

Abstract
Redox signalling comprises the biology of molecular signal transduction mediated by reactive oxygen (or nitrogen) species. By specific and reversible oxidation of redox-sensitive cysteines, many biological processes sense and respond to signals from the intracellular redox environment. Redox signals are therefore important regulators of cellular homeostasis. Recently, it has become apparent that the cellular redox state oscillates in vivo and in vitro, with a period of about one day (circadian). Circadian time-keeping allows cells and organisms to adapt their biology to resonate with the 24-hour cycle of day/night. The importance of this innate biological time-keeping is illustrated by the association of clock disruption with the early onset of several diseases (e.g. type II diabetes, stroke and several forms of cancer). Circadian regulation of cellular redox balance suggests potentially two distinct roles for redox signalling in relation to the cellular clock: one where it is regulated by the clock, and one where it regulates the clock. Here, we introduce the concepts of redox signalling and cellular timekeeping, and then critically appraise the evidence for the reciprocal regulation between cellular redox state and the circadian clock. We conclude there is a substantial body of evidence supporting circadian regulation of cellular redox state, but that it would be premature to conclude that the converse is also true. We therefore propose some approaches that might yield more insight into redox control of cellular timekeeping.
Keywords: biological clock, circadian timekeeping, cysteine oxidation, redox signalling
INTRODUCTION
Intracellular redox state determines the direction of redox reactions that occur in the cell, and is determined by the balance of pro-oxidants compared with antioxidants. Oxidising conditions result from an increase in levels of pro-oxidant reactive oxygen (or nitrogen) species (ROS, or RNS) or a decrease in reducing power. Although high levels of ROS (a state known as oxidative stress) are detrimental to the cellular environment, low levels of ROS are required for normal signalling in healthy cells (Schieber and Chandel, 2014).
One of the first examples of functional signalling initiated by ROS (aka redox signalling) was recognised in the mid-1970s, when it was observed that high levels of oxidising reagents induce an insulin-like response (Czech et al., 1974). Indications that ROS can also induce a direct transcriptional response came from studies on bacterial OxyR (Storz et al., 1990) and mammalian AP-1 (c-Fos and c-Jun) (Abate et al., 1990) transcription factors, which showed that oxidation of these proteins modulates their transcriptional activities. By the mid-1990s, Sundaresan and colleagues had noted that activation of the platelet derived growth factor (PDGF) signalling pathway induces an increase in intracellular hydrogen peroxide (H2O2) concentration that is required for functional PDGF signalling. Based on these findings, they proposed that H2O2 potentially functioned as a second messenger in this pathway (Sundaresan et al., 1995), a hypothesis that was subsequently confirmed and then expanded to include other signalling pathways (as exemplified below).
In contrast to other second messengers (like calcium or cAMP), H2O2 is not recognised by specialised binding (or effector) proteins. Instead, signalling through H2O2 proceeds via the oxidation of reactive cysteine residues. The response to a H2O2 transient is not binary however, and can lead to different molecular effects, depending on the concentration of H2O2, the identity of the oxidised cysteine, the degree of oxidation, and the nature of the oxidised cysteine’s subsequent reactions. Essential to this H2O2-mediated signal transduction therefore, is the principle of differential sensitivity of intracellular cysteines to oxidation. This is readily understood in the case of buried vs. surface-exposed cysteine residues but, as discussed below, also includes differential redox sensitivity of surface cysteines within the same protein.
FORMATION AND DECOMPOSITION OF REACTIVE OXYGEN SPECIES
Reactive oxygen species (ROS) is a generic term for many different oxygen-derivatives, including the extremely reactive hydroxyl radical (·OH), highly reactive superoxide anion (O2·−) and the less reactive but more abundant and diffusible H2O2; these latter features eminently suit H2O2 for its signalling role. As a result of aerobic metabolism, superoxide anions are constantly formed as natural by-products of mitochondrial respiration; indeed it has been estimated that about 1–2% of all acceptor oxygen escapes full reduction to water and exits the mitochondria as O2·− (Aon et al., 2003). Moreover, O2.− is actively produced by NADPH oxidases (NOXs) and xanthine oxidases (XO) (Dickinson, 2015) in response to growth factor signalling e.g. insulin and PDGF pathways.
Due to its high reactivity, an increase in O2.− levels induces damage to essential cellular components such as DNA, fatty acids and proteins. To protect themselves against ROS-induced damage, cells express antioxidant systems that decompose ROS. Mitochondrial manganese superoxide dismutase (MnSOD) and cytoplasmic copper/zinc superoxide dismutase (CuZnSOD) catalyse the dismutation of O2·− to less reactive H2O2 (McCord and Fridovich, 1969). Proteins that control cytoplasmic H2O2 levels are peroxiredoxin 1 and 2 (PRX1 and 2) and glutathione peroxidase (GPX) (Hanschmann et al., 2013). Followed protein oxidation, PRX and GPX are recycled via reduction by the thioredoxin (TXN) and glutaredoxin (GRX) systems, respectively. Both TXN and GRX are ultimately supplied with reducing equivalents in the form of NADPH – the cytosol’s master redox currency (see below).
CYSTEINE-DEPENDENT REDOX SIGNALLING
As noted above, redox signalling proceeds through the oxidation of specific cysteine residues in a wide variety of proteins. The sulphur atom in cysteine is sensitive to several degrees of oxidation by H2O2 (Fig. 1A). In its reduced state, it exists as a thiol (−SH) or as a deprotonated thiolate anion (−S−). The thiolate anion is most susceptible to oxidation, and upon successive encounters with H2O2, it reacts to form first a sulphenic acid (−SOH), then a sulphinic acid (−SO2H), then a fully oxidised sulphonic acid (−SO3H). Of these, a sulphinic acid can be actively reduced by sulphiredoxin1 (SRX) to form a sulphenic acid (Chang et al., 2004; Woo et al., 2003). Both sulphinic and sulphonic forms of cysteine may be referred to as “over-“ or “hyperoxidised”, but only the sulphonic form is irreversibly oxidised,
Fig. 1.
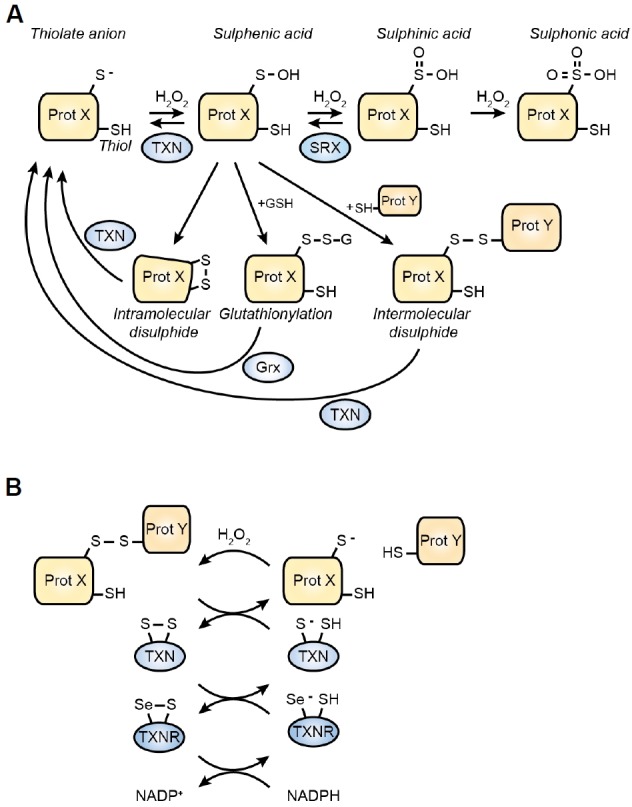
Oxidative cysteine modifications and their recycling. (A) Overview of oxidative cysteine modifications and their respective reduction procces. Abbreviations: Prot X: protein X – TXN: thioredoxin – SRX: sulfiredoxin – GRX: glutaredoxin. (B) Reduction of an inter- or intramolecular disulphide by the thioredoxin system. Thioredoxin reductase (TXNR) is a selenoprotein, containing a selenocysteine (Se) instead of a normal cysteine.
The initial protonation state of a cysteine residue is determined by its pKa. The pKa of free cysteine is 8.3–8.5, meaning that at cytosolic pH (∼7.4) free cysteines are fully protonated. Similarly, GSH has a pKa of 8.8 (Winterbourn and Metodiewa, 1999) and is fully protonated under physiological circumstances. Instead, redox sensitive proteins have a much lower pKa: the catalytic cysteine of TXN has a pKa of 6.5 (Goldman et al., 1995), the pKa of the catalytic cysteine in PTP1B is even lower at 5.4 (Monteiro and Stern, 1996), and PRX catalytic cysteines have a pKa between 5–6 (Peskin et al., 2007). Low pKa, however, is not sufficient to explain the higher reactivity of some cysteines compared with others. For example, although both PRX2 and PTP1B catalytic cysteines are fully deprotonated at physiological pH, the reaction rate of PRX2 with H2O2 is approximately six orders of magnitude higher than PTP1B. It is thought therefore, that residues surrounding highly redox-sensitive cysteines are critical for determining their activity, not only by facilitating an electrostatic microenvironment which favours deprotonation but also stabilising the transition state required for their high reactivity (Bindoli and Rigobello, 2012). For example, PRX2 and 3 contain a highly conserved arginine residue that is critical to their peroxidase function (Nagy et al., 2011).
Sulphenic acids are believed to be short-lived oxidation intermediates as they favour reaction with other cysteinethiols to form intra- or intermolecular disulphides (-S-S-) (Fig. 1A) (Rehder and Borges, 2010). Intramolecular disulphides potentially mediate conformational changes that facilitate cellular signal transduction. For example, formation of an intramolecular disulphide in Kelch-like ECH-associated protein 1 (KEAP1) inhibits its binding to the transcription factor NRF2 (Nuclear factor (erythroid-derived)-like 2). As KEAP1 facilitates NRF2 ubiquitination and degradation, oxidising conditions lead to activation of NRF2 and its transcriptional program. Being an important regulator of the cellular antioxidant response (with targets such as SOD, PRX and SRX) this allows for a proper cellular response to oxidative insults (Hayes and Dinkova-Kostova, 2014).
Intermolecular disulphides lead to covalently linked (homo- or hetero) dimers or multimers. Such a covalent interaction potentially stabilises otherwise transient interactions, thereby allowing changes in protein-protein interaction-mediated signalling (Cremers and Jakob, 2013; Putker et al., 2014a). Several observations indicate that such intermolecular disulphides can directly regulate cytosolic enzyme activity. For example, the activities of protein kinase A (PKA) (Burgoyne et al., 2015), protein kinase G (PKG) (Burgoyne et al., 2007; Prysyazhna et al., 2012) and apoptosis signal-regulating kinase 1 (ASK1) (Nadeau et al., 2007) are under disulphide-mediated redox control. Several transcription factors have also been shown to be regulated by disulphide-mediated interactions, with well-known examples being the Forkhead Box O tumour suppressor proteins, FOXO3 and FOXO4, both of which having been implicated in a plethora of disulphide-dependent complexes (Dansen et al., 2009; Putker et al., 2013; 2014b). ROS-induced nuclear accumulation and activation of both proteins is regulated in a (distinct) disulphide-dependent manner.
Alternatively, sulphenic acids can form intermolecular disulphides with the highly abundant tripeptide glutathione (Glu-Cys-Gly) (GSH), known as glutathionylation. Glutathionylation is considered an important posttranslational modification that serves several functions. First of all, it protects the sulphur from hyperoxidation, thereby increasing protein stability. Moreover, glutathionylation affects protein activity by altering cysteine function (see below) or may form new docking sites for protein-protein interactions (Grek et al., 2013).
Due to the nucleophilic character of the thiolate anion, cysteines often play important roles in protein activity or structure. Therefore, besides mediating disulphide-dependent signalling, oxidation of functional cysteines can greatly affect cellular signalling. Many enzymes feature catalytic cysteines in their active sites, with oxidation leading to enzymatic inhibition. Under physiological conditions, sulphenic acid formation has been shown to inhibit the activities of enzymes as diverse as phospho-tyrosine phosphatases [e.g. protein tyrosine phosphatase 1B (PTP1B) (Lee et al., 1998)], deubiquitinating enzymes [e.g. ubiquitin specific peptidase 1 (USP1), USP7 and A20 (Cotto-Rios et al., 2012; Kulathu et al., 2013)] and the glycolytic enzyme GAPDH (Peralta et al., 2015). In addition to catalytic cysteine oxidation, cysteines are often found at sites of structural significance, and oxidation has therefore been suggested to induce functional changes in protein structure. For example, cysteines in zinc finger motifs are crucial to secondary structure and DNA binding. Oxidation of zinc finger cysteines leads to Zn2+ release and subsequent conformational change/loss of DNA binding, and increased free Zn2+ itself can elicit further signalling responses. This redox control by Zn2+ exclusion has been demonstrated for the zinc finger transcription factors p53 (Rainwater et al., 1995) and SP-1 (Webster et al., 2001; Wu et al., 1996), for example.
Excepting –SO3, all cysteine derivatives described above are reversible. Inter- or intramolecular disulphides become reduced through the TXN and GRX systems (Hanschmann et al., 2013). Recycling of TXN is catalysed by thioredoxin reductase (TXNR) (Fig. 1B), whereas GRX recycles by oxidation transfer to a free GSH molecule. Oxidised GSH (GSSG) in turn is recycled by glutathione reductase (GR), with both oxidised TXNR and GR being ultimately reduced at the expense of NADPH. The intra-cellular GSH pool and NADP/NADPH ratio are thus critical determinants of cellular reduction potential.
SPECIFICITY OF CYSTEINE-DEPENDENT REDOX SIGNALLING
Cells possess extensive antioxidant systems to maintain intra-cellular ROS within a physiological range. GPX, PRX1 and 2 are highly abundant proteins, and their reaction rates with H2O2 are 4–6 orders of magnitude above that of most redox sensitive cysteines. For example, the rate constant of PRX2 for its reaction with H2O2 is ∼4*107 M−1s−1, whereas that of PTP1B is 20 M−1s−1. As the protein levels of PRX1 and 2 are high [20 μM versus e.g. 0.1 μM for PTP1B (Winterbourn and Hampton, 2008)], and their reaction rate with H2O2 is much higher than most protein thiols, it seems unlikely that PTP1B (or another similarly reactive protein thiol) would become oxidised upon physiological increases in H2O2 (i.e. those that mediate signalling, without inducing damage) (Fig. 2A). Oxidation and inhibition of PTP1B does occur however (Lee et al., 1998), as do the other protein oxidations described above. Although the sequence of molecular events during each oxidation are incompletely understood, in general the mechanism allowing cells to detect and respond to H2O2-induced cysteine oxidation is understood by (a combination of) two models.
Fig. 2.
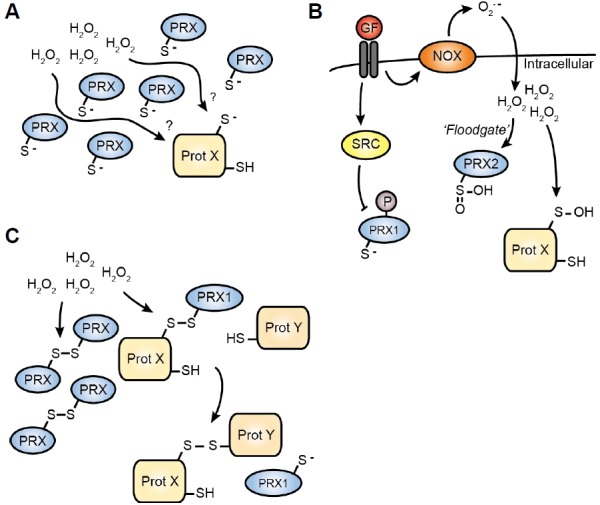
Models for how cysteine oxidation occurs in vivo. (A) High abundance and reactivity of PRX proteins makes it unlikely that any other cysteine in the cytoplasm could become oxidised. (B) Growth factor signalling induces a localised increase in H2O2 by inhibiting PRX proteins in at least two manners. The resulting increase in H2O2 allows the oxidation of less reactive thiols. (C) PRX-mediated redox relay. Oxidised PRX transfers its oxidation to other proteins. Interaction with PRX can mediate the oxidation of specific cysteines upon an increase of the H2O2 concentration.
In the first model, localised inactivation of PRX1/2 allows other proteins to become oxidised (Fig. 2B). The activity of PRX proteins is regulated by either oxidative or non-oxidative modifications. In the first case, localised increase in H2O2 levels (e.g. through NOX activation) overwhelms the “flood-gate” of the local antioxidant capacity leading to hyperoxidation of PRX peroxidatic cysteines, preventing their recycling. A localised pool of inactivated H2O2-scavenging enzymes results, allowing oxidation of less reactive thiol proteins (Wood et al., 2003). Upon cessation of H2O2 production, the sulphinic acids are (slowly) recycled via SRX (Chang et al., 2004), restoring normal redox balance. Besides oxidation-mediated regulation, tyrosine kinase-mediated phosphorylation of PRX1-tyr194 (e.g. by SRC) inactivates PRX1, contributing to localised inactivation of H2O2 scavenging enzymes. Interestingly, PRX2 is insensitive to such phosphorylation-dependent regulation. Instead, PRX2 is much more sensitive than PRX1 to hyperoxidation (Woo et al., 2010). Taken together, oxidation-dependent and -independent mechanisms lead to inactivation of PRX proteins, allowing H2O2 to oxidise other proteins and activate redox signalling.
The second model also uses PRX oxidation as a starting point. In this case, cysteines are not oxidised directly by H2O2, but through PRX-mediated oxidation relay processes (Fig. 2C). Instead of forming a homodimer with another PRX protein, oxidised PRX can transfer its oxidation state to specific target proteins, thereby inducing disulphide formation on its binding partner resulting in PRX reduction (Jarvis et al., 2012; Sobotta et al., 2014). This is functionally equivalent to the oxidative protein folding catalysed by ER-resident oxidoreductin1 and protein disulphide isomerase, which facilitate non-native structural disulphide formation in the secretory pathway (Bulleid and Ellgaard, 2011). First identified in yeast (Delaunay et al., 2002; Fourquet et al., 2008), such protein-mediated redox relays participate in several mammalian signalling pathways. For example, GPX4 transfers its H2O2-mediated oxidation to keratin-like proteins to stimulate disulphide bond formation in developing sperm (Maiorino et al., 2005). The first PRX-mediated redox relay was identified in the activation of ASK1. Here PRX1 plays an active and specific disulphide transfer role by facilitating ASK1 dimerisation, such that covalent, but transient, PRX1-ASK1 complexes (but not PRX2-ASK1) were observed upon an increase in ROS (Jarvis et al., 2012). Interestingly, besides explaining the occurrence of oxidation in the cytosol, the redox-relay model might also explain how specific redox signalling can occur through (scaffold-regulated) specific interactions between PRX1 and its target proteins.
CIRCADIAN TIMEKEEPING MECHANISMS DICTATE HOMEOSTASIS
The term “homeostasis” [homoios (similar) and stasis (standing still)] implies the existence of a biological steady state that is “preferred” and returned to, following a departure; however physiology is not as constant as suggested in some textbooks. Circadian (meaning “about daily”) timekeeping mechanisms allow organisms to adapt to the 24-hour daily cycle, allowing them to anticipate, and resonate with, the predictable environmental cycle of day and night (Dunlap, 1999). The mammalian “master clock”, resides in the hypothalamic suprachiasmatic nucleus (SCN), where it co-ordinates systemic adaptions to the day-night cycle; timing the temporal orchestration of overt physiology and behaviours, such as sleep/wake (Welsh et al., 2010). It is now well established however, that essentially every cell in the body is possessed with innate circadian timekeeping capacity (Welsh et al., 2004; Yoo et al., 2004). In vivo, these cell-autonomous oscillations are normally synchronised by the timing of endocrine signals, such as glucorticoids, and by other cues such as body temperature rhythms (Mohawk et al., 2012; Saini et al., 2015), but critically cellular circadian rhythms are not dependent upon any extracellular timing signals. Indeed using a bioluminescent reporter (PER2::LUC), under constant conditions, circadian gene expression rhythms in primary mouse fibroblasts were recently shown to persist for longer than a month ex vivo (Leise et al., 2012).
Circadian rhythms permeate mammalian cell biology, with ‘omics studies in tissues and isolated cells both revealing that significant proportions of gene expression and metabolism are circadian regulated (Asher and Schibler, 2011; Bass, 2012), with a commensurate impact upon biological function. For example, a recent report by (Zhang et al., 2014) found that more than a third of protein-coding genes in the mouse genome exhibit circadian regulation in one or more tissues. In consequence, it should not be surprising that from cell division (Bieler et al., 2014; Feillet et al., 2014; Matsuo et al., 2003) to signal transduction (O’Neill et al., 2008), from inflammation (Gibbs et al., 2014) to neuronal long-term potentiation (Chaudhury et al., 2005), circadian rhythms modulate cellular activity to support anticipated demand. A good example is the upregulation of transcripts and activities associated with gluconeogenesis and glycogenolysis at the beginning of the rest/inactive phase in mouse liver, which maintain blood glucose levels during the daily fast that occurs during sleep (Zhang et al., 2010).
The growing number of reports, discussed below, showing that cellular redox balance is circadian regulated should not come as any shock. Based on the evidence that redox signalling mediates many cellular functions, including the activity of specific transcription factors and metabolic enzymes, it is plausible the converse may also be true: that redox signalling regulates circadian timekeeping. A circadian output that also functions as a circadian input becomes indistinguishable from a core clock mechanism [or zeitnehmer (Roenneberg and Merrow, 2002)]. If cellular redox rhythms do constitute an important part of the circadian clock mechanism, then conceptual advances in both the redox and circadian field could readily be anticipated. In the rest of this review therefore, we wish to appraise the evidence supporting the hypothesis that cellular circadian rhythms reciprocally regulate cellular redox balance. To do so it is necessary to introduce some key paradigms and concepts from circadian research.
INPUTS, OUTPUTS AND MOLECULAR RHYTHMS – SEVERAL COGS IN ONE CLOCK
In eukaryotes the cell is the simplest level of biological scale that we know to possess circadian timekeeping. Cell biological approaches are therefore of utility for mechanistic investigations. The circadian vocabulary has largely developed from concepts based on behavioural and physiological measurements, but can be extended to define timekeeping at the cellular scale. At the organismal level the essential criteria defining a circadian rhythm are that it: persists with an approximately twenty-four hour period under constant conditions; is entrained by relevant external timing cues (zeitgebers, e.g. light, feeding); and is temperature-compensated (Pittendrigh, 1960). Refining this, we employ the term “cellular clock” to describe the complete set of cellular mechanisms and components that are necessary and sufficient to maintain a temperature-compensated ∼24 h oscillation in some biological process, that is sensitive to appropriate extracellular stimuli.
Whilst the functional contribution of assorted “clock genes” towards coordinating circadian physiology at the organismal level is proven beyond reasonable doubt (Bass and Takahashi, 2010), the mammalian cellular clock mechanism is not completely understood at this time. By this we mean that, to our knowledge, no extant hypothesis adequately explains in biochemical detail a causal chain of defined molecular events that give rise to a self-sustained oscillation of circadian period (O’Neill et al., 2013). Great progress has been made, however, in characterising many input signals to, and rhythmic outputs from, the cellular clock. Moreover many of its essential and auxiliary, transcriptional and post-translational components have been identified. The foremost of these are described below.
In the vast majority of contexts and mammalian cell types, several inter-connected delayed Gene Expression Feedback Loops (GEFLs; also known as Transcription-Translation Feedback Loops, TTFLs) regulate circadian gene expression. By binding to promoter regions in diverse clock-controlled genes, and acting in concert with tissue-specific promoters, cycling clock gene activity readily accounts for the majority of gene expression rhythms observed in vivo (Mohawk et al., 2012; Ueda, 2007; Zhang et al., 2014). The term “clock gene” itself is rather poorly defined, but in the broadest sense could be taken to mean a gene whose expression contributes to the fidelity of circadian timekeeping, and whose gene product directly or indirectly represses its own activity following a lengthy delay, leading to an oscillation in gene expression. As such a gene expression delayed negative feedback loop has been proposed as the essential mechanistic basis of the circadian rhythm in animals (Hardin et al., 1990; Rosbash, 2009) as well as plants and fungi (Dunlap, 1999); although it should be mentioned that the particular clock protein transcription factors identified in each eukaryotic kingdom share no similarity.
In short, the main mammalian GEFL (Fig. 3) begins with the activating bHLH-PAS (basic Helix-Loop-Helix-PER-ARNT-SIM) transcription factors BMAL1 (brain and muscle ARNT-like protein) and CLOCK (circadian locomotor output cycle kaput) or homologue NPAS2 (neuronal PAS-domain containing protein 2). Their activity at E-box promoters results in upregulation of many targets, amongst which are the period (encoding PER1, 2 and 3) and cryptochrome genes (encoding CRY1 and CRY2). PER and CRY proteins gradually accumulate and together form a repressive complex, which eventually is licensed for inhibition of nuclear BMAL1 and CLOCK activity - thereby repressing their own transcription. Due to the time required to assemble sufficient levels of this repressive complex (the negative feedback is delayed), accompanied by extensive daily changes in surrounding chromatin structure and histone modification, an oscillation of PER and CRY proteins (and of other CLOCK-BMAL1) target genes, results (Dunlap, 1999; Reppert and Weaver, 2002).
Fig. 3.
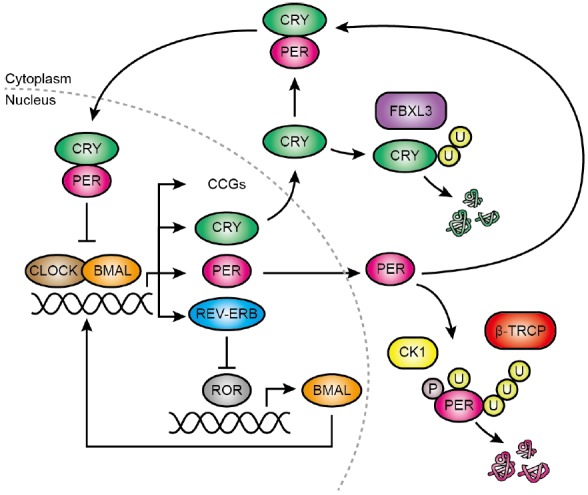
Models for gene expression feedback loops responsible for circadian gene expression. Simplified representation of the two major gene expression feedback loops. See text for details. CCG, Clock-controlled genes.
In a secondary loop, the levels of BMAL1 (and other ROR-regulated genes) are regulated in a circadian fashion. Here, ROR (Retinoic acid-related Orphan Receptor) proteins induce the expression of genes bearing promoter ROR-elements (RORE), including BMAL1. BMAL1 in turn regulates the (rhythmic) expression of REV-ERB proteins, which repress ROR-mediated gene expression by competing for RORE binding, forming another delayed negative feedback.
Important to note, and a repetitive theme in circadian biology, most of the “clock genes” (PER, CRY, BMAL, REV-ERB) may be considered both as inputs and outputs of the cellular clock: they contribute to the organisation of the clock, but also oscillate themselves as a result. Due to the overt oscillations of these proteins and their transcripts, clock gene expression is commonly used as a reporter of the molecular clockwork. In particular, the Bmal1 promoter, the Per2 promoter and the PER2 coding sequence, all coupled with firefly luciferase, are frequently used to measure the period, amplitude and phase of circadian rhythms in cultured cells/tissues and even in freely behaving animals (Saini et al., 2013; Sato et al., 2006; Yoo et al., 2004).
Both of these feedback loops rely on a positive input and a delayed feedback for their persistent oscillatory activity. Compared with the length of a day, transcription and translation of clock genes occurs quickly (0.25 – 1 h following activation), thus in the absence of extensive post-translational regulation, it is difficult to account for the delay constants that generate the slow kinetics of clock gene expression. Indeed, combined microarray and proteomic studies in mouse liver, show PER and CRY protein expression to peak roughly 6 hours later than peak mRNA expression of the same gene (Lee et al., 2001). As might be expected, this delay is predominantly regulated post-translationally, as overlap of the mRNA peak and ribosomal mRNA loading (Atger et al., 2015; Jang et al., 2015) suggests that translation of the mRNA occurs without any delay. Turnover of the PER and CRY proteins, for example, is tightly regulated by post-translational modifications. In particular CK1δ/ε (casein kinase 1 δ and ε) phosphorylates PER2 and target it for β-TRCP-mediated ubiquitination (Eide et al., 2005; Reischl et al., 2007), resulting in proteosomal degradation. Similarly, CRY is targeted for degradation by FBXL3-mediated ubiquitination (Godinho et al., 2007), which is counteracted by USP7 (HAUSP) (Papp et al., 2015). Interestingly, the PER-CRY binding interface blocks the binding site for FBXL3, and PER binding to CRY thus protects it from degradation (Nangle et al., 2014). In addition to CK1δ/ε, other ubiquitous kinases such as glycogen synthase kinase 3β and casein kinase 2 contribute to regulating the stability, activity and subcellular localisation of clock proteins (Maier et al., 2009; O’Neill et al., 2013) which are additionally regulated by a further host of post-translational modifications including acetylation, N-glycosylation and sumoylation (Asher et al., 2008; Cardone et al., 2005; Fogg et al., 2014; Kaasik et al., 2013; Ripperger and Schibler, 2006).
CELLULAR CLOCKS DO NOT REQUIRE (CYCLING) TRANSCRIPTION
Homozygous deletion of many of the core clock genes results in robust behavioural and cellular circadian phenotypes. These can be classified into three groups: (1) mild phenotype e.g. CLOCK (DeBruyne et al., 2006); (2) short or long period e.g. CRY1 and CRY2 knockout, respectively (Horst and Muijtjens, 1999); (3) no circadian rhythm detected (sometimes described as arrhythmic). In the last group, mice that lack either BMAL, or both CRY proteins, do not exhibit circadian rhythms in behaviour or clock gene transcription (Bunger et al., 2000; Horst and Muijtjens, 1999). These and other transgenic mouse models strongly suggest that the activity of clock proteins is necessary for the expression of normal circadian rhythms. Knockout mice do not demonstrate that clock gene expression is required to cycle per se though. Indeed when clock proteins are constitutively expressed the circadian phenotype is generally mild (Fan et al., 2007; Fujimoto et al., 2006; Okano et al., 2009), although overexpression can show more marked effects (Chen et al., 2009). It is therefore plausible that a certain level of clock protein abundance is simply permissive for normally functioning circadian rhythms, and that their oscillations in abundance play an auxiliary (rather than essential) role in terms of cellular clock mechanism, but do serve to increase the amplitude, robustness and fidelity of the oscillation (Hastings et al., 2008).
In support of this possibility, several observations have suggested that clock gene transcriptional cycles may not be absolutely required for circadian rhythms at the cellular level (O’Neill et al., 2013). For example, Paulose et al. (2012) observed circadian rhythms in glucose metabolism in mouse embryonic stem cells, which lack rhythms in clock gene expression. Similarly, separate groups have observed circadian rhythms in global, and clock protein, translation in isolated human cells, and mouse SCN ex vivo, respectively, that were homozygous null for the “essential” clock gene bmal1 (Ko et al., 2010; Lipton et al., 2015). Also PER2::LUC oscillations were found to persist in around 50% of SCN slices from mice that lack both CRY proteins, the “essential” repressors of the main GEFL (Maywood et al., 2011; Ono et al., 2013). Moreover, inspired by prior experiments performed upon algal and cyanobacterial clocks (Nakajima et al., 2005; O’Neill et al., 2011; Sweeney and Haxo, 1961; Tomita et al., 2005), several groups have now observed circadian rhythms in isolated human and mouse red blood cells. Mammalian erythrocytes lack a nucleus, or any other organelles, and therefore lack the capacity for nascent gene expression. Thus the erythrocyte cellular clockwork cannot conceivably rely upon GEFLs. The main marker for erythrocyte rhythms employed in these studies was PRX hyperoxidation (Cho et al., 2014; Homma et al., 2015; O’Neill and Reddy, 2011), which showed a temperature-compensated circadian oscillation that could be entrained by externally applied temperature cycles (mimicking body temperature rhythms). As might be anticipated, the PRX-SO2/3 rhythm was accompanied by a rhythm in total NA(P)DH levels (O’Neill and Reddy, 2011), and was dependent upon SOD1 activity, haemoglobin auto-oxidation and proteasomal degradation (Cho et al., 2014; Homma et al., 2015).
In our opinion though, whilst the evidence that the cellular clock does not absolutely require transcription, cycling or otherwise, has become quite compelling, it is also true that gene expression cycles facilitated by rhythmic clock protein activity play an essential cellular timekeeping role under almost all normal circumstances. Therefore in considering how the cellular clock interacts with redox signalling, we find it unhelpful to arbitrarily apportion timekeeping function between transcriptional and non-transcriptional mechanisms, since both are cogs of the same cellular clockwork and it is unlikely that either alone would sustain a circadian rhythm with any robustness.
CIRCADIAN CONTROL OF THE CELLULAR REDOX STATE
There is a substantial body of evidence demonstrating that the circadian clock regulates cellular redox state. First indications for this came from studies in the fungus Neurospora crassa, where it was shown that the oxidation state of nicotinamide adenine dinucleotide (the NAD/NADH ratio) exhibits daily rhythms (Brody and Harris, 1973). Subsequent studies in other model systems have also found the redox balance of pyridine nucleotides to be circadian-regulated. For example, nicotinamide adenine dinucleotide phosphate oxidation (NADP/NADPH) oscillations have been observed in plants (Zhou et al., 2015); protein glutathionylation and flavin adenine nucleotide (FAD)/NADPH oscillate in rodent SCN ex vivo (Wang and Gillette, 2012); NAD/NADH oscillates in the epidermis of living mice (Stringari et al., 2014); total NAD(P)H oscillates in human erythrocytes (O’Neill and Reddy, 2011); as do total NAD levels in mouse liver and cultured myoblasts (Peek et al, Science, 2013). Besides rhythmic abundance of dinucleotides, recently the oxidation state of PRX proteins have been a useful marker for circadian redox oscillations. In addition to isolated erythrocytes and the alga Ostreococcus tauri, cell-autonomous circadian oscillations in PRX-SO2/3 levels have now been observed in a variety of organisms, cell lines and tissues (Edgar et al., 2012; Kil et al., 2015; O’Neill and Reddy, 2011; O’Neill et al., 2011). The oscillation in PRX-SO2/3 abundance appears to be a cellular clock output only, since PRX activity is not required for cellular circadian rhythms (Causton et al., 2015). Rather it seems likely to be driven by an underlying rhythm in oxidative metabolism (Causton et al., 2015; Hoyle and O’Neill, 2014).
In line with the cellular clock being an important regulator of redox homeostasis, clock mutant mouse models exhibit ROS-associated phenotypes. Most pronounced is the premature ageing phenotype and shortened lifespan of BMAL1−/− mice, which was partially rescued by life-long administration of the GSH-precursor N-acetyl cysteine (NAC) (Kondratov et al., 2006, 2009). Fibroblasts and other tissues isolated from BMAL1−/− mice exhibit very high ROS loads (Lee et al., 2013a; Musiek et al., 2013), presumably due to another role of BMAL1 as a transcriptional regulator of cellular antioxidant expression via NRF2 (Lee et al., 2013a; Pekovic-Vaughan et al., 2014). Conversely, CRY1−/−/CRY2−/− cells seem to suffer from reductive stress (Jacobi et al., 2015; Peek et al., 2013).
Logically, oscillations in cellular redox state could either result from rhythmic ROS production, or from the rhythmic recycling or rhythmic expression of cellular antioxidant proteins. In fact, all three of these appear to be circadian regulated, as will be exemplified below.
First, cellular metabolism is under circadian control (Bass, 2012). As mitochondrial metabolism is the major source of cellular ROS in most cell types, rhythmic oxidative phosphorylation potentially contributes to cellular redox oscillations (excepting erythrocytes). As long ago as 1972 it was known that oxygen consumption of rat liver cells exhibit circadian oscillations (Langner and Rensing, 1972), suggesting some underlying circadian regulation of metabolic activity. Indeed, circadian oscillations in mitochondrial respiration have been found in mouse liver (Jacobi et al., 2015; Peek et al., 2013), skin (Stringari et al., 2014), and a myotube model (Peek et al., 2013). These oscillations coincide with diurnal BMAL1-dependent oscillations in mitochondrial biogenesis, protein expression and morphology. Liver cells from liver-conditional BMAL1−/− mice show diminished and arrhythmic respiration, swollen mitochondria and an increased ROS load, whereas these phenotypes are partially restored by expression of the fission protein FIS1, suggesting another role for BMAL1 in the regulation of mitochondrial quality control (Jacobi et al., 2015). An additional explanation for this phenotype is found in the circadian regulation of NAD biosynthesis. As well as being an important electron carrier required in glycolysis and the Krebs cycle, NAD functions as a co-factor for several enzymes, among which are the Sirtuin histone (and protein) deacetylase (HDAC) proteins (Vassilopoulos et al., 2011). The rate of NAD production is regulated, in part, by the abundance and activity of nicotinamide phosphoribosyltransferase (NAMPT), which is under transcriptional control the BMAL1:CLOCK complex (Nakahata et al., 2009; Ramsey et al., 2009). The circadian expression of NAMPT is thought to result in circadian abundance of NAD, coinciding with the rhythmic activity of mitochondrial SIRT3 and deacetylation of several SIRT3 targets involved in mitochondrial metabolism (Peek et al., 2013). In addition, nuclear SIRT6 is reported to be affected by NAD oscillations, remodelling chromatin structure to allow BMAL1:CLOCK DNA binding (thereby governing a positive feedback), and recruiting SREBP-1 to regulate the rhythmic expression of proteins involved in fatty acid and cholesterol metabolism (Masri et al., 2014). BMAL1-deficient fibroblasts exhibit reduced NAD levels, reduced oxidative phosphorylation/fatty acid oxidation rates and increased glycolytic rate, which is rescued by pharmacological restoration of NAD levels (Peek et al., 2013). Thus, via several different pathways, the BMAL1:CLOCK complex regulates mitochondrial metabolism and as such, mitochondrial ROS production.
Besides mitochondrial metabolism, NADPH oxidase (NOX) enzymes are implicated in the production of ROS. Clear circadian expression of NOX4 occurs in mouse lung, liver and kidney, for example, peaking in the opposite circadian phase to BMAL1 (CircaDB) (Pizarro et al., 2013). In agreement with this, NOX4 expression is significantly increased in arteries isolated from BMAL1−/− mice, as is the ROS load (Anea et al., 2013). We are not aware of studies directly testing the role of NOX proteins in cellular redox oscillations, but as the phase relationship of NOX4 with BMAL1 is similar to that of the NAD and oxidative phosphorylation peak (Peek et al., 2013), it seems plausible that NOX4 in some tissues contributes to circadian rhythms in ROS production.
Second, in addition to ROS production, cellular antioxidant capacity is also circadian regulated. For example, the NAMPT-mediated NAD oscillations described above allow for rhythmic activation of the nuclear HDAC function as well as rhythmic regulation of PER2 deacetylation and BMAL1:CLOCK activity (implicating NAD as both in- and outputs of cellular timekeeping) (Asher et al., 2008; Nakahata et al., 2009; Ramsey et al., 2009). SIRT1 in particular is also implicated in the regulation of several non-clock transcription factors. Among these, p53 (Vaziri et al., 2001) and FOXO (Brunet et al., 2004; van der Horst et al., 2004) are important regulators of the cellular redox state (de Keizer et al., 2011; Kruiswijk et al., 2015), and thus allow for circadian control of antioxidant protein expression. The contribution of p53 and FOXO to redox oscillations have yet to be studied to our knowledge, though we appreciate that their explicit contribution may be masked by direct regulation of the antioxidant response by BMAL:CLOCK. E-BOX mediated circadian regulation of NRF2 expression in the lung is required for rhythmic GSH production (Pekovic-Vaughan et al., 2014), and likely a plethora of other antioxidants (Gorrini et al., 2013). Oscillations in NRF2 expression are also found in several other tissues (Pizarro et al., 2013). In liver, a wide array of genes involved in the antioxidant response exhibit diurnal oscillations. Beyond NRF2 and KEAP1, the expression of NRF2 target genes NQO1, SOD1, CAT and GPX all exhibit circadian expression (Xu et al., 2012). NRF2 also regulates the expression of several enzymes involved in NADPH production (Mitsuishi et al., 2012), allowing BMAL1:CLOCK-NRF2 regulation of NADP/NADPH ratio, and therefore antioxidant recycling. In the brain, NRF2-independent oscillations of ALDH2 and NQO1 are associated with protection from redox-induced neurodegeneration (Musiek et al., 2013).
The above examples for circadian control of cellular redox state involve transcriptional regulation of either ROS or antioxidant production. Circadian oscillations of PRX oxidation (O’Neill and Reddy, 2011) and GSH:GSSG ratio (Radha et al., 1985) however, can occur in the absence of transcriptional cycles, clearly implicating transcription-independent mechanisms in circadian redox control. These remain largely uncharacterised to date, although several biochemical mechanisms have been described that could be relevant. For example, in the adrenal gland, lung, heart and brown adipose tissue, circadian oscillations in oxidation of (mitochondrial) PRX3 are proposed to be regulated through a redox feedback loop involving SRX. Here, mitochondrial ROS induces the oxidation and inactivation of PRX3, allowing H2O2 to leak into the cytosol. Cytosolic SRX becomes oxidised and forms a complex with HSP90, which allows for the import of SRX into the mitochondria where it reduces PRX3. Because free SRX in the mitochondria is sensitive to LON-mediated degradation, whereas PRX3-bound SRX is protected from this degradation, SRX becomes degraded when its job is done and the cycle begins afresh (Kil et al., 2015). The evidence supporting this sequence of molecular events is compelling, however it remains to be seen whether these oscillations are intrinsic to mitochondrial biology, or are driven by rhythmic cues from the surrounding cellular clockwork.
In summary a strong and growing body of evidence suggests that circadian rhythms in redox balance occur cell-autonomously as well as in vivo, and moreover that circadian disruption can be associated with deregulation of redox homeostasis.
REDOX CONTROL OF GEFL-MEDIATED TIMEKEEPING
As discussed above, redox signals impinge upon many signal transduction pathways. It is therefore plausible that GEFL-mediated timekeeping is also redox-regulated as has been suggested in non-mammalian models. For example, SOD-deficiency in Neurospora crassa increases the robustness of overt rhythmicity (Yoshida et al., 2011), and increased ROS concentrations induce shorter circadian period and phase advances. Interestingly, this was very much dependent on the source of ROS, since only metabolically generated ROS had this effect, whereas rhythmic ROS production by NOX did not (Gyöngyösi et al., 2013). Acute manipulation of redox state, e.g. using high extracellular H2O2, GSH, or diamide, does affect the phase and period in mammalian cells and cultured SCN (Tamaru et al., 2013; Wang and Gillette, 2012). Such manipulations certainly suggest that the cellular clock is susceptible exogenous oxidative and reductive stresses, but are not sufficient to demonstrate their relevance in the context of normal cellular timekeeping. Thus we are unaware of any convincing demonstration that oscillations in redox balance normally function to regulate circadian rhythms, although the principle that metabolism regulates the cellular clock is generally accepted (Asher and Schibler, 2011). It is the case however, that several possible redox-sensitive clock-relevant regulatory signalling nodes have been identified, as we shall describe.
First, we note that the metabolic oscillations described in the previous section have all been implicated in regulating transcriptional activity of the core clock genes e.g. oscillations in NAD regulate the activity of SIRT1, which in turn regulates PER2 deacetylation and BMAL1:CLOCK activity (Asher et al., 2008; Nakahata et al., 2009; Ramsey et al., 2009). The functional evidence that NAD oscillations make a significant contribution to cellular clock function is rather weak however (Sahar et al., 2011). Thus it remains possible that a certain range of cellular NAD is simply permissive for cellular clock function, and oscillations in steady state NAD levels result simply from a rhythm in consumption.
A second potential route from redox back into the clock is based upon biochemical observations that the affinity of CLOCK:BMAL1 (and NPAS2:BMAL1) heterodimers for DNA E-box elements is regulated by the redox state of NAD(P) cofactors, being favoured by high NAD(P)H, and opposed by high NAD(P) (Rutter et al., 2001; Yoshii et al., 2015). The biological relevance of these observations are unclear however, since this increased DNA-binding affinity was dependent upon dinucleotide concentrations within the low millimolar range, >10-fold higher than best estimates for total nuclear NAD(P)H (∼100 μM) (Zhang et al., 2002). Moreover to elicit circadian regulation of DNA binding would require a substantial oscillation either in ratio of free nuclear NAD(P)H:NAD(P), or else correctly timed, highly localized production of NAD(P)H in the immediate vicinity of the transcriptional complex. At present we are aware of no evidence supporting either scenario.
Rhythms in PRX oxidation suggest that the oxidation status of other proteinthiols might also oscillate and result, for example, in a concomitant release of KEAP1-NRF2 binding, thereby activating downstream NRF2-mediated antioxidant responses. NRF2 transcriptionally activates REV-ERBα expression (Yang et al., 2014), though the extent to which this contributes to normal circadian regulation of REV-ERBα is unknown. With respect to direct cysteine-mediated regulation of GEFL clock proteins several possible means of redox input have been described.
A recent x-ray crystal structure identified a tightly bound Zn2+ ion at the interface between PER2 and CRY1 (Schmalen et al., 2014). Zn2+ co-ordination was shown to be important for stabilising the PER2-CRY1 complex and was further proposed to facilitate the reduction of a nearby intramolecular disulphide bridge in CRY1. Moreover disulphide formation was shown to occur in CRY1 expressed in living cells. Whether or not CRY1 cysteine oxidation oscillates was not reported. A subsequent report by Nangle et al. (2014) confirmed this Zn2+-interface, but reported that Cys=>Ala mutation of the proposed redox-sensitive cysteine had little effect on cellular rhythms. These over-expression studies performed in CRY deficient mouse embryonic fibroblasts do not adequately address the contribution that CRY1 cysteine oxidation may or may not make to the cellular clock, however.
CRY proteins are also regulated by the nutrient-responsive adenosine monophosphate–activated protein kinase (AMPK), with AMPK stimulation leading to CRY phosphorylation and degradation (Lamia et al., 2009). AMPK is negatively regulated by cysteine oxidation in its α subunit and TRX-mediated AMPK reduction is essential for activation of AMPK during energy starvation (Shao et al., 2014). Moreover, CRY1 turnover upon DNA damage is regulated by the deubiquitinase protein USP7/HAUSP (Papp et al., 2015), which contains a redox-sensitive cysteine in its active site, that is inhibited by oxidation (Lee et al., 2013b). Perturbation of either AMPK or USP7 activity affects cellular rhythms, and thus these CRY-regulators introduce, additional plausible means whereby circadian rhythms in redox and metabolism might communicate with the GEFL through its essential CRY repressors. Again however, neither of these possibilities have been explicitly tested.
A further potential redox route into the clockwork lies in a redox-sensitive cysteine within the ligand-binding pocket of another GEFL protein, REV-ERBβ. Both REV-ERBα and β are nuclear receptors for the porphyrin haem, such that haem binding activates repressor activity against target genes (Raghuram et al., 2007). Cysteine oxidation abolishes haem-binding in vitro, and has been proposed to act as a “redox switch” in cells. The hypothesis that circadian REV-ERBβ oxidation feeds into the cellular clock has also not been tested however.
It is also perhaps worth mentioning that although cysteine is a relatively rare amino acid residue (∼2% content in mammalian proteins), the strongest point mutant circadian phenotypes in vivo and in vitro, with regards the speed at which the clock runs, result from missense mutations that involve a cysteine. A Cys => Ala mutation in CRY1 (Okano et al., 2009), and a Cys => Ser mutation in its E3 ligase F-box adaptor (FBXL3) (Godinho et al., 2007), both result in circadian periods that are 3–4 hours longer than wild type, whereas an Arg => Cys mutation in CK1ε results in a clock that runs 3–4 h shorter (Lowrey et al., 2000; Meng et al., 2008). Time will tell whether this correlation has any deeper significance.
From these examples it should be clear that whilst several mechanisms have been identified by which cell-autonomous circadian oscillations in redox balance might regulate GEFL activity, a clear demonstration that this actually occurs in mammalian cells is currently lacking.
CONCLUSION
Taken together, there is good evidence for cellular redox state being circadian regulated. Furthermore phenotypes of clock mutants suggest this has clear implications for systemic redox homeostasis. There are several biochemical indications that changes in cellular redox state may function as a circadian input (zeitgeber) and feedback into the cellular clock mechanism. We are not aware, however, of any compelling demonstration that the latter makes a significant contribution in a mammalian cellular context, let alone that it might be a prerequisite for cellular timekeeping. Therefore whilst it is most plausible that redox modulation of the cellular clock takes place, we think it is premature to conclude there is an important role for redox signalling in the mammalian cellular clock.
For redox oscillations to be considered as a mammalian cellular clock mechanism, as has Ca2+/cAMP-signalling (O’Neill and Reddy, 2012), it must be shown that: (1) a specific redox couple oscillates in a variety of cellular models; (2) that appropriate manipulation of this redox couple is sufficient to determine the phase, amplitude and period of cellular rhythms; and (3) that specific transduction pathways communicate cyclical redox signals to regulate GEFL function.
To approach the biological significance of redox control of the clock, we also need to understand the molecular underpinnings of PRX-SO2/3 oscillations – what is the timing mechanism that drives them? This question could be answered more readily with robust post-translational reporters for redox balance and ROS levels (Ezeriņa et al., 2014), rather than indirect markers such as PRX-SO2/3. In addition more sophisticated genetic tools are required than gene knockout. For example, SOD1 knockout does not affect timekeeping in mice (Homma et al., 2015). Is this because regulated ROS production is not relevant to circadian timekeeping or because compensatory mechanisms (e.g. increased SOD2) are employed? Inducible expression/repression models would be of great utility here.
Furthermore, it has not been strictly established that redox balance needs to oscillate for the cellular clock to function. Might there instead simply be a permissive range where cellular clocks operate? Indeed a similar argument has been made for BMAL1-deficient mice and tissues (Musiek, 2015; Musiek et al., 2013) i.e. it is plausible that BMAL1 abundance does not need to oscillate as such; rather that sufficient BMAL1 is required to activate antioxidant expression, and without BMAL1 cells succumb to chronic oxidative stress, outside the redox range where the cellular clock can function.
Until these issues have been resolved, we feel it inadvisable to use terms such as “redox oscillator” or “redox clock” since these phrases imply self-sufficiency. Until the underlying mechanism and mode(s) of coupling with GEFLs are firmly established it remains possible that circadian cycles of redox balance are exclusively clock outputs. If metaphors are required, we suggest that the image of a “metabolic cog” normally enmeshed with GEFLs, within the more complex cellular circadian machinery might suffice (Fig. 4). Indeed similarly, we are not aware of any evidence that a minimally reconstituted GEFL would be intrinsically circadian, as has been shown by in vitro reconstitution for the entirely post-translational circadian rhythm in the cyanobacteria Synechococcus elongatus (Nakajima et al., 2005).
Fig. 4.
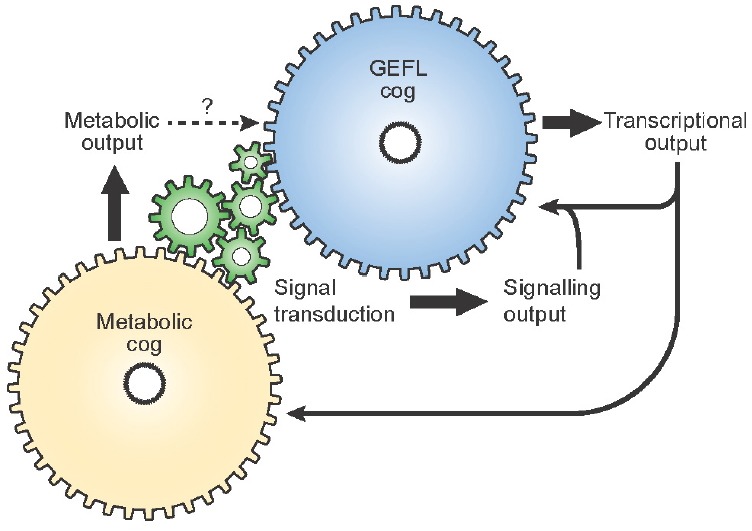
The role of GEFLs and metabolic oscillations in cellular timekeeping – multiple cogs in one clock. Cellular timekeeping involves several interconnected molecular systems, which are functionally interdependent under the vast majority of circumstances. At the level of the individual cell, there is no evidence for the existence of multiple clocks. It may instead be appropriate to consider the contribution that different cogs make to a single cellular clock mechanism. Some teeth of each cog make essential contributions to cellular clock function whereas others are auxiliary and/or mediate cell-type specific clock outputs. In addition, some cellular mechanisms, such as ATP generation for example, may simply be permissive for the cellular clock to tick rather than being a state variable of the oscillation. be permissive for the cellular clock to tick rather than being a state variable of the oscillation.
Even without understanding the detailed molecular underpinnings to circadian regulation of the cellular redox state, it is interesting to speculate about the functional consequences that redox oscillations might have upon normal cell signalling (Fig. 4). Just as the clock impacts upon cellular function through regulating the abundance of many key proteins involved in signal transduction and metabolic pathways (Mauvoisin et al., 2014; Robles et al., 2014), so might circadian rhythms in redox balance impact upon redox-sensitive signalling and metabolism. In mice and humans, for example, it has long been believed that synthesis/secretion of corticosteroids in the adrenal cortex are primarily regulated by circadian signaling from the SCN, through the hypothalamus–pituitary–adrenal (HPA) neuroendocrine axis. Recently however, (Kil et al., 2012) reported that circadian corticosterone production is accompanied by circadian oscillations in PRX3 and SRX abundance, that were required to protect the cell from increased mitochondrial ROS generation due to steroidogenesis during the latter half of the rest/sleep phase. In steroidogenic tissue-specific SRX-deficient mice, the circadian rhythm of adrenal corticosterone production was suppressed: severely blunting an important systemic cue that normally primes animal physiology for the active/wake phase.
Finally, of course it is quite plausible that circadian rhythms in redox may impact upon the efficacy of redox relay signalling (Jarvis et al., 2012), leading to the possibility that oxidation rhythms exist in other important redox-regulated cytosolic proteins such as GAPDH, KEAP1/NRF2, FOXO, ASK1, PKA, PKG, PTEN, PTP1B etc. If validated this would have far reaching, and potentially translational, consequences for the ways in which we understand the circadian clock machinery to interact with other cellular systems.
In conclusion, there exists great potential for cross fertilisation between research into redox signalling and circadian rhythms. From a basic research perspective it is of prime importance to establish explicitly whether crosstalk occurs bidirectionally and its mechanism(s), but this constitutes no barrier to investigating whether the cell interprets a redox signal differently, depending upon the (biological) time of day.
Acknowledgments
We wish to thank Jo Westmoreland of LMB Visual Aids. JON is supported by the Medical Research Council (MC_UP_1201/4) and the Wellcome Trust (093734/Z/10/Z). MP is funded by KWF BUIT 2014-6637. We apologise to the many colleagues whose work we did not discuss due to limitations of time and space.
REFERENCES
- Abate C., Patel L., Rauscher F.J., Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- Anea C.B., Zhang M., Chen F., Ali M.I., Hart C.M.M., Stepp D.W., Kovalenkov Y.O., Merloiu A.-M., Pati P., Fulton D., et al. Circadian clock control of Nox4 and reactive oxygen species in the vasculature. PLoS One. 2013;8:e78626. doi: 10.1371/journal.pone.0078626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon M. a., Cortassa S., Marbán E., O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- Asher G., Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Asher G., Gatfield D., Stratmann M., Reinke H., Dibner C., Kreppel F., Mostoslavsky R., Alt F.W., Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- Atger F., Gobet C., Marquis J., Martin E., Wang J., Weger B., Lefebvre G., Descombes P., Naef F., Gachon F. Circadian and feeding rhythms differentially affect rhythmic mRNA transcription and translation in mouse liver. Proc. Natl. Acad. Sci. USA. 2015;112:E6579–88. doi: 10.1073/pnas.1515308112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass J. Circadian topology of metabolism. Nature. 2012;491:348–356. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- Bass J., Takahashi J.S. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieler J., Cannavo R., Gustafson K., Gobet C., Gatfield D., Naef F. Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Mol. Syst. Biol. 2014;10:739. doi: 10.15252/msb.20145218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindoli A., Rigobello M.P. Principles in redox signaling: from chemistry to functional significance. Antioxid. Redox Signal. 2012;18:1–97. doi: 10.1089/ars.2012.4655. [DOI] [PubMed] [Google Scholar]
- Brody S., Harris S. Circadian rhythms in neurospora: spatial differences in pyridine nucleotide levels. Science. 1973;180:498–500. doi: 10.1126/science.180.4085.498. [DOI] [PubMed] [Google Scholar]
- Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y., Tran H., Ross S.E., Mostoslavsky R., Cohen H.Y., et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Bulleid N.J., Ellgaard L. Multiple ways to make disulfides. Trends Biochem. Sci. 2011;36:485–492. doi: 10.1016/j.tibs.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Bunger M.K., Wilsbacher L.D., Moran S.M., Clendenin C., Radcliffe L.a., Hogenesch J.B., Simon M.C., Takahashi J.S., Bradfield C. a. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne J.R., Madhani M., Cuello F., Charles R.L., Brennan J.P., Schröder E., Browning D.D., Eaton P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- Burgoyne J.R., Rudyk O., Cho H., Prysyazhna O., Hathaway N., Weeks A., Evans R., Ng T., Schröder K., Brandes R.P., et al. Deficient angiogenesis in redox-dead Cys17Ser PKARIα knock-in mice. Nat. Commun. 2015;6:7920. doi: 10.1038/ncomms8920. [DOI] [PubMed] [Google Scholar]
- Cardone L., Hirayama J., Giordano F., Tamaru T., Palvimo J., Sassone-Corsi P. Circadian clock control by SUMOylation of BMAL1. Science. 2005;309:1390–1394. doi: 10.1126/science.1110689. [DOI] [PubMed] [Google Scholar]
- Causton H.C., Feeney K.A., Ziegler C.A., O’Neill J.S. Metabolic cycles in yeast share features conserved among circadian rhythms. Curr. Biol. 2015;25:1056–1062. doi: 10.1016/j.cub.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T.-S., Jeong W., Woo H.A., Lee S.M., Park S., Rhee S.G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- Chaudhury D., Wang L.M., Colwell C.S. Circadian regulation of hippocampal long-term potentiation. J. Biol. Rhythms. 2005;20:225–236. doi: 10.1177/0748730405276352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Schirmer A., Lee Y., Lee H., Kumar V., Yoo S.-H., Takahashi J.S., Lee C. Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol. Cell. 2009;36:417–430. doi: 10.1016/j.molcel.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C.S., Yoon H.J., Kim J.Y., Woo H.A., Rhee S.G. Circadian rhythm of hyperoxidized peroxiredoxin II is determined by hemoglobin autoxidation and the 20S proteasome in red blood cells. Proc. Natl. Acad. Sci. USA. 2014;111:12043–12048. doi: 10.1073/pnas.1401100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotto-Rios X.M., Békés M., Chapman J., Ueberheide B., Huang T.T. Deubiquitinases as a signaling target of oxidative stress. Cell Rep. 2012;2:1–10. doi: 10.1016/j.celrep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers C.M., Jakob U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013;288:26489–26496. doi: 10.1074/jbc.R113.462929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech M.P., Lawrence J.C., Lynn W.S. Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proc. Natl. Acad. Sci. USA. 1974;71:4173–4177. doi: 10.1073/pnas.71.10.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansen T.B., Smits L.M.M., van Triest M.H., de Keizer P.L.J., van Leenen D., Koerkamp M.G., Szypowska A., Meppelink A., Brenkman A.B., Yodoi J., et al. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 2009;5:664–672. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- DeBruyne J.P., Noton E., Lambert C.M., Maywood E.S., Weaver D.R., Reppert S.M. A clock shock: mouse CLOCK is not required for circadian oscillator function. Neuron. 2006;50:465–477. doi: 10.1016/j.neuron.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Delaunay A., Pflieger D., Barrault M.B., Vinh J., Toledano M.B. A thiol peroxidase is an H2O2 receptor and redoxtransducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- Dickinson B.C. Plugging the leak Synergistic MRSA combinations. Nat. Publ. Gr. 2015;11:831–832. [Google Scholar]
- Dunlap J.C. Molecular bases for circadian clocks. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Edgar R.S., Green E.W., Zhao Y., van Ooijen G., Olmedo M., Qin X., Xu Y., Pan M., Valekunja U.K., Feeney K.A., et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485:459–464. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide E.J., Woolf M.F., Kang H., Woolf P., Hurst W., Camacho F., Vielhaber E.L., Giovanni A., Virshup D.M. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol. Cell. Biol. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeriņa D., Morgan B., Dick T.P. Imaging dynamic redox processes with genetically encoded probes. J. Mol. Cell. Cardiol. 2014;73:43–49. doi: 10.1016/j.yjmcc.2013.12.023. [DOI] [PubMed] [Google Scholar]
- Fan Y., Hida A., Anderson D.a., Izumo M., Johnson C.H. Cycling of CRYPTOCHROME proteins is not necessary for circadian-clock function in mammalian fibroblasts. Curr. Biol. 2007;17:1091–1100. doi: 10.1016/j.cub.2007.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feillet C., Krusche P., Tamanini F., Janssens R.C., Downey M.J., Martin P., Teboul M., Saito S., Levi F. a., Bretschneider T., et al. Phase locking and multiple oscillating attractors for the coupled mammalian clock and cell cycle. Proc. Natl. Acad. Sci. USA. 2014;111:9828–9833. doi: 10.1073/pnas.1320474111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogg P.C.M., O’Neill J.S., Dobrzycki T., Calvert S., Lord E.C., McIntosh R.L.L., Elliott C.J.H., Sweeney S.T., Hastings M.H., Chawla S. Class IIa histone deacetylases are conserved regulators of circadian function. J. Biol. Chem. 2014;289:34341–34348. doi: 10.1074/jbc.M114.606392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourquet S., Huang M.E., D’Autreaux B., Toledano M.B. The dual functions of thiol-based peroxidases in H2O2 scavenging and signaling. Antioxid. Redox Signal. 2008;10:1565–1576. doi: 10.1089/ars.2008.2049. [DOI] [PubMed] [Google Scholar]
- Fujimoto Y., Yagita K., Okamura H. Does mPER2 protein oscillate without its coding mRNA cycling?: post-transcriptional regulation by cell clock. Genes Cells. 2006;11:525–530. doi: 10.1111/j.1365-2443.2006.00960.x. [DOI] [PubMed] [Google Scholar]
- Gibbs J., Ince L., Matthews L., Mei J., Bell T., Yang N., Saer B., Begley N., Poolman T., Pariollaud M., et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat. Med. 2014;20:919–926. doi: 10.1038/nm.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho S.I.H., Maywood E.S., Shaw L., Tucci V., Barnard A.R., Busino L., Pagano M., Kendall R., Quwailid M.M., Romero M.R., et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- Goldman R., Stoyanovsky D.a., Day B.W., Kagan V.E. Reduction of phenoxyl radicals by thioredoxin results in selective oxidation of its SH-groups to disulfides. An antioxidant function of thioredoxin. Biochemistry. 1995;34:4765–4772. doi: 10.1021/bi00014a034. [DOI] [PubMed] [Google Scholar]
- Gorrini C., Harris I.S., Mak T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Grek C.L., Zhang J., Manevich Y., Townsend D.M., Tew K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013;288:26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyöngyösi N., Nagy D., Makara K., Ella K., Káldi K. Reactive oxygen species can modulate circadian phase and period in Neurospora crassa. Free Radic. Biol. Med. 2013;58:134–143. doi: 10.1016/j.freeradbiomed.2012.12.016. [DOI] [PubMed] [Google Scholar]
- Hanschmann E.-M., Godoy J.R., Berndt C., Hudemann C., Lillig C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013;19:1539–1605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin P.E., Hall J.C., Rosbash M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature. 1990;343:536–540. doi: 10.1038/343536a0. [DOI] [PubMed] [Google Scholar]
- Hastings M.H., Maywood E.S., O’Neill J.S. Cellular circadian pacemaking and the role of cytosolic rhythms. Curr. Biol. 2008;18:R805–R815. doi: 10.1016/j.cub.2008.07.021. [DOI] [PubMed] [Google Scholar]
- Hayes J.D., Dinkova-Kostova A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Homma T., Okano S., Lee J., Ito J., Otsuki N., Kurahashi T., Kang E.S., Nakajima O., Fujii J. SOD1 deficiency induces the systemic hyperoxidation of peroxiredoxin in the mouse. Biochem. Biophys. Res. Commun. 2015;463:1040–1046. doi: 10.1016/j.bbrc.2015.06.055. [DOI] [PubMed] [Google Scholar]
- Van Der Horst G.T.J., Muijtjens M. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. 1999;3495:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- Van der Horst A., Tertoolen L.G.J., de Vries-Smits L.M.M., Frye R.a., Medema R.H., Burgering B.M.T. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J. Biol. Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- Hoyle N.P., O’Neill J.S. Oxidation-reduction cycles of peroxiredoxin proteins and nontranscriptional aspects of timekeeping. Biochemistry. 2014;54:184–193. doi: 10.1021/bi5008386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi D., Liu S., Burkewitz K., Kory N., Knudsen N.H., Alexander R.K., Unluturk U., Li X., Kong X., Hyde A.L., et al. Hepatic Bmal1 regulates rhythmic mitochondrial dynamics and promotes metabolic fitness. Cell Metab. 2015;22:709–720. doi: 10.1016/j.cmet.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C., Lahens N.F., Hogenesch J.B., Sehgal A. Ribosome profiling reveals an important role for translational control in circadian gene expression. Genome Res. 2015;25:1836–1847. doi: 10.1101/gr.191296.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis R.M., Hughes S.M., Ledgerwood E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012;53:1522–1530. doi: 10.1016/j.freeradbiomed.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Kaasik K., Kivimäe S., Allen J.J.J., Chalkley R.J.J., Huang Y., Baer K., Kissel H., Burlingame A.L.L., Shokat K.M.M., Ptáček L.J.J., et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013;17:291–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keizer P.L.J., Burgering B.M.T., Dansen T.B. Forkhead box o as a sensor, mediator, and regulator of redox signaling. Antioxid. Redox Signal. 2011;14:1093–1106. doi: 10.1089/ars.2010.3403. [DOI] [PubMed] [Google Scholar]
- Kil I.S., Lee S.K., Ryu K.W., Woo H.A., Hu M.C., Bae S.H., Rhee S.G. Feedback Control of Adrenal Steroidogenesis via H2O2-Dependent, Reversible Inactivation of Peroxiredoxin III in Mitochondria. Mol. Cell. 2012;46:584–594. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Kil I.S., Ryu K.W., Lee S.K., Kim J.Y., Chu S.Y., Kim J.H., Park S., Rhee S.G. Circadian Oscillation of Sulfiredoxin in the Mitochondria. Mol. Cell. 2015;59:651–663. doi: 10.1016/j.molcel.2015.06.031. [DOI] [PubMed] [Google Scholar]
- Ko C.H., Yamada Y.R., Welsh D.K., Buhr E.D., Liu A.C., Zhang E.E., Ralph M.R., Kay S. a, Forger D.B., Takahashi J.S. Emergence of noise-induced oscillations in the central circadian pacemaker. PLoS Biol. 2010;8:e1000513. doi: 10.1371/journal.pbio.1000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov R. V., Kondratova A. a., Gorbacheva V.Y., Vykhovanets O. V., Antoch M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov R. V., Vykhovanets O., Kondratova A.a., Antoch M.P. Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging. 2009;1:979–987. doi: 10.18632/aging.100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F., Labuschagne C.F., Vousden K.H. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015;16:393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- Kulathu Y., Garcia F.J., Mevissen T.E.T., Busch M., Arnaudo N., Carroll K.S., Barford D., Komander D. Regulation of A20 and other OTU deubiquitinases by reversible oxidation. Nat. Commun. 2013;4:1569. doi: 10.1038/ncomms2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia K. a, Sachdeva U.M., DiTacchio L., Williams E.C., Alvarez J.G., Egan D.F., Vasquez D.S., Juguilon H., Panda S., Shaw R.J., et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langner R., Rensing L. Circadian rhythm of oxygen consumption in rat liver suspension culture: changes of pattern. Z. Naturforsch. B. 1972;27:1117–1118. doi: 10.1515/znb-1972-0945. [DOI] [PubMed] [Google Scholar]
- Lee S.-R., Kwon K.S., Kim S.R., Rhee S.G. Reversible Inactivation of Protein-tyrosine Phosphatase 1B in A431 Cells Stimulated with Epidermal Growth Factor. J. Biol. Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- Lee C., Etchegaray J.P., Cagampang F.R., Loudon aS., Reppert S.M. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- Lee J., Moulik M., Fang Z., Saha P., Zou F., Xu Y., Nelson D.L., Ma K., Moore D.D., Yechoor V.K. Bmal1 and β-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced β-cell failure in mice. Mol. Cell. Biol. 2013a;33:2327–2338. doi: 10.1128/MCB.01421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-G., Baek K., Soetandyo N., Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat. Commun. 2013b;4:1568. doi: 10.1038/ncomms2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leise T.L., Wang C.W., Gitis P.J., Welsh D.K. Persistent cell-autonomous circadian oscillations in fibroblasts revealed by six-week single-cell imaging of PER2::LUC bioluminescence. PLoS One. 2012;7:e33334. doi: 10.1371/journal.pone.0033334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton J.O., Yuan E.D., Boyle L.M., Ebrahimi-Fakhari D., Kwiatkowski E., Nathan A., Güttler T., Davis F., Asara J.M., Sahin M. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell. 2015;161:1138–1151. doi: 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey P.L., Shimomura K., Antoch M.P., Yamazaki S., Zemenides P.D., Ralph M.R., Menaker M., Takahashi J.S. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science. 2000;288:483–492. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B., Wendt S., Vanselow J.T., Wallach T., Reischl S., Oehmke S., Schlosser A., Kramer A. A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev. 2009;23:708–718. doi: 10.1101/gad.512209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorino M., Roveri A., Benazzi L., Bosello V., Mauri P., Toppo S., Tosatto S.C.E., Ursini F. Functional interaction of phospholipid hydroperoxide glutathione peroxidase with sperm mitochondrion-associated cysteine-rich protein discloses the adjacent cysteine motif as a new substrate of the selenoperoxidase. J. Biol. Chem. 2005;280:38395–38402. doi: 10.1074/jbc.M505983200. [DOI] [PubMed] [Google Scholar]
- Masri S., Rigor P., Cervantes M., Ceglia N., Sebastian C., Xiao C., Roqueta-Rivera M., Deng C., Osborne T.F., Mostoslavsky R., et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell. 2014;158:659–672. doi: 10.1016/j.cell.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T., Yamaguchi S., Mitsui S., Emi A., Shimoda F., Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–259. doi: 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- Mauvoisin D., Wang J., Jouffe C., Martin E., Atger F., Waridel P., Quadroni M., Gachon F., Naef F. Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Proc. Natl. Acad. Sci. USA. 2014;111:167–172. doi: 10.1073/pnas.1314066111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood E.S., Chesham J.E., Brien J.A.O., Hastings M.H. A diversity of paracrine signals sustains molecular circadian cycling in suprachiasmatic nucleus circuits. Proc. Natl. Acad. Sci. USA. 2011;108:14306–14311. doi: 10.1073/pnas.1101767108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord J.M., Fridovich I. Superoxide dismutase: and enzymatic function for erythrocuprein (hemocuprein) J. Biol. Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- Meng Q.-J., Logunova L., Maywood E.S., Gallego M., Lebiecki J., Brown T.M., Sládek M., Semikhodskii A.S., Glossop N.R.J., Piggins H.D., et al. Setting clock speed in mammals: the CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron. 2008;58:78–88. doi: 10.1016/j.neuron.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Mohawk J. a., Green C.B., Takahashi J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro H.P., Stern A. Redox modulation of tyrosine phosphorylation-dependent signal transduction pathways. Free Radic. Biol. Med. 1996;21:323–333. doi: 10.1016/0891-5849(96)00051-2. [DOI] [PubMed] [Google Scholar]
- Musiek E.S. Circadian clock disruption in neurodegenerative diseases: cause and effect? Front. Pharmacol. 2015;6:29. doi: 10.3389/fphar.2015.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek E.S., Lim M.M., Yang G., Bauer A.Q., Qi L., Lee Y., Roh J.H., Ortiz-gonzalez X., Dearborn J.T., Culver J.P., et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 2013;123:5389–5400. doi: 10.1172/JCI70317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau P.J., Charette S.J., Toledano M.B., Landry J. Disulfide Bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H(2)O(2)-induced c-Jun NH(2)-terminal kinase activation and apoptosis. Mol. Biol. Cell. 2007;18:3903–3913. doi: 10.1091/mbc.E07-05-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P., Karton A., Betz A., Peskin A.V, Pace P., O’Reilly R.J., Hampton M.B., Radom L., Winterbourn C.C. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J. Biol. Chem. 2011;286:18048–18055. doi: 10.1074/jbc.M111.232355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y., Sahar S., Astarita G., Kaluzova M., Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M., Imai K., Ito H., Nishiwaki T., Murayama Y., Iwasaki H., Oyama T., Kondo T. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science. 2005;308:414–415. doi: 10.1126/science.1108451. [DOI] [PubMed] [Google Scholar]
- Nangle S.N., Rosensweig C., Koike N., Tei H., Takahashi J.S., Green C.B., Zheng N. Molecular assembly of the period-cryptochrome circadian transcriptional repressor complex. Elife. 2014;15:e30674. doi: 10.7554/eLife.03674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J.S., Reddy A.B. Circadian clocks in human red blood cells. Nature. 2011;469:498–503. doi: 10.1038/nature09702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J.S., Reddy A.B. The essential role of cAMP / Ca2+ signalling in mammalian circadian timekeeping. Biochem. Soc. Trans. 2012;40:44–50. doi: 10.1042/BST20110691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J.S., Maywood E.S., Chesham J.E., Takahashi J.S., Hastings M.H. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science. 2008;320:949–953. doi: 10.1126/science.1152506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J.S., Van Ooijen G., Dixon L.E., Troein C., Corellou F., Bouget F.-Y., Reddy A.B., Millar A.J., van Ooijen G. Circadian rhythms persist without transcription in a eukaryote. Nature. 2011;469:554–558. doi: 10.1038/nature09654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J.S., Maywood E.S., Hastings M.H. Cellular mechanisms of circadian pacemaking: beyond transcriptional loops. Handb. Exp. Pharmacol. 2013;2013:67–103. doi: 10.1007/978-3-642-25950-0_4. [DOI] [PubMed] [Google Scholar]
- Okano S., Akashi M., Hayasaka K., Nakajima O. Unusual circadian locomotor activity and pathophysiology in mutant CRY1 transgenic mice. Neurosci. Lett. 2009;451:246–251. doi: 10.1016/j.neulet.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Ono D., Honma S., Honma K. Cryptochromes are critical for the development of coherent circadian rhythms in the mouse suprachiasmatic nucleus. Nat. Commun. 2013;4:1666. doi: 10.1038/ncomms2670. [DOI] [PubMed] [Google Scholar]
- Papp S.J., Huber A.-L., Jordan S.D., Kriebs A., Nguyen M., Moresco J.J., Yates J.R., Lamia K.A. DNA damage shifts circadian clock time via Hausp-dependent Cry1 stabilization. Elife. 2015;4 doi: 10.7554/eLife.04883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulose J.K., Rucker E.B., Cassone V.M. Toward the beginning of time: circadian rhythms in metabolism precede rhythms in clock gene expression in mouse embryonic stem cells. PLoS One. 2012;7:e49555. doi: 10.1371/journal.pone.0049555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek C.B., Affinati A.H., Ramsey K.M., Kuo H.-Y., Yu W., Sena L. a, Ilkayeva O., Marcheva B., Kobayashi Y., Omura C., et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekovic-Vaughan V., Gibbs J., Yoshitane H., Yang N., Pathiranage D., Guo B., Sagami A., Taguchi K., Bechtold D., Loudon A., et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 2014;28:548–560. doi: 10.1101/gad.237081.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta D., Bronowska A.K., Morgan B., Dóka É., Van Laer K., Nagy P., Gräter F., Dick T.P. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 2015;11:156–163. doi: 10.1038/nchembio.1720. [DOI] [PubMed] [Google Scholar]
- Peskin A.V., Low F.M., Paton L.N., Maghzal G.J., Hampton M.B., Winterbourn C.C. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- Pittendrigh C.S. Circadian rhythms and the circadian organization of living systems. Cold Spring Harb. Symp. Quant. Biol. 1960;25:159–184. doi: 10.1101/sqb.1960.025.01.015. [DOI] [PubMed] [Google Scholar]
- Pizarro A., Hayer K., Lahens N.F., Hogenesch J.B. CircaDB: A database of mammalian circadian gene expression profiles. Nucleic Acids Res. 2013;41:D1009–1013. doi: 10.1093/nar/gks1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prysyazhna O., Rudyk O., Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 2012;18:286–290. doi: 10.1038/nm.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putker M., Madl T., Vos H.R.R., de Ruiter H., Visscher M., van den Berg M.C.W., Kaplan M., Korswagen H.C.C., Boelens R., Vermeulen M., et al. Redox-dependent control of FOXO/DAF-16 by transportin-1. Mol. Cell. 2013;49:730–742. doi: 10.1016/j.molcel.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Putker M., Vos H.R., Dansen T.B. Intermolecular disulfide-dependent redox signalling. Biochem. Soc. Trans. 2014a;42:971–978. doi: 10.1042/BST20140097. [DOI] [PubMed] [Google Scholar]
- Putker M., Vos H., van Dorenmalen K., de Ruiter H., Duran A.G., Snel B., Burgering B.M., Vermeulen M., Dansen T.B. Evolutionary acquisition of cysteines determines FOXO paralog-specific redox signaling. Antioxid. Redox Signal. 2014b;22:15–28. doi: 10.1089/ars.2014.6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radha B.E., Hill T.D., Rao G.H.R., White J.G. GSH levels in human platelets display a circadian rythm in vitro. Trombos. Res. 1985;40:823–831. doi: 10.1016/0049-3848(85)90319-6. [DOI] [PubMed] [Google Scholar]
- Raghuram S., Stayrook K.R., Huang P., Rogers P.M., Nosie A.K., McClure D.B., Burris L.L., Khorasanizadeh S., Burris T.P., Rastinejad F. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat. Struct. Mol. Biol. 2007;14:1207–1213. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainwater R., Parks D., Anderson M.E., Tegtmeyer P., Mann K. Role of cysteine residues in regulation of p53 function. Mol. Cell. Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey K.M., Yoshino J., Brace S.C., Abrassart D., Kobayashi Y., Mercheva B., Hong H.-K., Chong J.L., Buhr E.D., Lee C., et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehder D.S., Borges C.R. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry. 2010;49:7748–7755. doi: 10.1021/bi1008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischl S., Vanselow K., Westermark P.O., Thierfelder N., Maier B., Herzel H., Kramer A. Beta-TrCP1-mediated degradation of PERIOD2 is essential for circadian dynamics. J. Biol. Rhythms. 2007;22:375–386. doi: 10.1177/0748730407303926. [DOI] [PubMed] [Google Scholar]
- Reppert S.M., Weaver D.R. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Ripperger J.A., Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat. Genet. 2006;38:369–374. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
- Robles M.S., Cox J., Mann M. In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS Genet. 2014;10:e1004047. doi: 10.1371/journal.pgen.1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roenneberg T., Merrow M. Life before the clock: modeling circadian evolution. J. Biol. Rhythms. 2002;17:495–505. doi: 10.1177/0748730402238231. [DOI] [PubMed] [Google Scholar]
- Rosbash M. The implications of multiple circadian clock origins. PLoS Biol. 2009;7:0421–0425. doi: 10.1371/journal.pbio.1000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter J., Reick M., Wu L.C., McKnight S.L. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science. 2001;293:510–514. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- Sahar S., Nin V., Barbosa M.T., Chini E.N., Sassone-Corsi P. Altered behavioral and metabolic circadian rhythms in mice with disrupted NAD+ oscillation. Aging. 2011;3:794–802. doi: 10.18632/aging.100368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini C., Liani A., Curie T., Gos P., Kreppel F., Emmenegger Y., Bonacina L., Wolf J.-P., Franken P., Schibler U. Real-time recording of circadian liver gene expression in freely moving mice reveals the phase-setting behavior of hepatocyte clocks. Genes Dev. 2013;27:1526–1536. doi: 10.1101/gad.221374.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini C., Brown S.A., Dibner C. Human peripheral clocks: applications for studying circadian phenotypes in physiology and pathophysiology. Front. Neurol. 2015;6:95. doi: 10.3389/fneur.2015.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.K., Yamada R.G., Ukai H., Baggs J.E., Miraglia L.J., Kobayashi T.J., Welsh D.K., Kay S.A, Ueda H.R., Hogenesch J.B. Feedback repression is required for mammalian circadian clock function. Nat. Genet. 2006;38:312–319. doi: 10.1038/ng1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M., Chandel N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalen I., Reischl S., Wallach T., Klemz R., Grudziecki A., Prabu J.R., Benda C., Kramer A., Wolf E. Interaction of circadian clock proteins CRY1 and PER2 is modulated by zinc binding and disulfide bond formation. Cell. 2014;157:1203–1215. doi: 10.1016/j.cell.2014.03.057. [DOI] [PubMed] [Google Scholar]
- Shao D., Oka S., Liu T., Zhai P., Ago T., Sciarretta S., Li H., Sadoshima J. A redox-dependent mechanism for regulation of AMPK activation by thioredoxin1 during energy starvation. Cell Metab. 2014;19:232–245. doi: 10.1016/j.cmet.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobotta M.C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A.N., Dick T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2014;11:64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- Storz G., Tartaglia L. a, Ames B.N. Transcriptional regulator of oxidative stress-inducible genes: direct activation by oxidation. Science. 1990;248:189–194. doi: 10.1126/science.2183352. [DOI] [PubMed] [Google Scholar]
- Stringari C., Wang H., Geyfman M., Crosignani V., Kumar V., Takahashi J.S., Andersen B., Gratton E. In vivo single-cell detection of metabolic oscillations in stem cells. Cell Rep. 2014;10:1–7. doi: 10.1016/j.celrep.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan M., Yu Z.X., Ferrans V.J., Irani K., Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- Sweeney B., Haxo F. Persistence of a photosynthetic rhythm in enucleated acetabularia. Science. 1961;134:1361–1363. doi: 10.1126/science.134.3487.1361. [DOI] [PubMed] [Google Scholar]
- Tamaru T., Hattori M., Ninomiya Y., Kawamura G., Varès G., Honda K., Mishra D.P., Wang B., Benjamin I., Sassone-Corsi P., et al. ROS stress resets circadian clocks to coordinate pro-survival signals. PLoS One. 2013;8:e82006. doi: 10.1371/journal.pone.0082006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita J., Nakajima M., Kondo T., Iwasaki H. No transcription-translation feedback in circadian rhythm of KaiC phosphorylation. Science. 2005;307:251–254. doi: 10.1126/science.1102540. [DOI] [PubMed] [Google Scholar]
- Ueda H.R. Systems biology of mammalian circadian clocks. Cold Spring Harb. Symp. Quant. Biol. 2007;72:365–380. doi: 10.1101/sqb.2007.72.047. [DOI] [PubMed] [Google Scholar]
- Vassilopoulos A., Fritz K.S., Petersen D.R., Gius D. The human sirtuin family: evolutionary divergences and functions. Hum. Genomics. 2011;5:485–496. doi: 10.1186/1479-7364-5-5-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H., Dessain S.K., Ng Eaton E., Imai S.I., Frye R.a., Pandita T.K., Guarente L., Weinberg R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Wang T.A., Gillette M.U. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science. 2012;337:839–842. doi: 10.1126/science.1222826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster K.A., Prentice H., Bishopric N.H. Oxidation of zinc finger transcription factors: physiological consequences. Antioxid. Redox Signal. 2001;3:535–548. doi: 10.1089/15230860152542916. [DOI] [PubMed] [Google Scholar]
- Welsh D.K., Yoo S.-H., Liu A.C., Takahashi J.S., Kay S.A. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr. Biol. 2004;14:2289–2295. doi: 10.1016/j.cub.2004.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh D.K., Takahashi J.S., Kay S.A. Suprachiasmatic nucleus: cell autonomy and network properties. Annu. Rev. Physiol. 2010;72:551–577. doi: 10.1146/annurev-physiol-021909-135919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn C.C., Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- Winterbourn C.C., Hampton M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Woo H.A., Chae H.Z., Hwang S.C., Yang K.-S., Kang S.W., Kim K., Rhee S.G. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- Woo H.A., Yim S.H., Shin D.H., Kang D., Yu D.-Y., Rhee S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Wood Z.A, Poole L.B., Karplus P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Wu X., Bishopric N.H., Discher D.J., Murphy B.J., Webster K.A., Wu X., Bishopric N.H., Discher D.J., Murphy B.J., Webster K.A. Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol. Cell. Biol. 1996;16:1035–1046. doi: 10.1128/mcb.16.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.-Q., Zhang D., Jin T., Cai D.-J., Wu Q., Lu Y., Liu J., Klaassen C.D. Diurnal variation of hepatic antioxidant gene expression in mice. PLoS One. 2012;7:e44237. doi: 10.1371/journal.pone.0044237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Wright C.J., Hinson M.D., Fernando A.P., Sengupta S., Biswas C., La P., Dennery P.A. Oxidative stress and inflammation modulate Rev-erbα signaling in the neonatal lung and affect circadian rhythmicity. Antioxid. Redox Signal. 2014;21:17–32. doi: 10.1089/ars.2013.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S.-H., Yamazaki S., Lowrey P.L., Shimomura K., Ko C.H., Buhr E.D., Siepka S.M., Hong H.-K., Oh W.J., Yoo O.J., et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. USA. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y., Iigusa H., Wang N., Hasunuma K. Crosstalk between the cellular redox state and the circadian system in Neurospora. PLoS One. 2011;6:e28227. doi: 10.1371/journal.pone.0028227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii K., Tajima F., Ishijima S., Sagami I. Changes in pH and NADPH regulate the DNA binding activity of neuronal PAS domain protein 2, a mammalian circadian transcription factor. Biochemistry. 2015;54:250–259. doi: 10.1021/bi5008518. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Piston D.W., Goodman R.H. Regulation of corepressor function by nuclear {NADH} Science. 2002;295:1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- Zhang E.E., Liu Y., Dentin R., Pongsawakul P.Y., Liu A.C., Hirota T., Nusinow D.A., Sun X., Landais S., Kodama Y., et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 2010;16:1152–1156. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Lahens N.F., Ballance H.I., Hughes M.E., Hogenesch J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA. 2014;111:16219–16224. doi: 10.1073/pnas.1408886111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Wang W., Karapetyan S., Mwimba M., Marqués J., Buchler N.E., Dong X. Redox rhythm reinforces the circadian clock to gate immune response. Nature. 2015;523:472–476. doi: 10.1038/nature14449. [DOI] [PMC free article] [PubMed] [Google Scholar]