nussbaum F. Platensimycin, a new antibiotic and ‘Superbug challenger’ from nature. ChemMedChem. 2006;1(9):951–954. doi: 10.1002/cmdc.200600145. [DOI] [PubMed] [Google Scholar]
- 14.Saleem M., Hussain H., Ahmed I., Van Ree T., Krohn K. Platensimycin and its relatives: a recent story in the struggle to develop new naturally derived antibiotics. Natural Product Reports. 2011;28(9):1534–1579. doi: 10.1039/c1np00010a. [DOI] [PubMed] [Google Scholar]
- 15.Jang K. P., Kim C. H., Na S. W., et al. 7-Phenylplatensimycin and 11-methyl-7-phenylplatensimycin: more potent analogs of platensimycin. Bioorganic & Medicinal Chemistry Letters. 2010;20(7):2156–2158. doi: 10.1016/j.bmcl.2010.02.037. [DOI] [PubMed] [Google Scholar]
- 16.McNulty J., Nair J. J., Capretta A. A synthesis of sulfonamide analogs of platensimycin employing a palladium-mediated carbonylation strategy. Tetrahedron Letters. 2009;50(28):4087–4091. doi: 10.1016/j.tetlet.2009.04.105. [DOI] [Google Scholar]
- 17.Wang J., Lee V., Sintim H. O. Efforts towards the identification of simpler platensimycin analogues-the total synthesis of oxazinidinyl platensimycin. Chemistry—A European Journal. 2009;15(12):2747–2750. doi: 10.1002/chem.200802568. [DOI] [PubMed] [Google Scholar]
- 18.Tiefenbacher K., Gollner A., Mulzer J. Syntheses and antibacterial properties of iso-platencin, Cl-iso-platencin and Cl-platencin: identification of a new lead structure. Chemistry—A European Journal. 2010;16(31):9616–9622. doi: 10.1002/chem.201000706. [DOI] [PubMed] [Google Scholar]
- 19.Waalboer D. C. J., Leenders S. H. A. M., Schülin-Casonato T., Van Delft F. L., Rutjes F. P. J. T. Total synthesis and antibiotic activity of dehydrohomoplatencin. Chemistry—A European Journal. 2010;16(37):11233–11236. doi: 10.1002/chem.201001744. [DOI] [PubMed] [Google Scholar]
- 20.Jang K. P., Kim C. H., Na S. W., Kim H., Kang H., Lee E. Isoplatensimycin: synthesis and biological evaluation. Bioorganic & Medicinal Chemistry Letters. 2009;19(16):4601–4602. doi: 10.1016/j.bmcl.2009.06.092. [DOI] [PubMed] [Google Scholar]
- 21.Nicolaou K. C., Tang Y., Wang J., Stepan A. F., Li A., Montera A. Total synthesis and antibacterial properties of carbaplatensimycin. Journal of the American Chemical Society. 2007;129(48):14850–14851. doi: 10.1021/ja076126e. [DOI] [PubMed] [Google Scholar]
- 22.Nicolaou K. C., Li A., Edmonds D. J., Tria G. S., Ellery S. P. Total synthesis of platensimycin and related natural products. Journal of the American Chemical Society. 2009;131(46):16905–16918. doi: 10.1021/ja9068003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J., Sintim H. O. Dialkylamino-2,4-dihydroxybenzoic acids as easily synthesized analogues of platensimycin and platencin with comparable antibacterial properties. Chemistry—A European Journal. 2011;17(12):3352–3357. doi: 10.1002/chem.201002410. [DOI] [PubMed] [Google Scholar]
- 24.Barykina O. V., Rossi K. L., Rybak M. J., Snider B. B. Synthesis and antibacterial properties of (-−)-nor-platencin. Organic Letters. 2009;11(22):5334–5337. doi: 10.1021/ol902194q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolaou K. C., Lister T., Denton R. M., Montero A., Edmonds D. J. Adamantaplatensimycin: a bioactive analogue of platensimycin. Angewandte Chemie—International Edition. 2007;46(25):4712–4714. doi: 10.1002/anie.200701548. [DOI] [PubMed] [Google Scholar]
- 26.Nicolaou K. C., Li A., Edmonds D. J. Total synthesis of platensimycin. Angewandte Chemie—International Edition. 2006;45(42):7086–7090. doi: 10.1002/anie.200603892. [DOI] [PubMed] [Google Scholar]
- 27.Sun M., Deng Y., Batyreva E., Sha W., Salomon R. G. Novel bioactive phospholipids: practical total syntheses of products from the oxidation of arachidonic and linoleic esters of 2-lysophosphatidylcholine. The Journal of Organic Chemistry. 2002;67(11):3575–3584. doi: 10.1021/jo0105383. [DOI] [PubMed] [Google Scholar]
- 28.Trost B. M., Toste F. D. Ruthenium-catalyzed cycloisomerizations of 1,6- and 1,7-enynes. Journal of the American Chemical Society. 2000;122(4):714–715. doi: 10.1021/ja993401r. [DOI] [Google Scholar]
- 29.Trost B. M., Surivet J.-P., Toste F. D. Ruthenium-catalyzed enyne cycloisomerizations. Effect of allylic silyl ether on regioselectivity. Journal of the American Chemical Society. 2004;126(47):15592–15602. doi: 10.1021/ja046824o. [DOI] [PubMed] [Google Scholar]
- 30.Ito Y., Hirao T., Saegusa T. Synthesis of α,β-unsaturated carbonyl compounds by palladium(II)-catalyzed dehydrosilylation of silyl enol ethers. The Journal of Organic Chemistry. 1978;43(5):1011–1013. doi: 10.1021/jo00399a052. [DOI] [Google Scholar]
- 31.Nicolaou K. C., Pappo D., Tsang K. Y., Gibe R., Chen D. Y.-K. A chiral pool based synthesis of platensimycin. Angewandte Chemie—International Edition. 2008;47(5):944–946. doi: 10.1002/anie.200705080. [DOI] [PubMed] [Google Scholar]
- 32.Molander G. A. Application of lanthanide reagents in organic synthesis. Chemical Reviews. 1992;92(1):29–68. doi: 10.1021/cr00009a002. [DOI] [Google Scholar]
- 33.Matsuo J.-I., Takeuchi K., Ishibashi H. Stereocontrolled formal synthesis of (±)-platensimycin. Organic Letters. 2008;10(18):4049–4052. doi: 10.1021/ol801584r. [DOI] [PubMed] [Google Scholar]
- 34.Kaliappan K. P., Ravikumar V. An expedient enantioselective strategy for the oxatetracyclic core of platensimycin. Organic Letters. 2007;9(12):2417–2419. doi: 10.1021/ol070848t. [DOI] [PubMed] [Google Scholar]
- 35.Stork G., Tang P. C., Casey M., Goodman B., Toyota M. Regiospecific and stereoselective syntheses of (±)-reserpine and (−)-reserpine. Journal of the American Chemical Society. 2005;127(46):16255–16262. doi: 10.1021/ja055744x. [DOI] [PubMed] [Google Scholar]
- 36.Janardhanam S., Shanmugam P., Rajagopalan K. Stereocontrolled synthesis of angularly fused tricyclic systems by Tin-mediated radical cyclization. The Journal of Organic Chemistry. 1993;58(27):7782–7788. doi: 10.1021/jo00079a023. [DOI] [Google Scholar]
- 37.Harada N., Sugioka T., Uda H., Kuriki T. Efficient preparation of optically pure Wieland-Miescher ketone and confirmation of its absolute stereochemistry by the CD exciton chirality method. Synthesis. 1990;1990(1):53–56. doi: 10.1055/s-1990-26786. [DOI] [Google Scholar]
- 38.Boyer F.-D., Ducrot P.-H. Synthesis of agarofuran antifeedants, part II: stereoselective construction of the tetrahydrofuran ring. Synthesis. 2000;2000(13):1868–1877. [Google Scholar]
- 39.Mandai T., Ueda M., Hasegawa S.-I., Kawada M., Tsuji J., Saito S. Preparation and rearrangement of 2-allyloxyethyl aryl sulfoxides; a mercury-free claisen sequence. Tetrahedron Letters. 1990;31(28):4041–4044. doi: 10.1016/s0040-4039(00)94494-0. [DOI] [Google Scholar]
- 40.Ohira S. Methanolysis of dimethyl (1-diazo-2-oxopropyl) phosphonate: generation of dimethyl (diazomethyl) phosphonate and reaction with carbonyl compounds. Synthetic Communications. 2006;19(3-4):561–564. doi: 10.1080/00397918908050700. [DOI] [Google Scholar]
- 41.Müller S., Liepold B., Roth G. J., Bestmann H. J. An improved one-pot procedure for the synthesis of alkynes from aldehydes. Synlett. 1996;1996(6):521–522. doi: 10.1055/s-1996-5474. [DOI] [Google Scholar]
- 42.Lalic G., Corey E. J. An effective enantioselective route to the platensimycin core. Organic Letters. 2007;9(23):4921–4923. doi: 10.1021/ol702323s. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi T., Yamasaki K. Rhodium-catalyzed asymmetric 1,4-addition and its related asymmetric reactions. Chemical Reviews. 2003;103(8):2829–2844. doi: 10.1021/cr020022z. [DOI] [PubMed] [Google Scholar]
- 44.Pucheault M., Darses S., Genêt J.-P. Potassium organotrifluoroborates in rhodium-catalyzed asymmetric 1,4-additions to enones. European Journal of Organic Chemistry. 2002;2002(21):3552–3557. doi: 10.1002/1099-0690(200211)2002:21<3552::aid-ejoc3552>3.0.co;2-4. [DOI] [Google Scholar]
- 45.Molander G. A., Ellis N. Organotrifluoroborates: protected boronic acids that expand the versatility of the Suzuki coupling reaction. Accounts of Chemical Research. 2007;40(4):275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 46.Nicolaou K. C., Gray D. L. F., Montagnon T., Harrison S. T. Oxidation of silyl enol ethers by using IBX and IBX. N-oxide complexes: a mild and selective reaction for the synthesis of enones. Angewandte Chemie—International Edition. 2002;41(6):996–1000. doi: 10.1002/1521-3773(20020315)41:6<996::aid-anie996>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 47.Li P., Payette J. N., Yamamoto H. Enantioselective route to platensimycin: an intramolecular robinson annulation approach. Journal of the American Chemical Society. 2007;129(31):9534–9535. doi: 10.1021/ja073547n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stork G., Shiner C. S., Winkler J. D. Stereochemical control of the internal Michael reaction. A new construction of trans-hydrindane systems. Journal of the American Chemical Society. 1982;104(1):310–312. doi: 10.1021/ja00365a067. [DOI] [Google Scholar]
- 49.Curran D. P., Chen M.-H. Radical-initiated polyolefinic cyclizations in condensed cyclopentanoid synthesis. Total synthesis of (±)-Δ9(12)-capnellene. Tetrahedron Letters. 1985;26(41):4991–4994. doi: 10.1016/s0040-4039(01)80834-0. [DOI] [Google Scholar]
- 50.Corey E. J., Weinshenker N. M., Schaaf T. K., Huber W. Stereo-controlled synthesis of prostaglandins F2α and E2 . Journal of the American Chemical Society. 1969;91(20):5675–5677. doi: 10.1021/ja01048a062. [DOI] [PubMed] [Google Scholar]
- 51.Hasegawa A., Ishikawa T., Ishihara K., Yamamoto H. Facile synthesis of aryl- and alkyl-bis(trifluoromethylsulfonyl)methanes. Bulletin of the Chemical Society of Japan. 2005;78(8):1401–1410. doi: 10.1246/bcsj.78.1401. [DOI] [Google Scholar]
- 52.Weinshenker N. M., Stephenson R. J. Basic hydrogen peroxide cleavage of a bicyclic ketone. A new procedure for a prostaglandin intermediate. The Journal of Organic Chemistry. 1972;37(23, article 3741) doi: 10.1021/jo00796a036. [DOI] [PubMed] [Google Scholar]
- 53.Curran D. P., Chen M.-H., Leszczweski D., Elliott R. L., Rakiewicz D. M. Regiocontrol in opening of 2H-cyclopenta[b]furanones with organocopper reagents. The Journal of Organic Chemistry. 1986;51(9):1612–1614. doi: 10.1021/jo00359a048. [DOI] [Google Scholar]
- 54.Yang C.-G., Reich N. W., Shi Z., He C. Intramolecular additions of alcohols and carboxylic acids to inert olefins catalyzed by silver(I) triflate. Organic Letters. 2005;7(21):4553–4556. doi: 10.1021/ol051065f. [DOI] [PubMed] [Google Scholar]
- 55.Rosenfeld D. C., Shekhar S., Takemiya A., Utsunomiya M., Hartwig J. F. Hydroamination and hydroalkoxylation catalyzed by triflic acid. Parallels to reactions initiated with metal triflates. Organic Letters. 2006;8(19):4179–4182. doi: 10.1021/ol061174+. [DOI] [PubMed] [Google Scholar]
- 56.Peng X., Bondar D., Paquette L. A. Alkoxide precoordination to rhodium enables stereodirected catalytic hydrogenation of a dihydrofuranol precursor of the C29-40 F/G sector of pectenotoxin-2. Tetrahedron. 2004;60(43):9589–9598. doi: 10.1016/j.tet.2004.06.142. [DOI] [Google Scholar]
- 57.Yang D., Zhang C. Ruthenium-catalyzed oxidative cleavage of olefins to aldehydes. The Journal of Organic Chemistry. 2001;66(14):4814–4818. doi: 10.1021/jo010122p. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y.-M., White S. W., Rock C. O. Inhibiting bacterial fatty acid synthesis. The Journal of Biological Chemistry. 2006;281(26):17541–17544. doi: 10.1074/jbc.r600004200. [DOI] [PubMed] [Google Scholar]
- 59.Wright H. T., Reynolds K. A. Antibacterial targets in fatty acid biosynthesis. Current Opinion in Microbiology. 2007;10(5):447–453. doi: 10.1016/j.mib.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brinster S., Lamberet G., Staels B., Trieu-Cuot P., Gruss A., Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature. 2009;458(7234):83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 61.Parsons J. B., Rock C. O. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery? Current Opinion in Microbiology. 2011;14(5):544–549. doi: 10.1016/j.mib.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jayasuriya H., Herath K. B., Zhang C., et al. Isolation and structure of platencin: a FabH and FabF dual inhibitor with potent broad-spectrum antibiotic activity. Angewandte Chemie—International Edition. 2007;46(25):4684–4688. doi: 10.1002/anie.200701058. [DOI] [PubMed] [Google Scholar]