

Summary
Establishment and maintenance of CNS glial cell identity ensures proper brain development and function, yet the epigenetic mechanisms underlying glial fate control remain poorly understood. Here we show that the histone deacetylase Hdac3 controls oligodendrocyte-specification gene Olig2 expression, and functions as a molecular switch for oligodendrocyte and astrocyte lineage determination. Hdac3 ablation leads to a significant increase of astrocytes with a concomitant loss of oligodendrocytes. Lineage-tracing indicates that the ectopic astrocytes originate from oligodendrocyte progenitors. Genome-wide occupancy analysis reveals that Hdac3 interacts with p300 to activate oligodendroglial lineage-specific genes, while suppressing astroglial differentiation genes including NFIA. Furthermore, we find that Hdac3 modulates the acetylation state of Stat3, and competes with Stat3 for p300 binding to antagonize astrogliogenesis. Thus, our data suggest that Hdac3 cooperates with p300 to prime and maintain oligodendrocyte identity while inhibiting NFIA and Stat3-mediated astrogliogenesis, and thereby regulates phenotypic commitment at the point of oligodendrocyte-astrocytic fate decision.
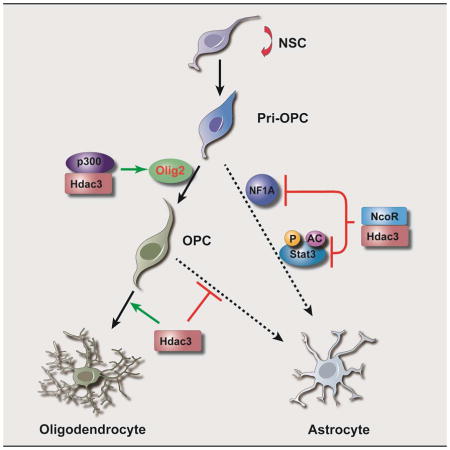
Graphical Abstract
INTRODUCTION
Oligodendrocyte (OL) and astrocytes, the major glial subtypes in the central nervous system (CNS), play key roles in brain development and homeostatic maintenance. They are derived developmentally from neural stem cells, that give rise to transit-amplifying intermediate progenitors. Yet the mechanisms that dictate OL and astrocyte fate choice, and the extent of phenotypic plasticity within these glial lineages, remain poorly understood.
OL precursors (OPCs) are specified from neural progenitors through intermediate primitive OL progenitors (designated as pri-OPC) expressing Olig1 and Olig2 (Lu et al., 2002; Zhou and Anderson, 2002), which precede the expression of the OPC markers PDGFRα and NG2. OL lineage commitment is critical for subsequent differentiation and axonal myelination. OL progenitors have been shown to exhibit multilineage competence, and can adopt an alternative fate such as type-II astrocytes under certain environmental and genetic conditions, both in vitro and in vivo (Kondo and Raff, 2000; Nunes et al., 2003; Raff et al., 1983; Zhu et al., 2012). Nonetheless, the molecules that control the fate choice of OL progenitors and maintain their identity have not yet been fully defined. Activation of the Jak-Stat pathway and BMP-Smad signaling promotes expression of astrocytic genes such as glial fibrillary acidic protein (GFAP), as well as astrogliogenesis from neural progenitors or OPCs in culture (He et al., 2005; Nakashima et al., 1999). The balance of OL- and astrocyte-promoting cues has been proposed to determine lineage specification and progression (Glasgow et al., 2014; He and Lu, 2013; Zuchero and Barres, 2013).
A series of transcriptional regulators have been identified to regulate OL development (Emery, 2010; He and Lu, 2013; Zuchero and Barres, 2013). For instance, Olig2 is a key regulator of OL lineage specification and differentiation (Lu et al., 2002; Takebayashi et al., 2002; Zhou and Anderson, 2002). It can direct Smarca4/Brg1-mediated SWI/SNF chromatin remodeling complex to the enhancers of OL lineage genes to initiate OPC differentiation (Yu et al., 2013). In addition, Olig2 also represses expression of GFAP, and regulates the developmental plasticity of NG2+ OL precursors in developing brain (Cai et al., 2007; Nakashima et al., 1999; Zhu et al., 2012). Although signaling pathways such as Shh and FGF (Gabay et al., 2003; Lu et al., 2000) have been shown to regulate Olig2 expression, the transcriptional and epigenetic events that directly target and activate Olig2 to establish the OL progenitor state are not fully understood.
Histone modifiers, histone acetyltransferases (HAT) and histone deacetylases (HDAC), shape chromatin conformations to control gene transcription during development (Haberland et al., 2009b; Yu et al., 2010). pan-HDAC inhibitors have been shown to block OPC differentiation in vitro (Marin-Husstege et al., 2002). Recent studies point to a critical role for class I HDACs in OL development and regeneration in vivo (Shen et al., 2008; Ye et al., 2009). Yet to date, the individual HDAC-mediated epigenetic machinery in control of glial fate choice and lineage identity has not been fully defined.
The existence of different class I HDAC complexes raises the question of potential specificity in their enzymatic activities and biological functions. In a screen of the effect of specific HDAC inhibitors on OPC differentiation, we found that Hdac3 inhibitors strongly suppress expression of the key OL specification gene Olig2, as compared with other HDAC inhibitors. Although structurally similar among class I HDAC, Hdac3 exerts distinct functions by forming a stable enzymatic complex with its co-factor NCoR1 or NCoR2/SMRT (silencing mediator of retinoic and thyroid receptors), in contrast to Hdac1/2, which primarily form complexes with Sin3, NuRD, or CoREST in mammalian cells (Haberland et al., 2009b).
Here we show that Hdac3 directly targets and activates Olig2 expression to control OL-astrocyte fate decision. Cell-type and temporally-specific fate mapping studies indicate that Hdac3 deletion causes a conversion of OL progenitors into astrocytes, with attendant oligodendrocytic loss and myelination defects. In addition, we find that Hdac3 sets a stage for OL lineage commitment through coordinating with a chromatin-remodeler p300 acetyltransferase while antagonizing Stat3 activity through modulating its acetylation state to inhibit astrogliogenesis. Our genome-wide occupancy study at different stages further offers a global view of gene expression programs regulated by Hdac3 during OL development and provides insights into key mechanisms that may underpin cellular plasticity and fate choice between glial subtypes in the CNS.
RESULTS
Hdac3 regulates Olig2 expression and is highly enriched in the OL lineage
To determine the effects of specific HDAC inhibition on expression of the OL specification factor Olig2, we treated OPCs isolated from P2 rat brains with inhibitors specific to Hdac1, Hdac3 and Hdac8. Treatment with Hdac1 or Hdac8 inhibitors (Hdac1i or Hdac8i), exerts minimal effects on Olig2 expression. However, the Hdac3 inhibitor RGFP966 or apicidin (Malvaez et al., 2013; Wells et al., 2013), which targets the Hdac3 and NCoR complex, strongly suppressed Olig2 expression assayed by immunocytochemistry, qRT-PCR and western blot analyses (Figure 1A–C). This suggests that targeted inhibition of Hdac3 activity blocks Olig2 expression.
Figure 1. Hdac3 regulates Olig2 expression and is expressed in the OL lineage.

A) Olig2 immunostaining in OPCs treated with DMSO, Hdac1i (2.5 μM), Hdac8i (5 μM), RGFP966 (10 μM) or apicidin (100 nM) and counterstained with Dapi.
B,C) Olig2 expression in OPCs treated different Hdac inhibitors over vehicle assayed by qRT-PCR (B) or western blotting (C). Experiments were performed 3 times for each treatment. *** p < 0.001; ANOVA with Newman–Keuls test.
D–F) Primary rat OPCs (D) and differentiated OLs (OL) (E, F) were immunostained for Hdac3, PDGFRα, Mbp and Olig2. Arrows: co-labeling cells.
G–I) immunostaining of Hdac3, CC1 and PDGFRα on the corpus callosum (G) and spinal white matter region (H) in wildtype mice at P14. Arrows and arrowheads indicate the Hdac3 co-labeling cells with CC1 and PDGFRα, respectively. panel I shows the percentage of CC1 or PDGFRα positive cells among Hdac3+ cells (n = 3 animals, > 600 cell counts).
J) Co-immunolabeling of GFAP-GFP with Hdac3 and CC1 in the corpus callosum at P14.
K) qRT-PCR analysis of Hdac3 expression in rat OPCs and differentiating OLs (OL) with T3 treatment for 24 hr (iOL) and 72 hr (mOL).
Scale bars in A, 50 μm; D–F, 25 μm; G, H and J, 50 μm.
See also Figure S1.
Hdac3 was detected in PDGFRα+ OPCs and mature MBP+ (myelin basic protein) OLs (Figure 1D,E), as well as Olig2-expressing OLs in culture (Figure 1F). In the corpus callosum and spinal white matter at P14, strong Hdac3 nuclear labeling was observed in CC1+ differentiating OLs (Figure 1G–H), in contrast to a weak labeling in PDGFRα+ OPCs. The proportions of CC1+ and PDGFRα+ cells among the Hdac3+ cells in the spinal white matter were approximately 85.2 ± 5.8% and 6.0 ± 3.5%, respectively (> 800 cell count; n = 3 animals; Figure 1I). Hdac3 exhibited a weak expression in a population of astrocytes in the corpus callosum marked by GFAP-GFP signals (Zhuo et al., 1997) (Figure 1J), in contrast to the intense Hdac3 signals in OLs (Figure 1J and Figure S1B) and neurons (Figure S1A). Similarly, Hdac3 is expressed weakly or absent in astrocytes marked by Aldolase C (AldoC) and glutamine synthetase (GS) (Figure S1C,D). qRT-PCR analysis indicated that Hdac3 expression level was elevated as OPCs became differentiated and mature OLs (Figure 1K), suggesting that Hdac3 expression increases during OL lineage progression.
Hdac3 mutants have fewer OLs and OPCs, exhibiting myelination defects
To assess the role of Hdac3 in OL development in vivo, we deleted Hdac3 floxed alleles in mice by crossing Hdac3lox/lox mice with an OL lineage-expressing Olig1-Cre line (Xin et al., 2005; Ye et al., 2009) (Figure 2A). The resulting mutant Hdac3lox/lox;Olig1Cre+/− mice (referred to as CKO-Olig1) were born at a normal Mendelian ratio and survived to adulthood. We observed that CKO-Olig1 mice but not their control Hdac3lox/+;Olig1Cre+/− littermates developed tremors from postnatal week 3 and abnormal limb clasping (Figure 2B), typical manifestations of widespread myelination defects in mice. The optic nerve, a myelinated white matter tract, from CKO-Olig1 mice was translucent (Figure 2C), indicative of a severe deficiency in myelin formation in the CNS.
Figure 2. Hdac3 ablation leads to OL loss and myelination deficits.

A) Cre-mediated excision of floxed Hdac3 exons encoding the nuclear import sequence and a carboxyterminal region that confers transcriptional activity.
B) Tail suspension test for control (Olig1Cre+/−;Hdac3lox/+) and CKO-Olig1 (Olig1Cre+/−;Hdac3lox/lox) mice at P60. Mutant mice developed abnormal limb clasps.
C) Appearance of the optic nerves of control and CKO-Olig1 mice at P21.
D) Immunostaining of Hdac3 in the corpus callosum of control and CKO-Olig1 mice at P21.
E–G) Mbp and Plp1 mRNAs or Mbp immunostaining on the brain (E,G) and spinal cord (F) from control and CKO-Olig1 mice at P21.
H–J) Electron microscopy of optic nerves from control and CKO-Olig1 mice at P14 and P62. Panel J shows the percentage of myelinated axons. n = three animals.
K,L) Expression of PDGFRα and Olig2 in the control and CKO-Olig1 brain at P14.
M) GFP signals in the P14 control and CKO-Olig1 brain carrying a PDGFRα-GFP reporter.
N) PDGFRα and BrdU labeling in cortices of control and CKO-Olig1 mice after 2 hr pulse-labeling at P14. Arrow and arrowhead indicate co-labeling cells.
O,P) Histograms depict the relative percentage of Olig2+ or PDGFRα+ (O), or BrdU+ cells among PDGFRα+ OPCs (P). n = 3 animals, > 600 cell counts. (*p < 0.05, Student’s t-test).
Scale bars in D, E–G, 100 μm; H,I, 1 μm. K,L, 100 μm; M,N, 50 μm.
See also Figure S2.
To characterize the myelin-deficient phenotype, we examined myelin gene expression in CKO-Olig1 mice. Immunostaining confirmed that Hdac3 expression was essentially eliminated in the corpus callosum of CKO-Olig1 mice (Figure 2D). At P21, in contrast to robust expression of myelin genes such as Mbp and Plp1 (proteolipid protein 1) in control mice, Mbp and Plp1 was substantially reduced in the cortex and spinal cord of Hdac3 mutants (Figure 2E,F). Similarly, fluorescent intensity of MBP was also diminished in the CKO-Olig1 mutants (Figure 2G).
In light of substantial reduction of myelin genes, we further examined myelin sheath assembly in the CNS of these mutants by electron microscopy. In contrast to the large number of myelinated axons in control mice at P14 (Figure 2H), the percentage of myelinated axons was greatly reduced in the optic nerve of CKO-Olig1 mutants (Figure 2H,J). Strikingly, by P62, myelinated axons were essentially undetectable in the Hdac3-mutant optic nerves (Figure 2I), raising the possibility that Hdac3 is required for myelin maintenance in the adult stage in addition to its role in OL differentiation. Myelinated axons were present in the spinal cord of Hdac3 mutants, however, the thickness of myelin sheaths was substantially reduced in the postnatal stage and adulthood (Figure S2A–D). These observations indicate that Hdac3 is required for OL myelination, and exhibits a region-selective function in myelinogenesis in the CNS.
We next investigated whether alterations of OPC development contribute to myelination defects. In the brain of CKO-Olig1 mutants at P14, PDGFRα mRNAs were detectable, however, their number was significantly reduced in the cortex and corpus callosum (Figure 2K,O). Similarly, Olig2+ cells were also diminished in the cortex (Figure 2L,O). To address the question of whether the presumptive OPCs were maintained but without OPC marker expression, we then bred PDGFRα promoter-driven H2bGFP reporter (PDGFRα-H2bGFP) (Hamilton et al., 2003) into the CKO-Olig1 mutants, and observed that the number of PDGFRα-H2bGFP+ cells decreased in the mutants, suggesting a reduction of PDGFRα+ OPCs in the absence of Hdac3 (Figure 2M).
Despite the reduction in number in the Hdac3 mutants, OPCs were able to proliferate as assayed by BrdU incorporation, though to a lesser extent as compared to the control at P14 (Figure 2N,P). Proliferating BrdU+/PDGFRα+ cells were mainly detected as OPCs with less complex processes (Figure 2N), compared with non-mitotic BrdU−/PDGFRα+ cells, which had extended cellular processes (Figure 2N, arrowhead). Similarly, a population of residual OPCs exhibited complex extended processes as shown by NG2+ immunostaining (Figure S2E). A few NG2+ cells displayed extended processes, which appeared to fill OL void space or make contacts with crossing axon fibers (Figure S2E). The NG2+ cells were co-labeled with PDGFRα but not GFAP (Figure S2F,G), indicating that they were OPCs or immature OLs, not astrocyte-like cells. Together, these observations suggest that Hdac3 deletion results in a loss of OLs and their progenitors, and blocks differentiation of remaining OPCs at a pre-myelinating stage.
Deletion of Hdac3 leads to ectopic astrocyte formation
The reduction of OPCs in Hdac3 deletion in Olig1+ progenitors could be due to death of OPCs or the adoption of an alternative cell fate. We did not detect any significant cell death in mutant brains assayed by the active form of Caspase-3 (Figure S3A). To examine whether deletion of Hdac3 affects other cell types in the CNS, we analyzed the expression of neuronal and astrocyte-specific markers in CKO-Olig1 mice. No substantial alterations of cortical neuron formation or lamination were observed, as marked by the pan-neuronal marker NeuN (Figure S3B). In control mice, expression of the astrocyte markers GFAP and Glast was barely detectable in the cortical gray matter postnatally or in adulthood (Figure 3A,B). In contrast, drastic upregulation of GFAP and Glast was observed in the developing cortex of CKO-Olig1 mice at the perinatal stage, and GFAP+ cells persist throughout adulthood (Figure 3A,B). Consistently, western blot analysis indicated that GFAP protein levels were upregulated in the Hdac3 mutant cortex, whereas Mbp expression was diminished, suggesting a reciprocal relationship of GFAP and Mbp expression (Figure 3C).
Figure 3. Excessive astrogliogenesis in the Hdac3 mutant brain.

A) The cortex of control and CKO-Olig1 mice at P14 and P67 was immunostained with GFAP.
B–D) Expression of Glast mRNA, GS or GFAP or MBP in control and CKO-Olig1 cortices at P14. Arrows indicate labeling cells. Panel C, Gapdh as loading control.
E) Quantification of the number of GS+ astrocytes in control and CKO-Olig1 cortices at indicated stages. n = 3 animals from each stage. (*p < 0.05, Student’s t-test).
F–H) The corpus callosum of control and CKO-Olig1 mutants at P14 was immunostained with NF1A (F) and Sox10 (G). Panel H shows the quantification of the number of NF1A+ or Sox10+ cells/area (0.2 mm2) (n = 3 animals/genotype). Data were presented as mean ± S.E.M. *p<0.05, Student’s t-test.
I) The cortex of CKO-Olig1 mice at P14 was immunostained with GFAP and Ki67 (Arrow).
J) GFAP and PDGFRα expression in cortices of control and Hdac3 CKO-mGFAP mice at P18.
K) The cortex of control and CKO-Olig2 mice at P28 was immunostained with GFAP.
L,M) The cortex of control and CKO-Olig2 mice at P28 was immunostained with Olig2 and S100β (L) and GFAP and S100β (M). Arrows indicate the co-labeling cells.
Scale bars, A,B,J,K;100 μm; D,F,G,I, L,M, 50 μm.
See also Figures S3 and S4.
To determine whether the number of astrocytes was altered in the mutants, we examined the expression of glutamine synthetase (GS), an astrocyte marker independent of GFAP expression (Cai et al., 2007), and detected that the density of GS+ astrocytes in the cortex of Hdac3 mutants significantly increased relative to control mice at perinatal stages and adulthood (Figure 3D,E). A recent study indicates that Sox10 and NFIA antagonize each other to modulate OL and astrocyte subtype differentiation in the spinal cord (Glasgow et al., 2014). Consistently, we observed a reduction of Sox10+ OLs while an increase of NFIA+ astrocytes in the corpus callosum of CKO-Olig1 mutants (Figure 3F–H). To determine whether the increase of ectopic astrocytes could be due to astrocyte proliferation, we then carried out co-immunostaining of GFAP with the proliferative marker Ki67 at P14. Essentially no Ki67 expression was detected in the GFAP+ cells, suggesting very little, if any, proliferation of GFAP+ astrocytes in Hdac3 mutants (Figure 3I).
It is possible that Hdac3 deletion might perturb neuronal formation or astrocyte development, leading to myelination defects and astrocytic reaction. To address these issues, we deleted Hdac3 alleles in developing neurons with a synapsin1-Cre line, in which the neuron-specific Cre activity commences at the early embryonic stage e12.5 (Zhu et al., 2001). In Hdac3lox/lox;Syn1-Cre (CKO-Syn1) mice, we did not observe drastic alterations in expression of the myelin gene Mbp, or the astrocytic marker GFAP (Figure S3C,D) in the brain and spinal cord at P14. In addition, to investigate the potential effects of Hdac3 deletion in the astrocyte lineage, we bred Hdac3lox/lox with a mouse GFAP promoter driven Cre line (mGFAP-Cre), where Cre selectively targets astrocytes (Figure S4A) (Garcia et al., 2004). In the brain of Hdac3lox/lox:mGFAP-Cre (CKO-mGFAP) mice, we did not detect substantial changes in astrocyte formation and GFAP expression (Figure 3J) or OL differentiation (Figure S4B). Together, these data suggest that the effects of Hdac3 ablation on OL development are cell-autonomous and, among neural cells, restricted to the oligodendroglial lineage.
To further confirm that the ectopic astrocyte phenotype arose during OL lineage specification in CKO-Olig1 mice, we crossed Hdac3lox/lox mice with another early OL lineage-expressing Cre line Olig2-Cre (Ligon et al., 2007). Olig2-Cre mediated Hdac3 deletion beginning at the primitive OPC stage, as in CKO-Olig1 mutants, yielded a substantial upregulation of expression of cortical GFAP in the resultant Hdac3lox/lox:Olig2-Cre (CKO-Olig2) mice (Figure 3K), as well as a myelination defect (Figure S4C). To explore this observation, we next assessed the expression of S100β in Hdac3 mutants, since this protein is normally expressed by both OPCs and astrocytes (Karram et al., 2008). In the normal developing cortex, we found that S100β was detected in a population of OPCs expressing Olig2, with few cellular processes (Figure 3L). In the cortex of CKO-Olig2 mutants, we observed a loss of Olig2 expression in these cells, which instead expressed robust S100β with extended bushy cellular processes, and exhibited both astrocytic morphology (Figure 3L) and GFAP expression (Figure 3M). These observations raised the possibility that Hdac3 ablation may cause a conversion of OPCs to astrocytes.
Lineage mapping reveal that Hdac3 ablation diverts OL progenitors to an astrocytic fate
Given the reciprocal decrease in OPCs and increase in ectopic astrocytes with Hdac3 ablation in Olig1+ or Olig2+ progenitors, we hypothesized that Hdac3-deficient Olig+ pri-OPCs or OPCs adopt an astrocytic fate when they fail to commit to the OL lineage. To trace the cell fate of Hdac3-ablated cells, we bred CKO-Olig1 mice with Z/EG reporter mice (Zhu et al., 2012) (Figure 4A). Cre-mediated loxP site excision simultaneously leads to Hdac3 deletion and activates EGFP expression. In the cortex of CKO-Olig1:Z/EG at P14, essentially all EGFP-reporter+ cells expressed GFAP in contrast to the control cortex (Figure 4B,C). The majority of EGFP+ cells exhibited highly branched “bushy” processes characteristic of protoplasmic astrocytes. This suggests that cortical astrocytes with GFAP upregulation or ectopically-induced astrocytes were derived from Hdac3-ablated Olig1+ progenitors.
Figure 4. Hdac3 ablation leads to a conversion of OPCs to astrocytes.

A) A diagram shows Cre-mediated EGFP expression in a Z/EG transgenic line.
B,C) EGFP signals and GFAP+ cells in the cortex of control Z/EG and CKO-Olig1:Z/EG mice. The box area in C was shown in the right. Arrows indicate a GFAP and EGFP co-labeled cell.
D) OPC from Hdac3lox/lox pups were transduced with lentiviral GFP or Cre-GFP and immunostained with Hdac3 and GFP three days post infection. Arrows indicate infected cells.
E) Hdac3lox/lox OPCs were transduced with lentiviral GFP or Cre-GFP and immunostained with GFP and Olig2 three days post infection. Arrows indicate infected cells.
Scale bars in B–D, 50 μm; E, 25 μm.
See also Figure S4.
To examine the fate of Hdac3-ablated OPCs in vitro, we carried out Cre-mediated Hdac3 floxed allele excision in cultures of purified PDGFRα+ OPCs. OPCs isolated from Hdac3lox/lox pups were transduced with the lentivirus expressing GFP or Cre-GFP. After three days post-transduction, GFP-transduced Hdac3lox/lox cells were maintained as Olig2+ OPCs, while the majority of Cre-GFP transduced cells, where Hdac3 expression was depleted, acquired GFAP expression and lost Olig2 expression (Figure 4D, E). Consistently, Hdac3 inhibition by apicidin in purified OPCs elevated GFAP+ astrocyte formation (Figures S5A,B), while causing a decrease in expression of Olig2 and myelination-associated genes (Figure S5C).
To determine whether deletion of Hdac3 could convert OPCs into astrocytes at the postnatal stage, we ablated Hdac3 in OPCs by breeding tamoxifen (TAM)-inducible OPC-expressing PDGFRα-CreERT2 (Rivers et al., 2008) and Rosa-tdTomato reporter lines with the floxed Hdac3 line to generate Hdac3lox/+ or Hdac3lox/lox:PDGFRα-CreERT2:Rosa-tdTomato mice, designated as Ctrl-PDGFRα and iCKO-PDGFRα, respectively. TAM was administered into Ctrl- and iCKO-PDGFRα pups once daily from P5 to P9, during the pre-myelinating stage. GFAP expression in the control and mutant brains harvested at P30 was examined by immunostaining. In the corpus callosum, virtually no GFAP+ cells were marked by tdTomato in Ctrl-PDGFRα mice, in contrast, we found that a population of tdTomato-labeled cells became GFAP+ astrocytes (Figure 5A,C). Similarly, in the cortex, we observed a sizable fraction of tdTomato reporter-labeled GS+ cells (22.8 ± 2.7%) in the Hdac3 mutants compared to only a marginal fraction of these cells (3.1 ± 0.9%) in the control TAM-treated animals (Figure 5B,D).
Figure 5. Fate-mapping analyses reveal that the ectopic astrocytes are derived from OPCs.

A,B) Ctrl-PDGFRα (Hdac3lox/+-PDGFRα-CreERT2:tdT) and iCKO-PDGFRα (Hdac3lox/lox-PDGFRα-CreERT2:tdT) mice were administrated with TAM from P5 to P9 for 5 days and harvested at P30. Arrows indicate GFAP+ (A) or GS+ (B) astrocytes in Hdac3-ablated cells marked by tdTomato expression.
C–F) The percentage of GFAP+ (C), GS+ (D) or CC1+ (E,F) cells among tdTomato+ cells in Ctrl-PDGFRα and iCKO-PDGFRα mice with TAM treatment from P5–P9 and harvested at P30, and TAM treatment from P35–45 and harvested at P90 in the corpus callosum or cortex as indicated.
G) The cortex of CKO-Olig1:PDGFRα-GFP mice at P14 was immunostained with Olig2 and GFAP. Arrows indicate GFAP+ H2bGFP+ Olig2− cells.
H) The optic nerve of control and CKO-Olig1 mice at P21 was immunostained with Olig2 and Aldh1l1 as indicated.
Data were presented as mean ± S.E.M. in the graphs C–F. At least 3 animals from each stage were analyzed. (*p < 0.05, **p < 0.01, ***p < 0.001, Student’s t-test). Scale bars in A,B,G, 25 μm; H, 50 μm.
See also Figure S5.
To investigate whether Hdac3 deletion could alter the fate of OPCs in adulthood, we next treated 5-week old young adult Ctrl- and iCKO-PDGFRα mutant mice with TAM for 10 days (P35-P45) and harvested at P90. In these adults, we detected a population of tdTomato reporter-labeled GFAP+ astrocytes in the white matter of Hdac3 mutant mice, but not in control mice (Figure 5C). Similarly, the percentage of reporter-expressing GS+ astrocytes in the cortex was substantially higher in mutant than control mice in adulthood, although the difference was less marked than that noted in the early postnatal stage (Figure 5D). Conversely, we detected a reduction of CC1+ OLs among tdTomato+ cells in the TAM-induced Hdac3 mutants at perinatal and adult stages (Figure 5E,F), suggesting that ectopic formation of astrocytes is at the expense of OLs in Hdac3 iCKO-PDGFRα mutants.
To further map the fate of OL progenitors in the absence of Hdac3, we took advantage of the stable histone H2b-GFP in CKO-Olig1 mutant mice carrying the PDGFRα-H2bGFP knock-in reporter to trace the fate of OPCs. In these mutant mice, we observed that a population of PDGFRα-H2bGFP+ cells, devoid of the OL lineage marker Olig2, acquired astrocytic GFAP expression in the cortex by P14 (Figure 5G). In contrast, no GFAP+ cells were identified among PDGFRα-H2bGFP+ cells in the control, suggesting that the ectopic astrocytes in the CKO-Olig1 mutant cortex were derived from PDGFRα-expressing OL precursors.
To determine the regional specificity of the astrocyte phenotype, we analyzed the optic nerve, a white matter tract in the CNS, by immunostaining for the astrocyte markers Aldh1l1 and GFAP. We observed a much stronger immunofluorescent signal of Aldh1l1 or GFAP in the CKO-Olig1 mutants than in the controls (Figure 5H and Figure S5D), with a concomitant loss of Olig2+ OLs, suggesting an increase of astrocytes in the optic nerve. Together, our data indicate that Hdac3 deletion induces Olig1/2+ pri-OPCs and PDGFRα+ OPCs to adopt an astrocytic fate in the CNS.
Hdac3 activates OL-lineage while repressing astrocyte-lineage transcriptional programs
To define the Hdac3-regulated transcriptional program that controls glial subtype development, we carried out RNA-seq profiling analysis of the transcriptome between control and CKO-Olig1 mutants using P12 optic nerves, which are highly enriched with OLs and astrocytes. Gene Ontology (GO) analysis of differentially expressed genes demonstrated that significantly downregulated genes in CKO-Olig1 mutants were correlated with lipid biosynthesis and myelination (Figure S6A). Approximately 80% OL-enriched genes in the neural cell type transcriptome database (Cahoy et al., 2008; Zhang et al., 2014) were downregulated in the CKO-Olig1 mutants, including myelin structural genes (e.g. Mbp, Plp1 and CNP) and OL transcription regulators (e.g. Sox10, Myrf/Gm98, Sip1/Zeb2 and Zfp191) (Figure 6A,B). In contrast, up-regulated genes in mutants are associated with astrocyte-enriched genes, including Gfap, Aqp9, Pla2g7 and Atp1a2 (Cahoy et al., 2008; Zhang et al., 2014) (Figure 6A,B). The gene expression alterations are consistent with OL myelination defects and an ectopic increase of astrocytes in CKO-Olig1 mutant animals.
Figure 6. Hdac3 cooperates with p300 to target differentially on the loci of OL and astrocyte-lineage specific genes.

A) RNA-seq transcriptome profiling changes between control and CKO-Olig1 optic nerves. Representative transcripts of OL (red) or astrocyte (blue)-enriched genes were shown.
B) The percentage of annotated astrocyte- and OL-enriched genes based on Transcriptome Database (Cahoy et al., 2008) among differentially expressed transcripts.
C) Heatmap of Hdac3-binding signals at OPCs and mOLs by ChIP-seq. The boxed areas using k-means clustering indicated unique Hdac3 binding loci in OPCs and mOL, respectively.
D) Temporal expression patterns of representative Hdac3-targeted genes in mOL were plotted against differentiation stages from an OL transcriptome database (Dugas et al., 2006).
E) A pie chart shows that Hdac3 targeting sites in OPCs are associated with both astrocyte- and OPC-enriched genes. The others represent Hdac3-targets that are not significantly enriched in OPCs or astrocytes based on the cell-type specific transcriptome profiles (Cahoy et al., 2008).
F,G) Heatmaps show expression patterns of representative OPC-enriched and astrocyte-enriched genes in the transcriptome of control and Hdac3 CKO-Olig1 optic nerves at P14 by RNA-seq.
H) ChIP-seq enrichment shows binding profiles of H3k27ac around Hdac3 peak summits in OPC and mOL.
I) ChIP-seq enrichment analysis shows binding profiles of Hdac3 and p300 around H3k27ac peak summits in OPCs.
J) The representative OPC-enriched gene loci that are targeted by Hdac3, P300 and H3k27ac marks.
K,L) The representative astrocyte-enriched gene loci that are targeted by Hdac3. See also Figure S6.
Genome occupancy analysis uncovers that Hdac3 cooperates with p300 and targets to distinct gene loci between OL and astrocyte lineages
To identify the direct target genes through which Hdac3 regulates oligodendroglial lineage development, we performed ChIP-seq analysis for genome-wide chromatin occupancy by Hdac3 in OPCs and maturing OLs (mOLs). Analysis of target loci revealed that Hdac3 has both common and distinct genomic binding sites in OPCs and mOLs (Figure 6C). The majority of Hdac3 targeting sites were present in evolutionarily conserved intergenic regions, or introns of the gene loci (Figure S6B). Genetic loci targeted uniquely by Hdac3 in mOL were enhancers linked to the genes that are elevated during OL maturation (Figure 6D and Figure S6C). In contrast, in OPCs, a cohort of Hdac3 targeted sites was distinct from those in mOL (Figure 6C). When compared with a transcriptome database of neural cell types (Zhang et al., 2014), Hdac3 targeting was situated within loci of the genes that were mainly enriched in either OPCs or astrocytes (Figure 6E), which were downregulated or upregulated in CKO-Olig1 mutants (Figure 6F,G), respectively.
Approximately 23% of Hdac3-binding sites in OPCs coincided with the enhancer sites marked by the activating histone 3 K27 acetylation mark (H3k27ac) (Creyghton et al., 2010; Yu et al., 2013) (Figure 6H). Since enhancer activities are often associated with p300 histone acetyltransferase binding (Visel et al., 2009), we analyzed p300 genomic occupancy in OPCs using ChIP-seq. Strikingly, we found that these Hdac3-targeted enhancers co-occupied by the transcriptional co-activator p300 were OPC-enriched genes (Figure 6I), including Olig2, Cspg4/Ng2, GPR17 and a chromatin remodeler Smarca2 (Figure 6J) (Zhang et al., 2014). Together with the down-regulation of these OPC-enriched genes in Hdac3 mutants (Figure 6F), these observations suggested that Hdac3 cooperates with p300 to target directly the enhancers of OPC-expressing genes to activate their expression.
In contrast, Hdac3-targeted regulatory elements with low or absent p300 were largely associated with astrocytes including astrocyte-regulatory genes such as NFIA (Figure 6K), a key factor in astroglial specification (Glasgow et al., 2014), and astrocyte-associated genes including Apg9, encoding the water channel aquaporin 9, Gulp1, encoding engulfment adaptor PTB domain containing 1 associated with phagocytosis activity, P4ha2, prolyl 4-hydroxylase alpha 2, and Gpam, encoding mitochondrial glycerol-3-phosphate acyltransferase, all of which are highly enriched in astrocytes (Figure 6L) (Zhang et al., 2014). Given that their expression was upregulated in CKO-Olig1 mutants (Figure 6G), it suggests that targeting by Hdac3 in the absence of the p300 co-activator inhibits expression of NFIA and other astrocyte regulatory genes to block astrogliogenesis.
Hdac3 inhibits Stat3 acetylation to antagonize Jak-Stat3 mediated astrogliogenesis
Activation of the Jak-Stat signaling pathway has been shown to promote astrogliogenesis (Bonni et al., 1997; He et al., 2005). Acetylation of Stat3 at Lys685, a core downstream effector of the Jak-Stat pathway, enhances Stat3 dimerization and its subsequent phosphorylation to activate transcription (Becker et al., 1998; Wang et al., 2005; Yuan et al., 2005). We then examined whether Hdac3 could modulate post-translational modifications of Stat3 in Hdac3 mutants to regulate astrogliogenesis. In the corpus callosum of both CKO-Olig1 and CKO-Olig2 mutants, we detected a drastic increase of Stat3 acetylation (Ac-Stat3) compared with control (Figure 7A). The phospho-Stat3 (P-Stat3 Y705) level, which is responsible for Stat3 nuclear translocation and DNA binding, was also substantially increased (Figure 7A), suggesting a role of Hdac3 for controlling the state of Stat3 post-translational modifications. We further detected a moderate increase of Stat3, consistent with a positive feedback loop mechanism whereby Stat3 activation induces the expression of Jak-Stat signaling components and promotes astrogliogenesis (He et al., 2005). In addition, we detected an upregulation of CNTF (ciliary neurotrophic factor) (Rajan and McKay, 1998) and CT1 (cardiotrophin 1) (Ochiai et al., 2001) in the optic nerve of Hdac3 mutants (Figure 7C). CNTF and CT1 have been shown to activate Stat3 phosphorylation and subsequent astrocytic differentiation through the CNTF/IL6-gp130 receptor-Jak/Stat signaling pathway (Ochiai et al., 2001; Rajan and McKay, 1998). Furthermore, treatment of an Hdac3/NcoR inhibitor apicidin, but not Hdac1 or Hdac8 inhibitors, in purified OPCs, increased P-Stat3 Y705 and acetylation levels (Figure 7B and Figure S7A) while promoting GFAP+ astrocyte formation (Figures S5A,B). These data suggest that Hdac3 inhibition relieves the repression of Stat3 acetylation or the Jak/Stat signaling pathway, permitting redirection of OPCs to astrocytic fate.
Figure 7. Hdac3 regulates Stat3 signaling activity post-transcriptionally to inhibit astrogliogenesis.

A) Western blot analysis of the cortices from control and Hdac3 mutants (CKO-Olig1 and CKO-Olig2) at P68 with Stat3, acetyl-Stat3 (Ac-Stat3), phospho-Stat3 (P-Stat3) and GAPDH.
B) Western blot analysis of Stat3, P-Stat3 and Ac-Stat3 in vehicle or apicidin treated rat OPCs for 8 hr. GAPDH was used as a control.
C) qRT-PCR analysis of CNTF and CT1 expression from mRNAs isolated from the optic nerves of control and CKO-Olig1 mutants at P21 (n = 3 animals, Student t-test; *p < 0.05)
D,E) The corpus callosum of the control and CKO-Olig1 mice at P60 was immunostained with Ac-Stat3, P-Stat3, and GFAP.
F) The luciferase activity from the 293T cells transfected with expression vectors harboring Stat3, p300, or Hdac3 together with a GFAP promoter–driven luciferase reporter after 48 h. * p < 0.05; ** p < 0.01; ANOVA with Newman–Keuls test.
G,H) Co-IP with an antibody to HA-tag p300 was carried out from 293T cells transfected with HA-tag p300 together with Flag-Stat3 and an increased amount of Flag-Hdac3 (H) or Flag-Hdac1, 2 or 3 (H) after 48 h. GAPDH, loading control.
I) Rat astrocytes were transfected with the control, Hdac3, and Hdac3-HEBI expressing vectors. Bar graphs depict that relative expression of myelination-associated genes over control. * p < 0.05; ANOVA with Newman–Keuls test.
J) A proposed model for Hdac3 control of OL-astrocyte fate switch. Hdac3 (ON) interacts with p300 to activate Olig2 and promotes OL lineage progression, while suppressing NFIA and Stat3 activity that otherwise sustains astrogliogenesis. Hdac3 loss (OFF) results in an upregulation of astrogenic programs and a conversion of OL progenitors, including Olig1/2+ pri-OPCs and PDGFRα+ OPCs, into astrocytes.
Data are presented as mean ± S.E.M. from three independent experiments. Scale bars in D, E; 50 μm.
See also Figure S7.
To verify activation of Stat3 in vivo, we immunostained control and Hdac3 mutant mice using Ac-Stat3 and P-Stat3 specific antibodies. Both Ac-Stat3 and P-Stat3 immunofluorescence were dramatically upregulated in the corpus callosum of CKO-Olig1 mutant brain (Figures 7D,E). Together with western blot analysis, these observations suggest that Hdac3 ablation leads to activation of the Stat3-mediated Jak-Stat pathway.
Hdac3 competes with Stat3 for p300 binding to suppress astrocyte phenotype
Stat3 was shown to activate the expression of astrocytic genes, such as GFAP, and to promote astrogliogenesis through cooperation with p300 (Nakashima et al., 1999). To determine the possible mechanisms of Hdac3-driven inhibition of astrogliogenesis, we examined the effect of Hdac3 on GFAP promoter activity. Co-expression of Stat3 and p300 transactivated GFAP-luciferase activity, while co-transfection of Hdac3 significantly blocked Stat3/p300-activated reporter activity (Figure 7F). To determine whether Hdac3 could compete with Stat3 for p300 binding and disrupt the Stat3/p300 complex, we co-transfected expression constructs carrying Stat3 and p300 as well as an increased amount of Hdac3, and performed co-immunoprecipitation with anti-p300 on cell lysates 48 hours after transfection. Consistent with previous studies (Nakashima et al., 1999), p300 was detected in the same complex containing Stat3 in the absence of Hdac3. However, in the presence of a low level of Hdac3, the amount of Stat3 was reduced in the co-immunoprecipitated p300 complex. With an increased level of Hdac3, essentially no Stat3 was detected in the p300 complex (Figure 7G); instead, p300 and Hdac3 were maintained in the complex (Figure 7G). These observations suggest that Hdac3 can hijack p300, effectively competing with Stat3 to bind p300, and thereby inhibit astrogliogenesis.
Strikingly, co-immunoprecipitation assay indicated that Hdac3, but not Hdac1 or Hdac2, interacts specifically with p300 (Figure 7H), suggesting that Hdac3 and p300 uniquely cooperate to provide a fate switch between oligodendrogenesis and astrogliogenesis. Furthermore, Hdac1/2 deletion by the Olig1-Cre line did not cause a substantial increase in GFAP+ astrocytes, or in Stat3 modifications including acetylation and phosphorylation (Figure S7B). In addition, deletion of another class I Hdac, Hdac8, with Olig1-Cre, did not appear to alter OL and GFAP+ astrocyte development (Figure S7C). These data suggest a unique activity for Hdac3 among Hdac family members in regulating astrogliogenesis and Stat3 activity.
To assess the possibility that Hdac3 overexpression might reprogram the transcriptional architecture of astrocytes, we transfected purified astrocytes with control and Hdac3-overexpressing vectors. Hdac3 overexpression induced the expression of OL-associated genes including Olig2, Sox10, Cgt and Mag (Figure 7I). In contrast, expression of a Hdac3 mutant form, Hdac3-HEBI, which lacks deacetylase activity and is unable to form a complex with NCoR/SMRT (Sun et al., 2013), did not alter the expression of OL-associated genes (Figure 7I). These observations suggest that Hdac3 activity is sufficient to activate transcriptional programs characteristic of the OL lineage in astrocytes.
DISCUSSION
Hdac3 is a cell-intrinsic epigenetic switch for the fate choice between OL and astrocyte lineages
The ability of OPCs to form OLs and type II astrocytes has been well established in vitro (Raff et al., 1983), however, there is a lack of convincing evidence showing that astrogliogenesis from OPCs could actually occur in vivo during normal CNS development (Kang et al., 2010; Rivers et al., 2008). This suggests that cell-intrinsic genetic and/or epigenetic factors maintain and restrict OPC fate in vivo. Our lineage tracing studies, using spatiotemporal lineage-specific Cre lines and fate mapping of PDGFRα-H2bGFP cells, suggest that the loss of Hdac3 led to the misdirection of OL progenitors to the astrocyte lineage. This effect was likely cell-autonomous, because a substantial increase of astrocytes was observed when Hdac3 was deleted in the Olig1/2+ pri-OPCs or OPCs, but not when it was deleted from GFAP+ astrocytes, or from neurons during early postnatal development. These findings demonstrate that the chromatin-modifying enzyme Hdac3 is critical for OPC specification and phenotypic maintenance and epigenetically governs oligodendrocyte and astrocyte fate decision.
Type-II astrocytes have been defined as astrocytes derived from OPCs, while type-I astrocytes arise from multipotent neural progenitor cells, but not from OPCs. We observe that a population of GFAP+ astrocytes are derived from pri-OPCs, OPCs as well as PDGFRα-H2bGFP-expressing OPCs in Hdac3 mutants. Hence, we provide an in vivo evidence that GFAP-astrocytes can be derived from OPCs in the absence of Hdac3, and they resemble the type-II astrocytes.
While class I HDACs have been shown to be involved in neural stem/progenitor cell development (Jiang and Hsieh, 2014; Montgomery et al., 2009; Norwood et al., 2014), the present study reveals a distinct role of Hdac3 in glial subtype switching, and in establishment and maintenance of OL identity. This fate switch mechanism is unique to Hdac3, as knockouts of Hdac1, Hdac2, or both together, or Hdac8, do not alter OPC fate or induce astrogliogenesis. Thus, the essential role of Hdac3 in OL-astrocyte fate switch is not shared by other HDACs. Our finding, which implicates a specific HDAC as a regulatory determinant at a well-defined developmental crossroad, challenges previous assumptions relegating class I HDACs to generic cellular machinery or veritable housekeepers involved in chromatin compaction.
Recent studies indicate that Olig2 is a critical regulator of developmental switch between NG2-expressing OPCs and astrocytes (Zhu et al., 2012) and functions as a negative regulator of the astrocytic differentiation pathway (Fukuda et al., 2004). We find that Hdac3 activates and maintains Olig2, which triggers expression of a downstream OL-promoting factor Sox10 (Yu et al., 2013), and primes the progenitor fate toward OL lineage. In addition, we show that Hdac3 can target directly and inhibit expression of NFIA (Figure 6G,K), which is a key transcription factor in the specification of astroglial identity and differentiation (Glasgow et al., 2014). Thus, our data demonstrate that Hdac3 controls the OL-astrocyte fate switch not only by activating an OL specification factor, Olig2, but also suppressing expression of the key astroglial regulator NFIA and Stat3 activity that otherwise sustains astrogliogenesis. Collectively, these data suggest that Hdac3 acts upstream in the regulatory network (Figure 7J) to control phenotypic commitment of OL and astrocyte lineages.
Control of OL-astrocyte lineage commitment by coordinate interaction of HDAC and HAT
Inhibition of class I Hdac activity results in a defect in OL differentiation (Marin-Husstege et al., 2002; Ye et al., 2009). Yet both genetic and RNA knockdown of the HAT Cbp/p300 also lead to decreased expression of OL genes and the inhibition of myelination (Wang et al., 2010). This suggests that both HDAC and HAT activities are required for proper OL development.
How does Hdac3 achieve cell type-specific functions to regulate OL vs. astrocyte lineage? Our data suggest that Hdac3 targets the regulatory regions of OPC-associated and astrocyte-enriched genes in a distinct fashion. Strikingly, Hdac3 appears to coordinate with a co-activator, p300 to target the enhancers of OPC-enriched genes and promote their expression, while Hdac3 targets the loci of astrocyte-enriched genes in the absence of p300, repressing their expression. Given the potential transcriptional repressive function of the Hdac3/NCoR complex (You et al., 2013), the cooperation of Hdac3 with distinct partners such as p300 or NCoR likely controls the selected expression of downstream targets, with the presence of p300 as an activating mark for OL lineage progression. Concurrently, Hdac3 can hijack p300 from the Stat3 complex to inhibit Stat3-mediated astrocytic gene expression (Figure 7J).
Hdac3 has been shown to form a complex with the NCoR1 or SMRT/NcoR2 for its repressive activity (Guenther et al., 2001). NCoR1 suppresses cytokine-induced astrocyte differentiation and inhibits astrocyte lineage-specific gene expression (Hermanson et al., 2002; Sardi et al., 2006). Since NCoR1 can be stably associated with Hdac3 (Guenther et al., 2001), Hdac3/NCoR1 likely acts as a co-repressor complex to repress astrocytic gene expression (Figure 7J). Hence, Hdac3 may possess diverse functions through interacting with distinct co-regulators to refine the chromatin landscape and thereby establish a specific transcriptional program for OL lineage commitment while suppressing astrocytic fate.
Hdac3 regulates Jak/Stat3 activity post-translationally to suppress astrogliogenesis
Besides regulating gene expression, Hdac3 also attenuate the activity of astrogliogenic factors through post-translational modifications. In particular, we find that Hdac3 can deacetylate Stat3, and thereby repress Stat3 activity for astrogliogenesis. Stat3 acetylation is critical for Stat3 dimerization, phosphorylation, and nuclear translocation (Yuan et al., 2005), leading to the activation of downstream transcriptional events. The exact mechanisms underlying the regulation of Stat3 hyper-phosphorylation remain to be determined. It is possible that the increase in acetylation of Stat3 in Hdac3 mutants facilitates Janus kinase (Jak) to phosphorylate Stat3 to promote an astrocytic fate. Alternatively, Hdac3 loss could lead to the activation of Jak/Stat3 signaling indirectly by elevating astrogliogenesis-inducing cues such as CNTF and CT1 (Figure 7C). These observations suggest that Hdac3 negatively regulates the Jak/Stat3 signaling pathway to block astrocytic differentiation.
Hdac3 appears to exert its function through both histone deacetylase-dependent and deacetylase-independent mechanisms (Sun et al., 2013). For histone deacetylase-independent functions, Hdac3 may act as a platform to maintain the integrity of co-repressor complexes, or to modify the activity of other transcriptional machinery (Alenghat et al., 2006; Sun et al., 2013). NCoR1 and SMRT consist of histone deacetylase activation domains (DAD) that enhance Hdac3 histone deacetylase activity. Intriguingly, we find that the deletion of DAD domains of both NCoR1 and SMRT in mutant mice (NS-DADm), which lack histone deacetylase activity in the NCoR/Hdac3 complex (You et al., 2013), does not lead to an OL-astrocyte fate switch and elevate Stat3 acetylation and astrogliogenesis (Figure S7D). This suggests a unique role of Hdac3 in Stat3 post-transcriptional modification. Thus, Hdac3 may function independent of NCoR1/SMRT-mediated histone deacetylase activity, and Hdac3 activity per se or Hdac3/NCoR-mediated transcriptional regulation is required for controlling OL-astrocyte fate switching.
Our data indicate that Hdac3 modulates the fate switch by controlling expression of OL specification gene Olig2 at the pivotal point of the OL vs. astrocyte fate decision. Hdac3 further inhibits activation of Jak/Stat3 activity, which promotes the commitment to an astrocytic fate and astrogliogenesis. These tandem activities of Hdac3 provide it a unique position from which to control the acquisition of glial phenotype, by silencing lineage-inappropriate genes and signaling pathways to ensure that OL identity is heritably maintained. Together, our studies demonstrate that Hdac3 regulates an OL-to-astrocyte fate switch through two levels of mechanisms: first by interacting with p300 to activate Olig2 expression while inhibiting NFIA transcriptionally during early fate specification, and second, by blocking JAK/Stat3 activity at a posttranscriptional level that otherwise promotes astrocytic differentiation.
Hdac3 in demyelinating diseases and CNS injury
Fate switch control is clinically significant, since a misguided fate switch of OPCs into astrocytes may cause depletion of OPC pools, leading to remyelination failure in MS lesions, which are typically pervaded by a dense astroglial milieu (Kotter et al., 2011). We observe a strong up-regulation of Hdac3 expression in the lesion penumbra following lysolecithin-mediated demyelination (Figure S7E), suggesting a potential role of Hdac3 in regulating OL regeneration, although Hdac3 function in remyelination remains to be determined. Inflammatory lesions in particular would be expected to raise cytokines that stimulate the Jak-Stat pathway to promote astrogliosis and glial scar formation at the expense of OLs, thus impeding myelin repair (Burda and Sofroniew, 2014; Gallo and Deneen, 2014). The induction of Hdac3 might permit to the dynamic reversal of astrocytic fate and re-acquisition of oligodendrocytic competence. In addition, our finding of the p300 function in OL fate specification has an implication on the pathogenesis of Rubinstein-Taybi syndrome. A type 2 form of the disease caused by a loss-of-function mutation in EP300 (p300) manifests, along with other systemic abnormalities, hypoplasia of the corpus callosum, reflecting congenital hypomyelination (Solomon et al., 2015). Given this critical role for Hdac3/p300 as a proximal regulator of the OL-astrocyte fate switch, it is conceivable that enhancing Hdac3/p300 interaction or activity might promote or maintain OL identity, while reprogramming astrocytes for OL regeneration. This capability would have broad and important applicability, both in vitro as a means of potentiating and accelerating the production of myelinogenic OPCs from pluripotential stem cells, and in vivo, as a means of directing therapeutically meaningful remyelination by parenchymal progenitors otherwise fated to gliosis.
EXPERIMENTAL PROCEDURES
Full details are provided in the Supplemental Experimental Procedures. Complete protocols available upon request from Richard.lu@cchmc.org
Animals and Experiments
Hdac3lox/lox (Montgomery et al., 2008) and Hdac8lox/lox mice (Haberland et al., 2009a) were from Dr. Eric Olson. Olig2-Cre (Stock No: 011103), Syn1-Cre (Stock No: 003966) and mGFAP-Cre mouse line 77.6 (Stock No. 024098) were from The Jackson Laboratory. PDGFRα-CreERT2 line (Rivers et al., 2008) was from Dr. W. D. Richardson. NS-DADm mice carrying deacetylase activating domain mutations in NCoR1 and SMRT were previously described (You et al., 2013). Phenotypic analyses were performed blindly without prior knowledge of animal genotypes. We used both male and female mice on a mixed C57Bl/6;129Sv;CD-1 background unless otherwise specified, for the study. Immunohistochemistry of tissue and electron microscopy were performed with standard methods. All animal use and studies were approved by the Institutional Animal Care and Use Committee of Cincinnati Children’s Hospital Medical Center, USA.
Primary Cell Culture and Transfection
Primary rat OPCs and astrocyte cultures have been described previously (Chen et al., 2007). Mouse OPCs were purified from transgenic mice by immunopanning as described (Chan et al., 2004). Rat OPCs or astrocytes were transfected with expression vectors by using Amaxa Nucleofector (Lonza) according to the manufacturer’s protocol. The detail information for standard cell culture, immunocytochemistry, qRT-PCR and western blotting and co-IP assays are described in the Supplemental Experimental Procedures.
ChIP-sequencing, Peak-calling and Data Analysis
ChIP-seq assays were performed in the nuclei isolated from OPCs and mOL (3 days after T3 treatment of OPCs) (~20 million cells) using antibodies: Anti-Hdac3 (Abcam ab7030), p300 (Santa Cruz sc-585), Olig2 (Millipore AB9610), Histone 3 Ac-K27 (Active Motif 39133) as previously described with minor modifications (Yu et al., 2013) with details in the Supplemental Experimental Procedures. All sequencing data were mapped to rat genome assembly version rn5 and peak calling was performed using MACS (Model-based Analysis of ChIP-Seq) version 1.4.2 (liulab.dfci.harvard.edu/MACS) with default parameters to get primary binding regions. These primary peak regions were further filtered using following criteria to define a more stringent protein-DNA interactome: 1) the p-value cutoff was set to <10−5; 2) an enrichment of 5 fold.
RNA-seq and Data analysis
RNAs isolated from the optic nerves of control mice and Hdac3 mutants at P12 were subject to RNA sequencing using a Illumina HiSeq 2500 system. RNA-seq reads were mapped using TopHat with default settings. TopHat output data was then analyzed by Cufflinks to (1) calculate FPKM values for known transcripts in mouse genome reference and (2) test for significant changes of gene expression between control and mutant tissues.
Lysolecithin-induced Injury
Lysolecithin-induced demyelination was carried out in the spinal cord of 8-week-old mice as detailed in Supplemental Experimental Procedures. Briefly, 0.5 μL of 1% lysolecithin (Sigma L4129) via a Hamilton syringe attached a glass micropipette was injected into the ventrolateral white matter via a stereotactic apparatus.
Statistical Analysis
All analyses were done using Microsoft Excel or Prism GraphPad 6.00 for Mac OS (www.graphpad.com). Quantifications were performed from at least three independent experimental groups. Data are presented as mean ± S.E.M. in the graphs. p-values are from Student’s two-tailed t test to compare two sets of data. To compare more than two sets, one-way analysis of variance analysis (ANOVA) with a Newman–Keuls multiple comparison test for post-hoc analysis. p < 0.05 is considered to be statistically significant.
Supplementary Material
Highlights.
Hdac3 controls fate decision between oligodendrocyte and astrocyte lineages
Hdac3 regulatory network controls glial subtype-specific transcriptional programs
Hdac3 competes with Stat3 for p300 to activates Olig2 for oligodendrocyte identity
Hdac3 inhibits Stat3 acetylation and antagonizes Jak-Stat3 mediated astrogliogenesis
Acknowledgments
Authors would like to thank Qianmei Li and Guojiao Huang for technical support, and Drs. Kenny Campbell, Masato Nakafuku, Sung Yoon, Ed Hurlock, Yiping Hou for critical comments, and Drs. Bill Richardson for PDGFRα-CreERT2 line, and Ben Deneen for NFIA antibody. This study was funded in part by grants from the US National Institutes of Health R01NS072427 and R01NS075243 to QRL, and the National Multiple Sclerosis Society (NMSS-4727) to QRL.
Footnotes
Accession Code: All the RNA-seq and ChIP-seq data are deposited in the NCBI Gene Expression Omnibus (GEO) GSE76412.
Supplemental Information includes Supplemental Experimental Procedures and seven figures can be found with this article online.
AUTHOR CONTRIBUTIONS
Q.R.L., L.Z., and X.H. designed the experiments, analyzed the data and wrote the manuscript with input from all authors. L.Z., X.H., L.L., M.J., C.Z., H.W., D.H., T.Z, X.Z., and Z.M. carried out the in vitro, in vivo, gene profiling or in silico studies. E.N.O. provided the Hdac3 and Hdac8 floxed mice. Z.S. and M.A.L. provided the NS-DADm mutant mice and expression vectors for Hdac3-HEBI. A.H., M.X., S.A.G. provided intellectual inputs and edited the manuscript. Q.R.L. supervised the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alenghat T, Yu J, Lazar MA. The N-CoR complex enables chromatin remodeler SNF2H to enhance repression by thyroid hormone receptor. EMBO J. 2006;25:3966–3974. doi: 10.1038/sj.emboj.7601280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Chen Y, Cai WH, Hurlock EC, Wu H, Kernie SG, Parada LF, Lu QR. A crucial role for Olig2 in white matter astrocyte development. Development. 2007;134:1887–1899. doi: 10.1242/dev.02847. [DOI] [PubMed] [Google Scholar]
- Chan JR, Watkins TA, Cosgaya JM, Zhang C, Chen L, Reichardt LF, Shooter EM, Barres BA. NGF controls axonal receptivity to myelination by Schwann cells or oligodendrocytes. Neuron. 2004;43:183–191. doi: 10.1016/j.neuron.2004.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, Lu QR. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nature protocols. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci. 2006;26:10967–10983. doi: 10.1523/JNEUROSCI.2572-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B. Transcriptional and post-transcriptional control of CNS myelination. Curr Opin Neurobiol. 2010;20:601–607. doi: 10.1016/j.conb.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Kondo T, Takebayashi H, Taga T. Negative regulatory effect of an oligodendrocytic bHLH factor OLIG2 on the astrocytic differentiation pathway. Cell Death Differ. 2004;11:196–202. doi: 10.1038/sj.cdd.4401332. [DOI] [PubMed] [Google Scholar]
- Gabay L, Lowell S, Rubin LL, Anderson DJ. Deregulation of dorsoventral patterning by FGF confers trilineage differentiation capacity on CNS stem cells in vitro. Neuron. 2003;40:485–499. doi: 10.1016/s0896-6273(03)00637-8. [DOI] [PubMed] [Google Scholar]
- Gallo V, Deneen B. Glial development: the crossroads of regeneration and repair in the CNS. Neuron. 2014;83:283–308. doi: 10.1016/j.neuron.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Glasgow SM, Zhu W, Stolt CC, Huang TW, Chen F, LoTurco JJ, Neul JL, Wegner M, Mohila C, Deneen B. Mutual antagonism between Sox10 and NFIA regulates diversification of glial lineages and glioma subtypes. Nat Neurosci. 2014;17:1322–1329. doi: 10.1038/nn.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Mokalled MH, Montgomery RL, Olson EN. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes & development. 2009a;23:1625–1630. doi: 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009b;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol. 2003;23:4013–4025. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8:616–625. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Lu QR. Coordinated control of oligodendrocyte development by extrinsic and intrinsic signaling cues. Neurosci Bull. 2013;29:129–143. doi: 10.1007/s12264-013-1318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson O, Jepsen K, Rosenfeld MG. N-CoR controls differentiation of neural stem cells into astrocytes. Nature. 2002;419:934–939. doi: 10.1038/nature01156. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Hsieh J. HDAC3 controls gap 2/mitosis progression in adult neural stem/progenitor cells by regulating CDK1 levels. Proc Natl Acad Sci U S A. 2014;111:13541–13546. doi: 10.1073/pnas.1411939111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karram K, Goebbels S, Schwab M, Jennissen K, Seifert G, Steinhauser C, Nave KA, Trotter J. NG2-expressing cells in the nervous system revealed by the NG2-EYFP-knockin mouse. Genesis. 2008;46:743–757. doi: 10.1002/dvg.20440. [DOI] [PubMed] [Google Scholar]
- Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells [see comments] Science. 2000;289:1754–1757. doi: 10.1126/science.289.5485.1754. [DOI] [PubMed] [Google Scholar]
- Kotter MR, Stadelmann C, Hartung HP. Enhancing remyelination in disease--can we wrap it up? Brain. 2011 doi: 10.1093/brain/awr014. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, McMahon AP, Stiles CD, Rowitch DH. Sonic hedgehog--regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron. 2000;25:317–329. doi: 10.1016/s0896-6273(00)80897-1. [DOI] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci U S A. 2013;110:2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Husstege M, Muggironi M, Liu A, Casaccia-Bonnefil P. Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J Neurosci. 2002;22:10333–10345. doi: 10.1523/JNEUROSCI.22-23-10333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci U S A. 2009;106:7876–7881. doi: 10.1073/pnas.0902750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M, Miyazono K, Taga T. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 1999;284:479–482. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- Norwood J, Franklin JM, Sharma D, D’Mello SR. Histone deacetylase-3 is necessary for proper brain development. J Biol Chem. 2014 doi: 10.1074/jbc.M114.576397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G, 2nd, Jiang L, Kang J, Nedergaard M, Goldman SA. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- Ochiai W, Yanagisawa M, Takizawa T, Nakashima K, Taga T. Astrocyte differentiation of fetal neuroepithelial cells involving cardiotrophin-1-induced activation of STAT3. Cytokine. 2001;14:264–271. doi: 10.1006/cyto.2001.0883. [DOI] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303:390–396. doi: 10.1038/303390a0. [DOI] [PubMed] [Google Scholar]
- Rajan P, McKay RD. Multiple routes to astrocytic differentiation in the CNS. J Neurosci. 1998;18:3620–3629. doi: 10.1523/JNEUROSCI.18-10-03620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G. Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell. 2006;127:185–197. doi: 10.1016/j.cell.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, Casaccia-Bonnefil P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008 doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Bodian DL, Khromykh A, Mora GG, Lanpher BC, Iyer RK, Baveja R, Vockley JG, Niederhuber JE. Expanding the phenotypic spectrum in EP300-related Rubinstein-Taybi syndrome. Am J Med Genet A. 2015;167A:1111–1116. doi: 10.1002/ajmg.a.36883. [DOI] [PubMed] [Google Scholar]
- Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, et al. Deacetylase-Independent Function of HDAC3 in Transcription and Metabolism Requires Nuclear Receptor Corepressor. Mol Cell. 2013;52:769–782. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima Y. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol. 2002;12:1157–1163. doi: 10.1016/s0960-9822(02)00926-0. [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Weaver IC, Gauthier-Fisher A, Wang H, He L, Yeomans J, Wondisford F, Kaplan DR, Miller FD. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell. 2010;18:114–125. doi: 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. 2005;280:11528–11534. doi: 10.1074/jbc.M413930200. [DOI] [PubMed] [Google Scholar]
- Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, Chagot B, Cortez D, Khabele D, Chazin WJ, Cooper A, et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One. 2013;8:e68915. doi: 10.1371/journal.pone.0068915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Yue T, Ma Z, Wu FF, Gow A, Lu QR. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J Neurosci. 2005;25:1354–1365. doi: 10.1523/JNEUROSCI.3034-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–187. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Casaccia P, Lu QR. Shaping the oligodendrocyte identity by epigenetic control. Epigenetics. 2010;5:124–128. doi: 10.4161/epi.5.2.11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LM, Mao M, et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 2013;152:248–261. doi: 10.1016/j.cell.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zuo H, Maher BJ, Serwanski DR, LoTurco JJ, Lu QR, Nishiyama A. Olig2-dependent developmental fate switch of NG2 cells. Development. 2012;139:2299–2307. doi: 10.1242/dev.078873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes & development. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Sun B, Zhang CL, Fine A, Chiu SY, Messing A. Live astrocytes visualized by green fluorescent protein in transgenic mice. Dev Biol. 1997;187:36–42. doi: 10.1006/dbio.1997.8601. [DOI] [PubMed] [Google Scholar]
- Zuchero JB, Barres BA. Intrinsic and extrinsic control of oligodendrocyte development. Curr Opin Neurobiol. 2013;23:914–920. doi: 10.1016/j.conb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.