

Abstract
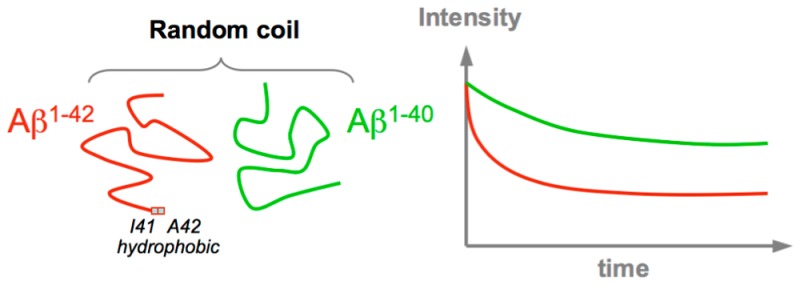
The pathogenesis of Alzheimer’s disease is characterized by the aggregation and fibrillation of amyloid peptides Aβ1–40 and Aβ1–42 into amyloid plaques. Despite strong potential therapeutic interest, the structural pathways associated with the conversion of monomeric Aβ peptides into oligomeric species remain largely unknown. In particular, the higher aggregation propensity and associated toxicity of Aβ1–42 compared to that of Aβ1–40 are poorly understood. To explore in detail the structural propensity of the monomeric Aβ1–40 and Aβ1–42 peptides in solution, we recorded a large set of nuclear magnetic resonance (NMR) parameters, including chemical shifts, nuclear Overhauser effects (NOEs), and J couplings. Systematic comparisons show that at neutral pH the Aβ1–40 and Aβ1–42 peptides populate almost indistinguishable coil-like conformations. Nuclear Overhauser effect spectra collected at very high resolution remove assignment ambiguities and show no long-range NOE contacts. Six sets of backbone J couplings (3JHNHα, 3JC′C′, 3JC′Hα, 1JHαCα, 2JNCα, and 1JNCα) recorded for Aβ1–40 were used as input for the recently developed MERA Ramachandran map analysis, yielding residue-specific backbone ϕ/ψ torsion angle distributions that closely resemble random coil distributions, the absence of a significantly elevated propensity for β-conformations in the C-terminal region of the peptide, and a small but distinct propensity for αL at K28. Our results suggest that the self-association of Aβ peptides into toxic oligomers is not driven by elevated propensities of the monomeric species to adopt β-strand-like conformations. Instead, the accelerated disappearance of Aβ NMR signals in D2O over H2O, particularly pronounced for Aβ1–42, suggests that intermolecular interactions between the hydrophobic regions of the peptide dominate the aggregation process.
Amyloid-β (Aβ) peptides are the 39–43-residue cleavage products of the amyloid precursor protein and represent the main component of senile plaques, which are neuropathological hallmarks of Alzheimer’s disease (AD).1,2 Although the aggregation of Aβ is considered a key step in the development of AD, the nature of the molecular species exerting the neurotoxicity remains a matter of debate.3,4 Accumulating evidence supports the hypothesis that assembly of Aβ into neurotoxic oligomers, and not into mature fibrils, is the seminal event in AD pathogenesis.5−8 From such a perspective, preventing the folding of nascent Aβ monomers into toxic conformers or oligomers could be of great therapeutic benefit.
Many studies have aimed to describe the structures transiently formed by the monomeric soluble Aβ peptides in solution.9−12 The two most hydrophobic regions (L17–A21 and A30–V40) were generally found to have an elevated propensity for β-conformations, while a turn propensity in the central hydrophilic region (E22–G29) has been put forward as the mechanism for bringing the two transient β-strands together.11 In the solid-state nuclear magnetic resonance (NMR) structure of the amyloid fibril, the only charged side chains in the core of the fibril are those of D23 and K28, forming a salt bridge and stabilizing the formation of a turn at G25–G29.13 It is therefore tempting to hypothesize that the formation of fibrils occurs by joining Aβ monomers in their transiently folded forms. Various structural propensities of the amyloid peptides have also been deduced from temperature-induced transitions. In NMR studies of Aβ1–40, J couplings14 and relaxation parameters15 were found to be temperature-dependent while Yamaguchi et al.16 reported that an increase in temperature induces a loss of 15N–1H and 1Hα–13Cα HSQC signal intensities that was most pronounced in the central hydrophilic D23–A30 region. This loss of signal intensity was attributed to chemical exchange line broadening, associated with transient hairpin-like conformations involving residues D23–K30.16 Interestingly, Lazo et al. reported that the A21–A30 region of Aβ1–40 was highly resistant to proteolytic cleavage and that the V24–K28 region of the decapeptide Aβ21–30 adopted a turn conformation.11
A β-hairpin with two β-strands (L17–D23 and G29–V36) connected by a short loop (V24–K28) was also found in a monomeric Aβ1–40 bound to the affibody ZAβ3.17 Subsequently, this group introduced an engineered double-cysteine mutant (AβCC) in which the β-hairpin is stabilized by an intramolecular disulfide bond that was designed on the basis of the structure of Aβ1–40 in complex with ZAβ3.18 Aβ40CC and Aβ42CC both spontaneously form stable oligomeric species with distinct molecular weights and secondary structure content, with both being unable to convert into amyloid fibrils.18 Considering all of these observations, it appears to be generally accepted that the amyloid peptides in solution are in a dynamic equilibrium between random coil conformations and a folded structure with a turn in the D23–A30 region. This scenario is also supported by the finding that oxidation of M35, which reduces the β-structure propensity of Aβ monomers, reduces the level of aggregation and fibril formation.19
Although the Aβ1–40 and Aβ1–42 peptides both are ubiquitous in biological fluids (at an approximate ratio of 9:1), the longer Aβ1–42 is generally considered to be more pathogenic, a conclusion reached on the basis of its higher fractional presence in the amyloid plaques of sporadic AD patients and the stronger in vitro tendency of Aβ1–42 to aggregate and precipitate.20,21 However, most studies have been performed on Aβ1–40 (or even smaller Aβ fragments), because of their greater stability in solution compared to that of Aβ1–42. Thus, less information about the behavior of the longer and more neurotoxic peptide is available. Analysis of the 15N relaxation properties led to the conclusion that the C-terminus of Aβ1–42 is more rigid than that of Aβ1–40, which has been interpreted by Yan and Wang as a sign of β-conformation preordering at the C-terminus of Aβ1–42.22 These authors hypothesized that the C-terminus of Aβ1–42 may thereby serve as an internal seed for aggregation. A more structured C-terminus of Aβ1–42, compared to that of Aβ1–40, was also observed in replica exchange molecular dynamics (REMD) simulations.23
In the study presented here, we use solution NMR to systematically compare the structural propensities of Aβ1–40 and Aβ1–42 peptides at neutral pH. On the basis of comparisons of the backbone 1H, 15N, and 13C chemical shift as well as 3JHNHα values, we find that the monomeric forms of these two peptides are virtually indistinguishable. Analysis of the secondary chemical shifts shows that both peptides are highly disordered under our conditions. Two-dimensional (2D) nuclear Overhauser effect spectroscopy (NOESY) spectra collected at very high resolution and sensitivity show no unambiguous long-range NOE contacts that would be indicative of transiently populated ordered species. Analysis of the residue-specific backbone angles of Aβ1–40 in terms of Ramachandran maps, using the recently developed MERA program, shows only modest deviations from random coil library distributions, without a strongly elevated propensity for β-conformations in the hydrophobic region of the peptide over what would be expected on the basis of nearest neighbor effects.24
Materials and Methods
Sample Preparation
Uniformly 15N-labeled and 15N- and 13C-labeled Aβ1–40 and Aβ1–42 peptides were purchased from Alexotech (Umea, Sweden) and used without further purification. In this study, all experiments were performed on samples containing 150 μM peptides in 20 mM sodium phosphate buffer at pH 7.0. To dissolve the peptide, we followed the protocol of Teplow and co-workers:25 The chilled powder peptide is first dissolved in 10 mM NaOH (2 mg/mL) and sonicated in a cold-water bath for 1 min. The sample is then diluted 5-fold with pH 6.6 buffer to reach a final pH of 7.0 and sonicated for an additional 1 min.
Acquisition of NMR Data
All NMR data were collected at 4 °C using Aβ1–40 and Aβ1–42 samples at a peptide concentration of 150 μM in 20 mM sodium phosphate buffer at pH 7.0. All NMR data were processed using NMRpipe26 and analyzed with NMRDraw26 and Sparky.27 Resonance assignments were obtained from three-dimensional (3D) HNCA and HNCO spectra recorded on a 600 MHz Bruker Avance III spectrometer equipped with a z-axis gradient QCI cryogenic probe. The 15N indirect dimension was acquired in the mixed-time manner28 for both experiments, and the 13C dimension in the HNCA experiment was recorded using a 28 ms constant-time evolution period, thereby eliminating 1JCαCβ splittings.
The 3JHNHα couplings were determined from WATERGATE-optimized 15N–1H TROSY-HSQC spectra, as recently described,29 recorded at 800 MHz with an acquisition time in the direct dimension of 252 ms. The 1JCαHα couplings were measured from a 13C–1H HSQC spectrum recorded at 900 MHz using a 56 ms constant-time evolution period. The 2JNCα and 1JNCα couplings were measured using the sensitivity-enhanced experiment described by Ding and Gronenborn,30 conducted at a 1H frequency of 800 MHz. The 3JC′C′ couplings were derived from a 3D HN(COCO)NH spectrum,31 recorded at 500 MHz. 3JC′Hα couplings were measured at 600 MHz from a four-dimensional (4D) HACANH[C′] E.COSY spectrum.32 Nonuniform sampling with 5% sparsity was employed for a total acquisition time of 4 days.
3D NOESY-HSQC spectra were recorded with a mixing time of 250 ms on a Bruker Avance III 900 MHz spectrometer equipped with a z-axis gradient TCI cryogenic probe. In addition to the 3D NOESY-HSQC spectra, a 2D Hα–HN NOESY spectrum with band-selective homonuclear (BASH) decoupling33 in the indirect Hα dimension was recorded at 900 MHz, using a mixing time of 200 ms. The Hα band-selective pulse for the BASH decoupling had a duration of 2 ms, centered at the water resonance, and a REBURP profile.34
The aggregation kinetics of the Aβ1–42 peptide in either H2O or D2O solvent was determined from a series of 28 ms constant-time 1H–13C spectra recorded periodically at 500 MHz.
ThT-Detected Experiments
The lyophilized peptides were first dissolved in 10 mM NaOH (2 mg/mL), sonicated in a cold-water bath for 15 min, and then diluted with a sodium phosphate buffer to form a stock solution with a peptide concentration of 20 μM in 25 mM sodium phosphate at pH 7.4 (or pD, uncorrected pH meter reading using a glass electrode) in either H2O or D2O solvent. The samples used for these experiments contained 5 μM Aβ1–40 and Aβ1–42 in 25 mM sodium phosphate at pH (or pD) 7.4 (H2O or D2O solvent) with 100 μM thioflavin T (ThT) and were filtered with an Amicon centrifugal filter unit (cutoff of 100 kDa) just before the experiments were conducted. A Tecan Magellan microplate reader was used for these experiments, with excitation at 415 nm and detection at 480 nm. The plate was maintained at 37 °C and continuously shaken at 434 rpm. Four replicas of each sample were disposed in the same microplate, and results were averaged.
Results and Discussion
Although there have been extensive prior solution NMR studies of both Aβ1–40 and Aβ1–42 peptides, both the raw data and the interpretation of these data varied substantially. For this reason, these earlier measurements were repeated for both peptides using standardized conditions and the most robust experimental schemes currently available, and the data were supplemented by multiple types of J couplings that have not yet been reported.
Chemical Shift Comparison
Both the monomeric Aβ1–40 and Aβ1–42 peptides yield well-dispersed 15N–1H HSQC spectra with no significant resonance overlap at 277 K and pH 7.0 (Figure 1A). Because of rapid amide hydrogen exchange with water, the amide cross-peaks of residues D1 and A2 are not visible, while those of H6, H13, and H14 are considerably attenuated. The 1H, 15N, and 13C backbone chemical shift assignments were completed using 3D NOESY-HSQC, HNCO, and HNCA spectra. Except for several small outliers, mostly for His residues and reflecting small pH differences, the secondary 13Cα chemical shifts, often considered to be most indicative of secondary structure, are in closest agreement with literature values of Yamaguchi et al.16 for Aβ1–40 [root-mean-square deviation (rmsd) of 0.064 ppm (Figure S1A)] and Waelti et al.35 for Aβ1–42 [rmsd of 0.083 ppm (Figure S1B)]. The differences between the observed chemical shifts and the corresponding residue-specific random coil values, often termed secondary chemical shifts, Δδ, are commonly used as sensitive indicators of local secondary structure. With rmsd’s of 0.018, 0.048, and 0.007 ppm for the 13C′, 13Cα, and 1Hα nuclei, respectively (Figure 1B–D), Aβ1–40 and Aβ1–42 show very similar secondary chemical shifts for the first 34 residues. Small differences become apparent only when the C-termini of the two peptides are approached, starting with a 0.1 ppm difference in Δδ(Hα) for M35. Our chemical shift values closely match those reported by Hou et al. for the nonoxidized state of M35,19 and indeed, inspection of 1H–15N HSQC spectra (Figure 1A) shows the absence of any cross-peaks at positions that would correspond to those reported by Hou et al.19 for the oxidized form of the peptide, indicating an ∼2% upper limit for the presence of the oxidized form.
Figure 1.

(A) Overlay of the 15N–1H HSQC spectra recorded at 800 MHz for monomeric Aβ1–40 (red) and Aβ1–42 (black) peptides at 277 K. Assignments of the backbone amide cross-peaks are colored gray for residues with nearly identical chemical shifts in the two peptides, while labels in red and black (for Aβ1–40 and Aβ1–42, respectively) correspond to residues with significantly different chemical shifts in the two peptides. Secondary chemical shifts for (B) 13C′, (C) 13Cα, and (D) 1Hα nuclei of Aβ1–40 (red) and Aβ1–42 (black) were derived using random coil values and correction factors of Poulsen and co-workers.36,37
The small Δδ(Hα) differences therefore reflect subtle differences in the distribution of backbone angles sampled by M35 in the two peptides, also reflected in a small difference in their respective 3JHNHα values (see below), but the differences in the other M35 backbone chemical shifts are remarkably small between the two peptides. The absence of chemical shift differences between Aβ1–40 and Aβ1–42 prior to M35 suggests that the two additional C-terminal residues, I41 and A42, are not substantially engaged in long-range interactions with the 34 N-terminal residues of the peptide. Typical chemical shift changes for a random coil peptide upon adoption of a stable interaction are on the order of several parts per million for 13C, and the observed chemical shift differences between the two peptides are ∼2 orders of magnitude smaller, indicating that the long-range interactions involving the C-terminal residues of Aβ1–42 are unlikely to be populated at a level much greater than a few percent. Importantly, the C-terminal residues of Aβ1–42 also show only minimal deviations from random coil chemical shift values, suggesting the absence of any particular propensity for secondary structure for this region (Figure 1B–D).
Overall, with root-mean-square (rms) values of only 0.36, 0.32, and 0.083 ppm for Δδ(13C′), Δδ(13Cα), and Δδ(1Hα), respectively, for residues 2–39 of Aβ1–40, these three types of secondary chemical shifts are remarkably small. Notably, by using the random coil values of Poulsen and co-workers, adjusted for pH, ionic strength, and temperature, the rms value of only 0.32 ppm we obtained for Δδ(13Cα), which is generally considered the best marker of local secondary structure, is very close to that calculated from the 13Cα chemical shifts reported previously by Hou et al. [rmsΔδ(13Cα) = 0.33 ppm].19,38 These chemical shift data therefore strongly suggest, but do not conclusively prove, that the population of any ordered structural elements is very small.
Any differences in secondary chemical shifts between Aβ1–40 and Aβ1–42 are yet another order of magnitude smaller than the secondary chemical shifts themselves. If one of the two peptides would transiently adopt an ordered structure, not populated by the other peptide, the contribution of this transiently ordered conformer to the chemical shifts would be proportional to its population. Ordered structural elements, such as α-helices, β-sheets, or turns, typically exhibit secondary chemical shifts of ca. 0.3–1 ppm for 1HN and 1Hα and 1–4 ppm for 13Cα, 13C′, and 15N.39 The largest chemical shift differences (excluding the highly pH-sensitive His residues) between the two peptides are more than 20-fold smaller than these values, indicating an upper limit of ∼5% for the population of any transiently ordered conformer present for one peptide but not the other. This result suggests that the difference in aggregation kinetics of the two peptides is unlikely to be dominated by their differences in secondary structure propensity. Nevertheless, we will attempt to make a quantitative interpretation of the weak local structural preferences that both peptides have in common, which may or may not contribute to their shared ability to form amyloid.
Analysis of Three-Bond J Couplings
Three-bond 3JHNHα couplings are related to backbone torsion angles ϕ by the empirically parametrized Karplus equation.40,41 In particular, when protein structures are refined by residual dipolar couplings, resulting in backbone dihedral angles that are known at high accuracy, very tight correlations between predicted and observed 3JHNHα couplings can be obtained, yielding rmsd values between observed and predicted values of <0.4 Hz.42,43
In the past, 3JHNHα values have been used extensively to study structural preferences in the Aβ peptides35,38,44,45 and to validate conformational ensembles.46,47 However, the spread in 3JHNHα couplings in disordered systems such as the Aβ peptides is much smaller than in folded proteins, making precise measurement of these values more important. Although the 3JHNHα values can be measured at very high precision (<0.05 Hz) from the cross-peak intensity modulation in a series of constant-time 1H–15N HMQC spectra48,49 or related three-dimensional NMR spectra,50 we here use a simpler method in which the splitting is measured directly from a slightly modified 15N–1H TROSY-HSQC spectrum,51 recorded with a long 1H acquisition time (>250 ms) to take advantage of the favorable transverse relaxation properties of the 1HN TROSY signal at high field (800 MHz).52,53 This approach yields well-resolved doublets for nearly all amide protons (Figure 2A and Table S1), from which 3JHNHα values can be measured at a precision that is limited by only the available signal-to-noise ratio. Indeed, the weaker cross-peaks of residues experiencing rapid hydrogen exchange result in higher uncertainties for the extracted 3JHNHα values (see, for example, the larger error bar for H13 in Figure 2B). As expected,54,55 the smallest 3JHNHα couplings are observed for Ala residues and the largest values for β-branched residues, notably, V18, I31, V39, and V40.
Figure 2.
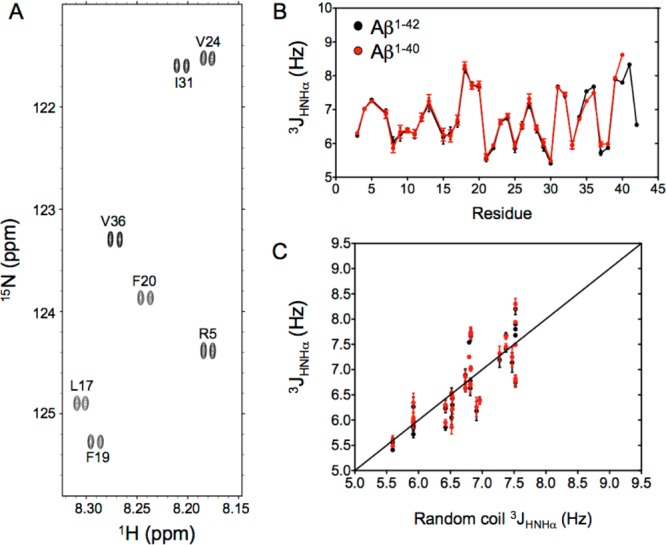
3JHNHα couplings measured in the Aβ peptides. (A) Small expanded region of the 15N–1H TROSY spectrum recorded at a 1H frequency of 800 MHz for the Aβ1–42 peptide showing the well-resolved doublets of cross-peaks arising from the J coupling between the 1HN and 1Hα protons. (B) 3JHNHα coupling values measured for the Aβ1–40 (red) and Aβ1–42 (black) peptides at 277 K. (C) Plot of 3JHNHα measured for Aβ1–40 (red) and Aβ1–42 (black) against residue-specific random coil values, derived from α-synuclein.55
With a pairwise rmsd of only 0.10 Hz, the reproducibility between the Aβ1–40 and Aβ1–42 values is considerably higher than that seen previously (Figure S2) and approaches the intrinsic precision of the measurement. This observation indicates that there are no meaningful differences for the first 34 residues of the two Aβ peptides, a result that is perhaps not surprising given the high degree of similarity in chemical shifts. By contrast, a small increase of ∼0.45 Hz in the 3JHNHα value of M35 in Aβ1–42 over its value in Aβ1–40 is well outside the measurement uncertainty and is indicative of a change in the distribution of the ϕ angles of this residue, also reflected in a distinct upfield change of 0.1 ppm in the 1Hα chemical shift. Overall, the 3JHNHα coupling constants measured here for Aβ1–40 and Aβ1–42 show an rmsd of only 0.41 Hz from their residue-specific random coil values (Figure 2C), nearly 40% lower than that previously reported relative to a replica exchange molecular dynamics study of Aβ1–42,46 and 25–58% lower compared to a number obtained for other free or experimentally restrained ensemble models generated for Aβ1–40 and Aβ1–42.47
For disordered systems, such as the Aβ peptides, the 3JHNHα couplings correspond to the time average of the values sampled over the duration of the measurement, i.e., on the time scale of seconds. To a first approximation, the ϕ distribution for residue i may be considered Gaussian, with an average ⟨ϕi⟩ and a standard deviation σi. Clearly, with the 3JHNHα coupling being dependent on both ⟨ϕi⟩ and its standard deviation, σi,56 this coupling alone cannot distinguish between a static ϕ value and a dynamic ensemble. However, we have recently shown that 3JC′C′ couplings represent a valuable complement to 3JHNHα and their combined use can define both ⟨ϕ⟩ and σ.57 Although the range of 3JC′C′ couplings is much smaller than that for 3JHNHα, their rmsd from a best-fit Karplus equation in proteins of known structure is correspondingly smaller too, making these couplings at least as valuable as 3JHNHα in defining molecular structure.31 A plot of 3JHNHα versus 3JC′C′ couplings yields both ⟨ϕ⟩ and σ (Figure 3A) and shows that all residues undergo quite large ϕ angle fluctuations, ranging from σ ≈ 23° for β-branched residues V18, I31, and V39 to ∼40° for S8 and A21. At first glance, this finding appears to contradict the presence of highly populated regions of secondary structure such as helices and turns, reported in previous studies.23,38,44,47,58,59 However, transient population of such secondary structure elements is not necessarily inconsistent with our new data, provided that the population of each such element falls well below 50%. The most negative ⟨ϕ⟩ values, ca. −110° (Figure 3A), are observed for V18, F19, and F20, indicating that even though this short stretch of residues, located in the central hydrophobic cluster (CHC, residues L17–A21), is quite dynamic, it also is more extended than the remainder of the peptide. The two flanking hydrophobic residues, L17 and A21, exhibit less negative ⟨ϕ⟩ and large σ values, indicating that if the CHC has a propensity for β-strand formation, its length is restricted to only the center three residues.
Figure 3.
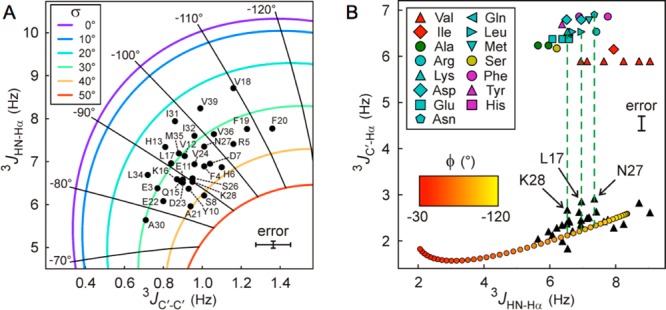
Analysis of residue-specific Aβ1–40 ϕ angles from combinations of 3J couplings. (A) ⟨ϕ⟩ and its standard deviation, σ, are obtained from 3JHNHα and 3JC′C′ values. Black dots with labels correspond to pairs of experimental 3J couplings. Radial spokes and colored contours correspond to iso-ϕ and iso-σ lines, respectively, with the ϕ value labeled at the end of each spoke and the color code of σ values displayed in the inset. The average measurement uncertainties based on signal to noise are ±0.08 and ±0.09 Hz for 3JHNHα and 3JC′C′ couplings, respectively. (B) Fractional population of positive ϕ angles (P+) obtained from 3JC′Hα and 3JHNHα coupling values. Black triangles indicate the measured values, and notable residues are labeled. The red to yellow bottom line shows the expected correlation between 3JC′Hα and 3JHNHα Karplus equations assuming σ ≈ 30° for IDPs, if no positive values of ϕ were sampled. The top markers represent predicted 3JC′Hα values when using only the ϕ > 0 fraction of each residue-specific coil library. Interpolation of the data points between the top markers and the bottom orange line yields the residue-specific P+. Residues with poor reproducibility in two independent measurements (H6, H13, and S26, because of lower signal to noise) are excluded.
In strict terms, the 3JHNHα versus 3JC′C′ analysis described above is valid only under the assumption that the population of conformers with positive ϕ angles is vanishingly small,57 i.e., precluding the presence of type I′, type II, or type II′ β-turns. However, as we recently demonstrated, the fraction of time any given residue samples the positive ϕ region of Ramachandran space is readily quantified from the combination of 3JHNHα and 3JC′Hα values.32 Such an analysis confirms that indeed, with the possible exceptions of L17, N27, and K28, all non-Gly residues in Aβ1–40 have vanishingly small populations of positive ϕ angles (Figure 3B). The ∼13% population of a positive ϕ angle population seen for both L17 and K28 is too small to have a significant impact on the ⟨ϕ⟩/σ analysis of Figure 3A but suggests that these residues are located at the center of transient β-turns. The same applies for N27, but the ∼13% population of positive ϕ angles seen for this residue is less surprising given the elevated propensity for Asn residues to adopt such values in random coil libraries.60,61 Interestingly, N27 is the only residue adopting a positive ϕ angle in an Aβ1–42 fibril structure, determined from hydrogen bonding restraints derived from quenched hydrogen/deuterium exchange NMR and side-chain packing restraints that were obtained from pairwise mutagenesis studies [Protein Data Bank (PDB) entry 2BEG].62
Analysis of NOESY Spectra
Several types of NOESY spectra were collected, using the same conditions for Aβ1–40 and Aβ1–42. A very high resolution for the HN–Hα region of the 2D spectrum was obtained by using band-selective homonuclear decoupling in the F1 dimension of the spectrum.33,63 Combined with the advantage of high field (900 MHz), this yielded a fingerprint region with much reduced spectral overlap compared to that of prior such measurements, thereby removing much of the ambiguity in the spectral interpretation. As can be readily seen (Figure 4A), not only the cross-peak positions but also their relative intensities are very similar in the two peptides. For all residues, we find that the sequential Hα–HN(i–1,i) NOE intensity, daN(i–1,i), is considerably stronger than the intraresidue daN(i,i) NOE, as expected for the mostly extended backbone conformations seen in coil libraries.60,61 The daN(i,i)/daN(i–1,i) ratios obtained for Aβ1–40 average 0.29 ± 0.10 (Figure 4C), values comparable to those reported for α-synuclein,64 widely considered a prototypical IDP. Although, with the exception of the exchange-broadened His residues, sequential HN–HN NOEs are observed for virtually every pair of amides (Figure 4B), these NOEs are ∼3-fold weaker than daN(i,i), again typical of what is seen in α-synuclein or short unstructured peptides, and excluding the possibility of high fractional populations of type I or type II′ β-turns that should give rise to strong dNN(i,i+1) NOEs.
Figure 4.

Nuclear Overhauser data recorded for the Aβ peptides. (A) Expanded region of the 2D NOESY spectrum recorded at 900 MHz for the Aβ1–40 (red) and Aβ1–42 (black) peptides at 277 K. The cross-peaks correspond to sequential and intraresidue interactions between the 1HN and 1Hα protons. Significant chemical shift differences between Aβ1–40 and Aβ1–42 are seen in this region for the intraresidue HN–Hα(i,i) M35 and sequential Hα–HN(i,i+1) M35-V36 cross-peaks. (B) Expanded region of the 2D projection from the 3D NOESY-HSQC spectrum recorded at 900 MHz for Aβ1–40, showing the HN–HN region. (C) Ratio between the intraresidue daN(i,i) NOE intensity and the sequential Hα–HN(i–1,i) NOE measured for Aβ1–40 and reported as a function of residue number. (D) Correlation between the intraresidue daN(i,i) NOE intensity and the 15N transverse relaxation rate measured at a 1H frequency of 600 MHz.
Interestingly, as was previously reported for α-synuclein,55 a strong correlation is seen between the intraresidue daN(i,i) NOE intensity and the transverse relaxation rate of 15N (Figure 4D). The latter is dominated by J(0) spectral density, whereas daN(i,i) is proportional to the product of J(0) and rHNHα–6. Considering that the intraresidue rHNHα distance varies relatively little in the most populated region of the coil library, a strong correlation with J(0) is not surprising, but clearly this result highlights that quantitative interpretation of NOE intensities, for example, through eNOE analysis,35,65 is a challenging undertaking. A second factor complicating the quantitative interpretation of NOE intensities relates to the high degree of motional anisotropy in the dynamics of a random coil, where motions orthogonal to the Cα–Cα chain direction are much faster than reorientation of the Cα–Cα vector itself.55 These considerations highlight the fact that quantitative interpretation of NOE intensities for highly dynamic systems such as the Aβ peptides remains a difficult problem. A potential solution to this time scale dependence for IDPs considers both the time and distance dependence of the 1H–1H dipolar interaction autocorrelation function, which is accessible when analyzing a molecular dynamics trajectory.38,47,66,67 Although elegant, we note that this latter solution transfers the burden of the variable time dependence of the autocorrelation function to the accuracy of the molecular dynamics calculations and the rates at which conformational transitions take place. The latter tends to remain one of the most challenging problems when generating optimal force fields, and further development appears to be needed before quantitative analysis of IDP molecular dynamics trajectories becomes suitable for routine quantitative interpretation of NOE intensities.
Although the presence of long-range NOEs, between residues more than five apart in the sequence of the peptide, have been inferred from analysis of the 2D NOESY spectra of Aβ1–40 and Aβ1–42, often these NOEs could not be uniquely assigned because of the lack of sufficient resonance dispersion in the spectra of these disordered peptides.38 Only very few nonsequential NOEs in that study were found to be consistent with the analysis of their molecular dynamics trajectory, and for Aβ1–40, all of these corresponded to i to i + 2 NOEs. A much larger number of “false-negative long-range NOEs” were reported by the same group in an earlier study,58 referring to NOEs identified in the spectrum but absent in the dynamics simulation. Many of these long-range NOE cross-peaks fall in very crowded regions of the spectrum, but searching for those that should be unambiguously identifiable in our 900 MHz spectrum did not reveal support for the presence of these interactions, despite quite good spectral quality (see, e.g., Figures S3 and S4). In other studies, the 21–30 peptide fragment of Teplow and co-workers had shown a weak NOE between A30 HN and E22 Hα, partly overlapping with an approximately equally weak NOE between A30 HN and K28 Hα, but a higher-field study by Fawzi et al. of the same peptide revealed only the A30 HN to K28 Hα interaction.67 Indeed, both our 2D NOESY spectra and the 3D 15N-separated NOESY spectra show a weak but clear A30 HN to K28 Hα NOE, ∼6-fold weaker than the intraresidue A30 daN(i,i) NOE, and the proposed A30 HN and E22 Hα interaction, which would be well resolved in our 900 MHz spectrum, falls below the noise threshold; i.e., it must be >3-fold weaker than the already weak A30 HN to K28 Hα NOE.
Comparison of these prior NMR results with our newly acquired data made it abundantly clear that such an analysis is very difficult without access to explicitly annotated spectra. Although inclusion of such data is no longer common practice in the biological NMR literature, a relatively recent 900 MHz 2D NOESY study of Aβ1–40 did identify a number of long-range NOE interactions and included detailed annotated spectral regions to support these observations.59 On the basis of their data, these authors concluded that Aβ1–40 at least transiently adopts a compact, partially folded structure with a long helical segment spanning H13–D23, and long-range interactions between the F4 aromatic ring protons and hydrophobic side chains in the V18–V24 segment driving the compaction of the N-terminal segment on this region, presumed to be helical on the basis of local NOEs. An NOE between F19 and G38 Hα protons resulted in compaction of the C-terminal segment against this center helical segment. These spectra were recorded on the synthetic peptide at an ionic strength (50 mM NaCl) higher than that of our data, which can impact the structural distribution of the dynamic Aβ1–40 peptide.35 However, inspection of the annotated spectrum of Vivekanandan et al. showed chemical shifts very similar to those seen in our spectrum and also revealed alternate short-range assignments that better matched the NOE cross-peak positions in our spectrum (Figure S4). For example, the partial overlap of F4 Hδ and F19 Hδ resonances converted the prior long-range NOEs between F4 and CHC residues L16–A21 to intra-CHC NOEs, involving F19. Similarly, G38 Hα overlaps with V18 Hα, and the putative NOE between F19 Hβ/δ and G38 Hα better matches the position and multiplet structure of V18 Hα. Therefore, even though we can positively identify a substantial number of short- and medium-range NOEs, we were unable to uniquely identify even a single long-range NOE between residues more than five positions apart in the sequence. In addition to this dearth of long-range NOE restraints, the problem of defining the peptide’s structure is compounded by its dynamic character, which would require an ensemble refinement and therefore more restraints than a static structure,68,69 an analysis that is further complicated by the strong variation in dynamics along the sequence impacting the NOE quantification (cf. Figure 4D). As mentioned above, in principle, the latter problem could be solved by directly calculating the relevant autocorrelation functions from the MD trajectory, but in practice, this proves challenging because of limitations in the force field, which can give rise to stable structural features for which no clear evidence exists in the experimental data.23,38,46,47
To investigate whether differences between these results and earlier literature data could be the result of differences in ionic strength, which may potentially affect the conformation adopted by the amyloid peptides in solution, we compared the Aβ1–40 CD spectra at 0 and 100 mM NaCl70 (Figure S5A). We found that the two CD spectra were essentially indistinguishable and fully consistent with random coil behavior. Similarly, comparing the backbone NMR chemical shifts and the intraresidue daN(i,i) NOE intensities from 3D NOESY-HSQC experiments, recorded at either 0 or 30 mM NaCl,59 again shows no significant differences (Figure S5B).
MERA Analysis of Backbone Torsion Angles
Despite the clear absence of a significantly populated folded state for Aβ1–40 and Aβ1–42, weak medium-range NOEs provide strong evidence of the transient presence of locally compact structures such as those mentioned above for residues in the CHC region. To gain further insight into the distribution of backbone angles sampled by Aβ1–40, we also measured three additional types of J couplings (1JHαCα, 2JNCα, and 1JNCα) that are sensitive to the backbone torsion angles, in addition to 3JHNHα, 3JC′C′, 3JC′Hα, and the backbone chemical shifts. The measurement of these additional couplings was limited to the Aβ1–40 peptide because the greater stability in solution of this shorter peptide is a prerequisite for measurement of the smaller couplings at the requisite very high experimental precision. Moreover, as discussed above, indistinguishable NOE patterns and virtually identical chemical shifts and 3JHNHα values for the first 34 residues of Aβ1–40 and Aβ1–42 indicate that no significant differences relative to Aβ1–40 will be detectable for the longer sequence within the limits of experimental precision.
With up to 12 measured parameters for most residues, including three types of NOEs [dNN(i,i+1), daN(i,i), and daN(i,i+1)], three types of chemical shifts (15N, 13Cα, and 13C′), and the six types of J couplings mentioned above, the available experimental data (Table S2) provide a reasonable set of restraints for probing the ϕ/ψ Ramachandran map populations of each individual residue. For this purpose, we previously developed the MERA program (Maximum Entropy Ramachandran map Analysis from NMR data), which generates residue-by-residue Ramachandran map distributions for disordered proteins or disordered regions in folded proteins on the basis of experimental NMR parameters.55,61 The Ramachandran map distributions are reported in terms of populations of their 15° × 15° voxels, and a maximum entropy regularizer is used to ensure that the obtained distributions deviate minimally from the residue-specific random coil library Ramachandran distributions, i.e., not more than required for obtaining agreement with the experimental data. Without such a regularizer, the 12 parameters provide insufficient information to uniquely determine the relative populations of the ∼120 voxels that exhibit nonvanishing probabilities in the coil library. The entropy term is defined as S = −∑kwk ln(wk/wklib), where the summation extends over all voxels, k, and wk is the MERA-derived fractional population of voxel k, with wklib being the corresponding population in the coil library for a given residue type.
The minimum rmsd, χ, between the experimental input data and the calculated values obtained for the MERA Ramachandran map distribution initially increases only very slowly when the weight, θ, of the entropy term is increased in a stepwise manner but typically starts rising more rapidly for θ ≥ ∼1 (Figure S6). For all distributions shown here, we have chosen a θ = 0.8 value, which yields normalized χ2 values of ≤1.5 for all residues analyzed (Figure S7). Without stereospecific assignments of the Gly Hα resonances, which frequently have nearly identical chemical shifts, the NMR parameters cannot distinguish between right-handed and left-handed structures, and Gly residues therefore are not included in the MERA analysis. Figure 5 displays the MERA-derived ϕ/ψ distributions for selected residues of Aβ1–40: D7 as a representative residue of the N-terminus, V18 and F19, located in the CHC and proposed to adopt a β-conformation in most studies,9 E22 and K28, both in the central hydrophilic region (E22–G29), and A30, L34, and M35 as representative residues of the C-terminal hydrophobic region (A30–I40), which have also been described as adopting a β-conformation.9 MERA maps of the remaining residues are included in Figure S7. The population of each voxel in these maps is depicted by the size of the colored circles, whereas the color represents the fractional deviation from the random coil distribution after nearest neighbor correction.
Figure 5.

Examples of ϕ/ψ distributions derived for Aβ1–40 residues D7, V18, F19, E22, K28, A30, L34, and M35. The surface area of each circle is proportional to the population of its 15° × 15° voxel, and the color of each circle reflects the ratio relative to that of the population seen in the coil database for that residue type, from 0.2 (blue) to 5 (red). Green boxes mark secondary structure regions: β, PPII, αL, type I β-turn (β-I), and αR. An entropy weight factor of 0.8 was used as well as a diffusion anisotropy parameter61k = 0.3 for the analysis of NOEs. The full set of residues and the corresponding χ2 vs S plots are presented in Figures S7 and S6, respectively.
As can be seen from the mostly yellow voxel colors obtained for D7, results for this residue fall close to those seen in the coil library distribution for Asp residues. Indeed, with an entropy S of −0.15, this residue is among the closest to the coil library distribution. Note that –S is often also termed the Kullback–Leibner information divergence.71 For comparison, residues in well-ordered regions of folded proteins typically exhibit S values lower than ca. −0.8.61 The MERA maps of residues V18 and F19 both show an elevated propensity for extended β conformations, as exemplified by F19, but with S values of ca. −0.4, the backbone angles of these residues remain closer to random coil (S = 0) than to values seen in folded proteins, consistent with their large σ values mentioned above. No clear propensity for secondary structure is found in the central hydrophilic region (E22–G29), although N27 and L28 show elevated populations of both αR and αL conformations (Figure S7). Both residues I31 and I32 show an elevated population of the β-region over the already higher random coil β-propensity of these Cβ-branched residues, whereas a slightly elevated propensity for turn formation is seen for M35. We note, however, that with the possible slight exception of I32, all residues in this C-terminal region show near-zero values of entropy, meaning that all of these match the coil library distribution of these residues rather closely (Figure S7).
Comparison with Previous Studies
Although the solution behavior of the Aβ1–40 and Aβ1–42 peptides has been the subject of numerous studies, both by NMR spectroscopy9,19,45 and by other biophysical methods,72 our present study adds a substantial number of additional parameters to the discussion. In particular, our measurements of the structurally important 3JHNHα couplings are essentially complete, whereas 25–50% of these were missing in earlier studies. The high accuracy of our 3JHNHα measurements is implied by the very close agreement between values seen for the first 34 residues of the two Aβ peptides, where the nearly indistinguishable chemical shifts already point to very similar structural distributions. Small differences seen for residues 35–40 between the two peptides appear to reflect slightly different structural propensities, but with no significant changes in chemical shift or 3JHNHα for residues preceding M35 these cannot be attributed to a stable long-range interaction for Aβ1–42, proposed in earlier studies23,38,46 or differences in long-range interactions involving the N-terminal residues.72
Our results indicate that both peptides are void of distinct highly populated structural features. Significant occupancy of a β-sheet for V18–F20 appears to be excluded as we do not find a matching set of residues with which to pair these residues, and intermolecular β-sheet formation is excluded by the complete absence of a concentration dependence of chemical shifts. We note that the more extended, β-like character seen for V18–F20 and the C-terminal region (I31–V36) is largely a direct effect of the types of amino acids of which these regions are composed, as β-branched and aromatic residues are known to have a more extended backbone propensity.24 A number of previous studies are in general agreement that the central hydrophilic region (E22–G29) has a tendency to adopt turn or bendlike structures, presumed to be a requirement for allowing an interaction between the two main hydrophobic regions (L17–A21 and A30–V40), therefore mimicking the loop conformations found in prior NMR structures of Aβ amyloid fibrils.9−11,13,73 Small variations in sample conditions may impact the average structure. In particular, variations in pH, temperature, ionic strength, and in particular the nature of the anions can impact the partitioning of the Aβ peptide between its monomeric state in solution and an aggregated oligomeric form.70,74,75 However, we note that chemical shift changes with varying ionic conditions are very small, strongly suggesting that the conformational distribution of the free, monomeric peptides is little affected. Indeed, we also find that variations in ionic strength had no measurable impact on the conformational propensity of monomeric Aβ1–40 in solution as measured by NOEs and circular dichroism (Figure S5). This result suggests that the salt bridge interaction between D23 and K28,76 which is believed to drive the formation of a turn in the hydrophilic region (E22–G29), is not significantly populated in the monomeric states of the Aβ peptides. Previous NMR measurements typically were conducted at near-neutral pH values. Note that aggregation is strongly enhanced at pH values close to the isoelectric point (pI = 5.3) and the requisite signature amide HN signals disappear at elevated pH values (ca. ≥8) due to rapid exchange with solvent. However, comparison of the secondary 13Cα chemical shifts recorded in this study at pH 7.0 with those reported by Yamaguchi et al.16 at pH 6.5 and Waelti et al.35 at pH 7.4 (Figure S1) shows that small variations around neutral pH do not significantly impact the secondary structure propensity of the Aβ peptides.
Of particular interest is the atomic model of Aβ1–40 reported by Vivekanadan et al.,59 which represents the only study to date reporting a semifolded full-length monomeric Aβ structure in solution, with a central 310-helix extending from H13 to D23. In this collapsed structure, the central helical fragment makes long-range contacts to the N- and C-termini, driving the collapse in the structural modeling. Aided by the higher resolution available in our 2D NOESY spectra (Figures S3 and S4), the vast majority of these long-range contacts can be attributed unambiguously to short-range interactions. However, we note that even though the chemical shifts observed in our study match very closely those of the Vivekanadan study, the relative intensities of a number of the non-intraresidue, nonsequential NOEs differ significantly between our NOESY spectrum and the earlier study. In particular, the NOESY cross-peaks observed for the V24(Hγ)–F20(Hε), V24(Hγ)–F19(Hε), V24(Hγ)–F20(Hδ), and V24(Hγ)–F19(Hδ) contacts are much weaker in our NOESY spectrum, presumably resulting from small differences in experimental conditions (pH 7.2 vs pH 7.0, 50 mM NaCl vs 20 mM sodium phosphate, or 288 K vs 277 K) or differences in sample preparation (synthetic vs recombinant peptide). In addition, and as noted by Vivekanadan et al., the possibility that some of these NOEs actually represent transferred NOEs resulting from exchange between a high-molecular weight, NMR-invisible, aggregated state of Aβ1–40, also observed in the NMR study by Narayanan and Reif,70 cannot be excluded and is perhaps even likely.
Relation between Monomer Structure and Protofibril Formation
Monomeric amyloid peptides are capable of oligomerization through different pathways, involving both primary and secondary nucleation, which then can propagate into the growth of long, regular fibrillar structures.77−82 The initial nucleation process is believed to be thermodynamically unfavorable, explaining the existence of a lag phase in the kinetics of amyloid fibril formation.83 Characterization of the nucleation structures has been the focus of numerous studies. Although it may be considered plausible that the secondary structures found in fibril-associated β-sheets are already transiently adopted by the monomeric Aβ in solution, the data presented here show very low propensities for stable secondary structure elements in the monomeric peptide. In particular, we cannot identify any significantly elevated β-conformation propensity for the hydrophobic region of residues 30–40 or a clear turn propensity in the G22–K28 region, with the possible exception of low (∼13%) turn propensities at N27 and K28. The absence of previously reported long-range NOE contacts highlights how difficult the task of identifying transiently populated structures in a dynamic ensemble really is. We note that our chemical shift values agree very closely with those of prior studies of Aβ1–40 and Aβ1–42 peptides.16,35 Perhaps even more importantly, we find that the backbone secondary chemical shift values of these two peptides are very similar over the first 34 residues, with pairwise rmsd values of 0.018, 0.048, and 0.007 ppm for the 13C′, 13Cα, and 1Hα nuclei, respectively (Figure 1B–D), suggesting that any structural differences between the two peptides cannot be very large. Considering the large differences in amyloid propensity of the two peptides, this conclusion appears to be at odds with the assumption that the aggregation is initiated by transient interaction of peptides in a fibril-prone structural state.
If under the quiescent, low-temperature (4 °C) NMR conditions the peptide is in exchange between a monomeric structure and an oligomeric species, such an exchange process must be slow considering that there is no concentration dependence of the chemical shifts. Indeed, above a threshold concentration of ∼150 μM, Fawzi et al. found positive evidence of slow chemical exchange between the monomer and a stable, NMR-invisible large (2–80 MDa) protofibrillar state,84−86 with a pseudoequilibrium between the two forms. From the observed relaxation behavior, these authors conclude that the first eight residues of the exchanging peptides exist predominantly in a mobile tethered state when bound, whereas the largely hydrophobic central regions are in direct contact with the protofibril surface for a significant fraction of time.
Our data are perhaps the most extensive and detailed at probing the monomeric solution behavior of the Aβ peptides to date and show no pronounced secondary structure propensities for either peptide, or any significant differences between Aβ1–40 and Aβ1–42 at 4 °C. Note that any secondary structure propensity of the monomeric peptide is expected to further decrease when the temperature is increased to 37 °C. We therefore interpret our results as evidence that primary nucleation of the monomeric peptides likely is driven primarily by nonspecific interactions between hydrophobic segments (residues L17–A21 and I31–V40 or I31–A42 for Aβ1–42) rather than by transient interactions between preformed β-strands. Interestingly, the large difference in fibril formation propensities of Aβ1–40 and Aβ1–42 has been linked to their difference in primary nucleation rate.87 In combination, these results therefore suggest that the higher aggregation propensity of Aβ1–42 compared to that of Aβ1–40 is simply the result of the longer stretch of hydrophobic residues at the peptide’s C-terminus rather than the result of a shift in propensity to aggregation-prone β-conformations in the free peptide.
The degree of peptide order in the aggregated state generated under NMR conditions, generally believed to be protofibrillar,86 remains a matter of debate.35,70,88 The observation that under our quiescent, low-temperature conditions the disappearance of free monomer peptide NMR signals is seen only above a threshold concentration (∼150 μM for Aβ1–40 in H2O)85 could be interpreted as evidence of the presence of a peptidic micelle with a critical micelle concentration of 150 μM. However, such a model is unlikely to be correct. First, circular dichroism data show a very high fraction of β-sheet in the aggregated state obtained for such samples (Figure S8A). Second, if a sample that has evolved to contain a significant fraction of aggregated state is subsequently diluted 2-fold, the monomer signal does not grow back in intensity (Figure S8B), contrary to what would be expected for a monomer–micelle equilibrium. This result indicates that the aggregation process is unidirectional also under our NMR conditions, even though individual peptides are in dynamic exchange between a free state and a state in which they are not yet irreversibly anchored to the protofibril, with the latter process being responsible for the NMR relaxation effects observed by Fawzi et al.84−86 Remarkably, we find that NMR signal loss over time for the free peptides in a solution of D2O is larger than for H2O, an effect seen for both peptides but most pronounced for Aβ1–42 (Figure 6A). This observation is consistent with the conclusion that primary nucleation is being driven by the hydrophobic effect described above, as D2O is known to be a poorer solvent for hydrophobic molecules than H2O.89−91
Figure 6.

(A) Kinetics of aggregation measured at 4 °C from the loss of the normalized cross-peak intensity of methyl groups in a time series of NMR spectra recorded for 150 μM samples of Aβ1–42 solubilized in either H2O (black) or D2O (red), or 300 μM samples of Aβ1–40 solubilized in either H2O (blue) or D2O (green), all in 20 mM sodium phosphate at pH (or pD, uncorrected meter reading using a glass electrode) 7.0. Fitted curves correspond to I = A + (1 – A) exp(−t/T), where A = 0.24 ± 0.01 and T = 65 ± 4 h for the H2O Aβ1–42 sample, A = 0.43 ± 0.02 and T = 31.5 ± 0.7 h for the D2O Aβ1–42 sample, A = 0.85 ± 0.01 and T = 72.1 ± 5.7 h for the H2O Aβ1–40 sample, and A = 0.65 ± 0.01 and T = 24.9 ± 1.6 h for the D2O Aβ1–40 sample. (B) Aggregation kinetics monitored by ThT fluorescence of 5 μM samples of Aβ1–40 and Aβ1–42 in 20 mM sodium phosphate at pH (or pD, uncorrected meter reading) 7.4. The ThT fluorescence experiments were conducted at 37 °C. For each sample, the fluorescence signal was averaged over four replicas.
To further investigate the effect of solvent on fibril formation, we complemented our NMR observations by recording the aggregation kinetics of both Aβ1–40 and Aβ1–42 peptides in solvents H2O and D2O through conventional measurements of ThT fluorescence (Figure 6B). The figure shows that the use of D2O strongly decreases the lag time for both peptides but increases the growth rate for only Aβ1–42. These observations suggest that primary nucleation of both peptides is significantly impacted by hydrophobic effects, but that the nature of the interactions that govern secondary nucleation and/or the growth of larger aggregates or fibrils is likely to differ for Aβ1–40 and Aβ1–42. Meisl et al. showed that the presence of the two additional hydrophobic residues in Aβ1–42 mainly affects the primary nucleation rate (by decreasing the lag time) and has little impact on the elongation rate.87 Their result appears to be consistent with our findings, as hydrophobically driven primary nucleation will be accelerated both by the increased hydrophobicity of the peptide sequence and by a poorer solvent of hydrophobic residues.89 The poorer solvent properties toward hydrophobic substances can be considered as squeezing the solute out of the stronger hydrogen bonded network of the D2O solvent.91,92 The strong effect of D2O seen on the steepness of the growth phase for Aβ1–42 (Figure 6B) suggests that elongation and/or secondary nucleation of this peptide is also significantly impacted by hydrophobic interactions, whereas for Aβ1–40 this is not the case, implying a difference in the underlying mechanisms. The existence of different mechanisms involved in the elongation of nascent Aβ1–40 and Aβ1–42 fibrils appears to be consistent with the observation by Cukalevski et al. that even though the two peptides fibrillize synergistically from a mixed solution of the two peptides, the resulting fibrils are homomolecular.93 Their result indicates that highly sequence-specific interactions underlie fibril elongation, a process that on the basis of our results appears to be significantly impacted by the D2O/H2O solvent composition for Aβ1–42 but not for Aβ1–40.
Concluding Remarks
It is well recognized that the presence of transient structure, in particular transient helix formation, in an otherwise disordered protein can be an important determinant for target binding.94−96 However, comparison of a large set of NMR parameters, collected by us for Aβ1–40 and Aβ1–42, shows no evidence of significant transient populations of long-range order in these peptides under conditions where they are strictly monomeric. Although very weak medium-range NOEs suggest the transient formation of locally ordered structures beyond what is expected on the basis of nearest neighbor effects, no significant population of stable β-strand, α-helix, or β-turn is observed in the NOE or J coupling data, with the possible exception of a weak turn propensity at residues N27 and K28, reflected in elevated 3JC′Hα values and daN(i,i)/daN(i–1,i) NOE ratios. Many of the previously identified long-range NOE interactions are absent in our highly resolved 900 MHz NOESY spectra or can be confidently assigned to short-range pairs of protons. Nevertheless, small differences relative to prior NOESY spectra are also seen, which are likely attributable to transferred NOE effects, associated with transient binding to aggregated species in samples that were not strictly controlled to be free of aggregated species.
An analysis of the Ramachandran map distribution of each residue shows very low Kullback–Leibler divergences from the corresponding coil library distributions. It should be noted, however, that the MERA program used to derive these Ramachandran map distributions searches for distributions that deviate minimally from coil library distributions, without significantly violating the NMR restraints. Somewhat different distributions with higher Kullback–Leibler divergences that agree equally well with the experimental data can also be created. However, considering the very high degree of similarity in the chemical shift and J coupling parameters for the first 34 residues of the Aβ1–40 and Aβ1–42 peptides, the presence of a substantial difference in the conformations sampled by these two peptides appears to be excluded. Root-mean-square 3JHNHα deviations of only 0.41 Hz (Aβ1–40) and 0.42 Hz (Aβ1–42) relative to random coil values are considerably lower than for ensembles generated previously on the basis of advanced, NMR-guided multiconformer models, suggesting that even though the presence of transiently structured species is not excluded by our data, their populations must be low and their structures quite heterogeneous. Our result contrasts somewhat with those of prior studies in which binding of IDP to a functional target frequently involves elements of conformational selection. Although our results cannot exclude the involvement of such processes, our NMR data indicate that the upper limit population of transiently ordered species for the Aβ peptides is considerably lower than those seen in other cases.94−96
Indeed, the very high degree of similarity in NMR characteristics of the 34 N-terminal residues of Aβ1–40 and Aβ1–42 in terms of chemical shifts, 3JHNHα, and NOEs strongly argues against the notion that a substantial structural difference in the unfolded states of these two peptides is responsible for their nearly 10-fold difference in aggregation kinetics. Instead, it appears that the increase in hydrophobicity caused by the additional C-terminal Ile41 and Ala42 residues is responsible for the faster primary nucleation observed by Meisl et al.,87 a finding supported by our observation of shorter nucleation delays in D2O over H2O solutions. Our observation that the kinetics of secondary nucleation and/or fibril growth of only Aβ1–42 is impacted by solvent composition suggests that hydrophobic packing is different in the two types of fibrils.
Acknowledgments
We thank Dennis Torchia for useful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b01259.
Experimental procedures and additional data (PDF)
This work was funded by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH), and the Intramural AIDS-Targeted Antiviral Program of the Office of the Director, NIH.
The authors declare no competing financial interest.
Supplementary Material
References
- Lesne S.; Koh M. T.; Kotilinek L.; Kayed R.; Glabe C. G.; Yang A.; Gallagher M.; Ashe K. H. (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. (2009) Amyloid beta-Protein Assembly and Alzheimer Disease. J. Biol. Chem. 284, 4749–4753. 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M.; Spires-Jones T. L.; Prada C.; Garcia-Alloza M.; de Calignon A.; Rozkalne A.; Koenigsknecht-Talboo J.; Holtzman D. M.; Bacskai B. J.; Hyman B. T. (2008) Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–U5. 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I.; Karran E.; De Strooper B. (2012) The toxic A beta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Rahimi F.; Shanmugam A.; Bitan G. (2008) Structure-function relationships of pre-fibrillar protein assemblies in Alzheimer’s disease and related disorders. Curr. Alzheimer Res. 5, 319–341. 10.2174/156720508784533358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C.; Steiner H. (2001) Protofibrils, the unifying toxic molecule of neurodegenerative disorders?. Nat. Neurosci. 4, 859–860. 10.1038/nn0901-859. [DOI] [PubMed] [Google Scholar]
- Klein W. L.; Krafft G. A.; Finch C. E. (2001) Targeting small A beta oligomers: the solution to an Alzheimer’s disease conundrum?. Trends Neurosci. 24, 219–224. 10.1016/S0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- Kirkitadze M. D.; Bitan G.; Teplow D. B. (2002) Paradigm shifts in Alzheimer’s disease and other neuro degenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res. 69, 567–577. 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- Abelein A.; Abrahams J. P.; Danielsson J.; Graslund A.; Jarvet J.; Luo J.; Tiiman A.; Warmlander S. K. T. S. (2014) The hairpin conformation of the amyloid beta peptide is an important structural motif along the aggregation pathway. JBIC, J. Biol. Inorg. Chem. 19, 623–634. 10.1007/s00775-014-1131-8. [DOI] [PubMed] [Google Scholar]
- Kotler S. A.; Walsh P.; Brender J. R.; Ramamoorthy A. (2014) Differences between amyloid-beta aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 43, 6692–6700. 10.1039/C3CS60431D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo N. D.; Grant M. A.; Condron M. C.; Rigby A. C.; Teplow D. B. (2005) On the nucleation of amyloid beta-protein monomer folding. Protein Sci. 14, 1581–1596. 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant M. A.; Lazo N. D.; Lomakin A.; Condron M. M.; Arai H.; Yamin G.; Rigby A. C.; Teplow D. B. (2007) Familial Alzhemier’s disease mutations alter the stability of the amyloid beta-protein monomer folding nucleus. Proc. Natl. Acad. Sci. U. S. A. 104, 16522–16527. 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. (2015) Amyloid Polymorphism: Structural Basis and Neurobiological Relevance. Neuron 86, 632–645. 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson J.; Jarvet J.; Damberg P.; Graslund A. (2005) The Alzheimer beta-peptide shows temperature-dependent transitions between left-handed 3(1)-helix, beta-strand and random coil secondary structures. FEBS J. 272, 3938–3949. 10.1111/j.1742-4658.2005.04812.x. [DOI] [PubMed] [Google Scholar]
- Danielsson J.; Andersson A.; Jarvet J.; Graslund A. (2006) N-15 relaxation study of the amyloid beta-peptide: structural propensities and persistence length. Magn. Reson. Chem. 44, S114–S121. 10.1002/mrc.1814. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T.; Matsuzaki K.; Hoshino M. (2011) Transient formation of intermediate conformational states of amyloid-beta peptide revealed by heteronuclear magnetic resonance spectroscopy. FEBS Lett. 585, 1097–1102. 10.1016/j.febslet.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Hoyer W.; Gronwall C.; Jonsson A.; Stahl S.; Hard T. (2008) Stabilization of a beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. U. S. A. 105, 5099–5104. 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg A.; Luheshi L. M.; Sollvander S.; Pereira de Barros T.; Macao B.; Knowles T. P. J.; Biverstal H.; Lendel C.; Ekholm-Petterson F.; Dubnovitsky A.; Lannfelt L.; Dobson C. M.; Hard T. (2010) Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. U. S. A. 107, 15595–15600. 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L. M.; Shao H. Y.; Zhang Y. B.; Li H.; Menon N. K.; Neuhaus E. B.; Brewer J. M.; Byeon I. J. L.; Ray D. G.; Vitek M. P.; Iwashita T.; Makula R. A.; Przybyla A. B.; Zagorski M. G. (2004) Solution NMR studies of the A beta(1–40) and A beta(1–42) peptides establish that the met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 126, 1992–2005. 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- Jarrett J. T.; Berger E. P.; Lansbury P. T. (1993) The Carboxy Terminus of the Beta-Amyloid Protein Is Critical for the Seeding of Amyloid Formation - Implications for the Pathogenesis of Alzheimers-Disease. Biochemistry 32, 4693–4697. 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- El-Agnaf O. M. A.; Mahil D. S.; Patel B. P.; Austen B. M. (2000) Oligomerization and toxicity of beta-amyloid-42 implicated in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 273, 1003–1007. 10.1006/bbrc.2000.3051. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Wang C. (2006) A beta 42 is more rigid than A beta 40 at the C terminus: Implications for A beta aggregation and toxicity. J. Mol. Biol. 364, 853–862. 10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Sgourakis N. G.; Yan Y.; McCallum S. A.; Wang C.; Garcia A. E. (2007) The Alzheimer’s peptides A beta 40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 368, 1448–1457. 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S. R.; Sharman G. J.; Maynard A. J.; Searle M. S. (1998) Modulation of intrinsic phi,psi propensities of amino acids by neighbouring residues in the coil regions of protein structures: NMR analysis and dissection of a beta-hairpin peptide. J. Mol. Biol. 284, 1597–1609. 10.1006/jmbi.1998.2264. [DOI] [PubMed] [Google Scholar]
- Fezoui Y.; Hartley D. M.; Harper J. D.; Khurana R.; Walsh D. M.; Condron M. M.; Selkoe D. J.; Lansbury P. T.; Fink A. L.; Teplow D. B. (2000) An improved method of preparing the amyloid beta-protein for fibrillogenesis and neurotoxicity experiments. Amyloid 7, 166–178. 10.3109/13506120009146831. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRpipe - a multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 6, 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G. (2008) Sparky 3, University of California, San Francisco. [Google Scholar]
- Ying J. F.; Chill J. H.; Louis J. M.; Bax A. (2007) Mixed-time parallel evolution in multiple quantum NMR experiments: sensitivity and resolution enhancement in heteronuclear NMR. J. Biomol. NMR 37, 195–204. 10.1007/s10858-006-9120-z. [DOI] [PubMed] [Google Scholar]
- Roche J.; Ying J.; Bax A. (2016) Accurate measurement of 3JHNHa couplings in small or disordered proteins from WATERGATE-optimized TROSY spectra. J. Biomol. NMR 10.1007/s10858-015-0004-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K. Y.; Gronenborn A. M. (2004) Protein backbone H-1(N)-C-13(alpha) and N-15-C-13(alpha) residual dipolar and J couplings: New constraints for NMR structure determination. J. Am. Chem. Soc. 126, 6232–6233. 10.1021/ja049049l. [DOI] [PubMed] [Google Scholar]
- Li F.; Lee J. H.; Grishaev A.; Ying J.; Bax A. (2015) High Accuracy of Karplus Equations for Relating Three-Bond J Couplings to Protein Backbone Torsion Angles. ChemPhysChem 16, 572–578. 10.1002/cphc.201402704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Ying J.; Bax A. (2015) Quantitative evaluation of positive phi angle propensity in flexible regions of proteins from three-bond J couplings. Phys. Chem. Chem. Phys. 10.1039/C5CP04542H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying J.; Roche J.; Bax A. (2014) Homonuclear decoupling for enhancing resolution and sensitivity in NOE and RDC measurements of peptides and proteins. J. Magn. Reson. 241, 97–102. 10.1016/j.jmr.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geen H.; Freeman R. (1991) Band-selective radiofrequency pulses. J. Magn. Reson. 93, 93–141. 10.1016/0022-2364(91)90034-Q. [DOI] [Google Scholar]
- Waelti M. A.; Orts J.; Voegeli B.; Campioni S.; Riek R. (2015) Solution NMR Studies of Recombinant A beta(1–42): From the Presence of a Micellar Entity to Residual beta-Sheet Structure in the Soluble Species. ChemBioChem 16, 659–669. 10.1002/cbic.201402595. [DOI] [PubMed] [Google Scholar]
- Kjaergaard M.; Poulsen F. M. (2011) Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution. J. Biomol. NMR 50, 157–165. 10.1007/s10858-011-9508-2. [DOI] [PubMed] [Google Scholar]
- Kjaergaard M.; Brander S.; Poulsen F. M. (2011) Random coil chemical shift for intrinsically disordered proteins: effects of temperature and pH. J. Biomol. NMR 49, 139–149. 10.1007/s10858-011-9472-x. [DOI] [PubMed] [Google Scholar]
- Ball K. A.; Phillips A. H.; Wemmer D. E.; Head-Gordon T. (2013) Differences in beta-strand Populations of Monomeric A beta 40 and A beta 42. Biophys. J. 104, 2714–2724. 10.1016/j.bpj.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Bax A. (2012) Identification of helix capping and beta-turn motifs from NMR chemical shifts. J. Biomol. NMR 52, 211–232. 10.1007/s10858-012-9602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi A.; Billeter M.; Wüthrich K. (1984) Calibration of the angular dependence of the amide proton-Cα proton coupling constants, 3JHNα, in a globular protein: Use of 3JHNα for identification of helical secondary structure. J. Mol. Biol. 180, 741–751. 10.1016/0022-2836(84)90035-4. [DOI] [PubMed] [Google Scholar]
- Vuister G. W.; Bax A. (1993) Quantitative J correlation: A new approach for measuring homonuclear three-bond J(HNHα) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 115, 7772–7777. 10.1021/ja00070a024. [DOI] [Google Scholar]
- Vogeli B.; Ying J. F.; Grishaev A.; Bax A. (2007) Limits on variations in protein backbone dynamics from precise measurements of scalar couplings. J. Am. Chem. Soc. 129, 9377–9385. 10.1021/ja070324o. [DOI] [PubMed] [Google Scholar]
- Maltsev A. S.; Grishaev A.; Roche J.; Zasloff M.; Bax A. (2014) Improved Cross Validation of a Static Ubiquitin Structure Derived from High Precision Residual Dipolar Couplings Measured in a Drug-Based Liquid Crystalline Phase. J. Am. Chem. Soc. 136, 3752–3755. 10.1021/ja4132642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R.; Guntert P.; Dobeli H.; Wipf B.; Wuthrich K. (2001) NMR studies in aqueous solution fail to identify significant conformational differences between the monomeric forms of two Alzheimer peptides with widely different plaque-competence, A beta(1–40) (ox) and A beta(1–42) (ox). Eur. J. Biochem. 268, 5930–5936. 10.1046/j.0014-2956.2001.02537.x. [DOI] [PubMed] [Google Scholar]
- Yan Y.; McCallum S. A.; Wang C. (2008) M35 oxidation induces A beta 40-like structural and dynamical changes in A beta 42. J. Am. Chem. Soc. 130, 5394–5395. 10.1021/ja711189c. [DOI] [PubMed] [Google Scholar]
- Sgourakis N. G.; Merced-Serrano M.; Boutsidis C.; Drineas P.; Du Z.; Wang C.; Garcia A. E. (2011) Atomic-Level Characterization of the Ensemble of the A beta(1–42) Monomer in Water Using Unbiased Molecular Dynamics Simulations and Spectral Algorithms. J. Mol. Biol. 405, 570–583. 10.1016/j.jmb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball K. A.; Wemmer D. E.; Head-Gordon T. (2014) Comparison of Structure Determination Methods for Intrinsically Disordered Amyloid-beta Peptides. J. Phys. Chem. B 118, 6405–6416. 10.1021/jp410275y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboniwa H.; Grzesiek S.; Delaglio F.; Bax A. (1994) Measurement of HN-Hα J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J. Biomol. NMR 4, 871–878. 10.1007/BF00398416. [DOI] [PubMed] [Google Scholar]
- Maltsev A. S.; Ying J. F.; Bax A. (2012) Impact of N-Terminal Acetylation of α-Synuclein on Its Random Coil and Lipid Binding Properties. Biochemistry 51, 5004–5013. 10.1021/bi300642h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendel C.; Damberg P. (2009) 3D J-resolved NMR spectroscopy for unstructured polypeptides: fast measurement of (3)J(HNH alpha) coupling constants with outstanding spectral resolution. J. Biomol. NMR 44, 35–42. 10.1007/s10858-009-9313-3. [DOI] [PubMed] [Google Scholar]
- Roche J.; Ying J.; Bax A. (2015) Accurate measurement of 3JHNHa couplings in small or disordered proteins from WATERGATE-optimized TROSY spectra. J. Biomol. NMR 10.1007/s10858-015-0004-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K.; Riek R.; Wider G.; Wuthrich K. (1997) Attenuated T-2 relaxation by mutual cancellation of dipole- dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U. S. A. 94, 12366–12371. 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K. V.; Wider G.; Wuthrich K. (1998) Single transition-to-single transition polarization transfer (ST2-PT) in [N15, H1]-TROSY. J. Biomol. NMR 12, 345–348. 10.1023/A:1008268930690. [DOI] [PubMed] [Google Scholar]
- Smith L. J.; Fiebig K. M.; Schwalbe H.; Dobson C. M. (1996) The concept of a random coil - Residual structure in peptides and denatured proteins. Folding Des. 1, R95–R106. 10.1016/S1359-0278(96)00046-6. [DOI] [PubMed] [Google Scholar]
- Mantsyzov A. B.; Maltsev A. S.; Ying J.; Shen Y.; Hummer G.; Bax A. (2014) A maximum entropy approach to the study of residue-specific backbone angle distributions in alpha-synuclein, an intrinsically disordered protein. Protein Sci. 23, 1275–90. 10.1002/pro.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruschweiler R.; Case D. A. (1994) Adding Harmonic Motion To The Karplus Relation For Spin-Spin Coupling. J. Am. Chem. Soc. 116, 11199–11200. 10.1021/ja00103a062. [DOI] [Google Scholar]
- Lee J. H.; Li F.; Grishaev A.; Bax A. (2015) Quantitative residue-specific protein backbone torsion angle dynamics from concerted measurement of 3J couplings. J. Am. Chem. Soc. 137, 1432–1435. 10.1021/ja512593s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball K. A.; Phillips A. H.; Nerenberg P. S.; Fawzi N. L.; Wemmer D. E.; Head-Gordon T. (2011) Homogeneous and Heterogeneous Tertiary Structure Ensembles of Amyloid-beta Peptides. Biochemistry 50, 7612–7628. 10.1021/bi200732x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekanandan S.; Brender J. R.; Lee S. Y.; Ramamoorthy A. (2011) A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 411, 312–316. 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzkee N. C.; Fleming P. J.; Rose G. D. (2005) The protein coil library: A structural database of nonhelix, nonstrand fragments derived from the PDB. Proteins: Struct., Funct., Genet. 58, 852–854. 10.1002/prot.20394. [DOI] [PubMed] [Google Scholar]
- Mantsyzov A. B.; Shen Y.; Lee J. H.; Hummer G.; Bax A. (2015) MERA: A webserver for evaluating backbone torsion angle distributions in dynamic and disordered proteins from NMR data. J. Biomol. NMR 63, 85–95. 10.1007/s10858-015-9971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Dobeli H.; Schubert D.; Riek R. (2005) 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. U. S. A. 102, 17342–17347. 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström A.; Otting G. (1994) Improved spectral resolution in 1H NMR spectroscopy by homonuclear semiselective shaped pulse decoupling during acquisition. J. Am. Chem. Soc. 116, 8847–8848. 10.1021/ja00098a070. [DOI] [Google Scholar]
- Maltsev A. S.; Grishaev A.; Bax A. (2012) Monomeric alpha-Synuclein Binds Congo Red Micelles in a Disordered Manner. Biochemistry 51, 631–642. 10.1021/bi201435d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogeli B.; Kazemi S.; Guntert P.; Riek R. (2012) Spatial elucidation of motion in proteins by ensemble-based structure calculation using exact NOEs. Nat. Struct. Mol. Biol. 19, 1053–1058. 10.1038/nsmb.2355. [DOI] [PubMed] [Google Scholar]
- Peter C.; Daura X.; van Gunsteren W. F. (2001) Calculation of NMR-relaxation parameters for flexible molecules from molecular dynamics simulations. J. Biomol. NMR 20, 297–310. 10.1023/A:1011241030461. [DOI] [PubMed] [Google Scholar]
- Fawzi N. L.; Phillips A. H.; Ruscio J. Z.; Doucleff M.; Wemmer D. E.; Head-Gordon T. (2008) Structure and dynamics of the A ss(21–30) peptide from the interplay of NMR experiments and molecular simulations. J. Am. Chem. Soc. 130, 6145–6158. 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgi R.; Pitera J.; van Gunsteren W. F. (2001) Assessing the effect of conformational averaging on the measured values of observables. J. Biomol. NMR 19, 305–320. 10.1023/A:1011295422203. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Camilloni C.; Vendruscolo M. (2013) Molecular dynamics simulations with replica-averaged structural restraints generate structural ensembles according to the maximum entropy principle. J. Chem. Phys. 138, 094112. 10.1063/1.4793625. [DOI] [PubMed] [Google Scholar]
- Narayanan S.; Reif B. (2005) Characterization of chemical exchange between soluble and aggregated states of beta-amyloid by solution-state NMR upon variation of salt conditions. Biochemistry 44, 1444–1452. 10.1021/bi048264b. [DOI] [PubMed] [Google Scholar]
- Kullback S.; Leibler R. A. (1951) On Information and Sufficiency. Ann. Math. Stat. 22, 79–86. 10.1214/aoms/1177729694. [DOI] [Google Scholar]
- Teplow D. B.; Lazo N. D.; Bitan G.; Bernstein S.; Wyttenbach T.; Bowers M. T.; Baumketner A.; Shea J.-E.; Urbanc B.; Cruz L.; Borreguero J.; Stanley H. E. (2006) Elucidating amyloid beta-protein folding and assembly: A multidisciplinary approach. Acc. Chem. Res. 39, 635–645. 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Iwata K.; Lachenmann M. J.; Peng J. W.; Li S.; Stimson E. R.; Lu Y.; Felix A. M.; Maggio J. E.; Lee J. P. (2000) The Alzheimer’s peptide A beta adopts a collapsed coil structure in water. J. Struct. Biol. 130, 130–141. 10.1006/jsbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- Abelein A.; Graslund A.; Danielsson J. (2015) Zinc as chaperone-mimicking agent for retardation of amyloid beta peptide fibril formation. Proc. Natl. Acad. Sci. U. S. A. 112, 5407–5412. 10.1073/pnas.1421961112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghalebani L.; Wahlstrom A.; Danielsson J.; Warmlander S. K. T. S.; Graslund A. (2012) pH-dependence of the specific binding of Cu(II) and Zn(II) ions to the amyloid-beta peptide. Biochem. Biophys. Res. Commun. 421, 554–560. 10.1016/j.bbrc.2012.04.043. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. (2002) A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U. S. A. 99, 16742–16747. 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J.; Vendruscolo M.; Dobson C. M. (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. (2012) From Macroscopic Measurements to Microscopic Mechanisms of Protein Aggregation. J. Mol. Biol. 421, 160–171. 10.1016/j.jmb.2012.02.031. [DOI] [PubMed] [Google Scholar]
- Wogulis M.; Wright S.; Cunningham D.; Chilcote T.; Powell K.; Rydel R. E. (2005) Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J. Neurosci. 25, 1071–1080. 10.1523/JNEUROSCI.2381-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers E. T.; Powers D. L. (2008) Mechanisms of protein fibril formation: Nucleated polymerization with competing off-pathway aggregation. Biophys. J. 94, 379–391. 10.1529/biophysj.107.117168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S.-H.; Legname G.; Serban A.; Prusiner S. B.; Wright P. E.; Dyson H. J. (2009) Prion Proteins with Pathogenic and Protective Mutations Show Similar Structure and Dynamics. Biochemistry 48, 8120–8128. 10.1021/bi900923b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo H. P.; Moore R. C.; Cohen F. E.; Westaway D.; Prusiner S. B.; Wright P. E.; Dyson H. J. (2001) Two different neurodegenerative diseases caused by proteins with similar structures. Proc. Natl. Acad. Sci. U. S. A. 98, 2352–2357. 10.1073/pnas.051627998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W.-F.; Homans S. W.; Radford S. E. (2008) Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. U. S. A. 105, 8926–8931. 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi N. L.; Ying J.; Ghirlando R.; Torchia D. A.; Clore G. M. (2011) Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature 480, 268–U161. 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi N. L.; Ying J.; Torchia D. A.; Clore G. M. (2010) Kinetics of Amyloid beta Monomer-to-Oligomer Exchange by NMR Relaxation. J. Am. Chem. Soc. 132, 9948–9951. 10.1021/ja1048253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi N. L.; Libich D. S.; Ying J.; Tugarinov V.; Clore G. M. (2014) Characterizing Methyl-Bearing Side Chain Contacts and Dynamics Mediating Amyloid beta Protofibril Interactions Using C-13(methyl)-DEST and Lifetime Line Broadening. Angew. Chem., Int. Ed. 53, 10345–10349. 10.1002/anie.201405180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisl G.; Yang X.; Hellstrand E.; Frohm B.; Kirkegaard J. B.; Cohen S. I. A.; Dobson C. M.; Linse S.; Knowles T. P. J. (2014) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the A beta 40 and A beta 42 peptides. Proc. Natl. Acad. Sci. U. S. A. 111, 9384–9389. 10.1073/pnas.1401564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L. P.; Edalji R.; Harlan J. E.; Holzman T. F.; Lopez A. P.; Labkovsky B.; Hillen H.; Barghorn S.; Ebert U.; Richardson P. L.; Miesbauer L.; Solomon L.; Bartley D.; Walter K.; Johnson R. W.; Hajduk P. J.; Olejniczak E. T. (2009) Structural Characterization of a Soluble Amyloid beta-Peptide Oligomer. Biochemistry 48, 1870–1877. 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- Efimova Y. M.; Haemers S.; Wierczinski B.; Norde W.; van Well A. A. (2007) Stability of globular proteins in H2O and D2O. Biopolymers 85, 264–273. 10.1002/bip.20645. [DOI] [PubMed] [Google Scholar]
- Kresheck G. C.; Schneider H.; Scheraga H. A. (1965) Effect of D2O on Thermal Stability of Proteins. Thermodynamic Parameters for Transfer of Model Compounds from H2O to D2O. J. Phys. Chem. 69, 3132–3144. 10.1021/j100893a054. [DOI] [PubMed] [Google Scholar]
- Hummer G.; Garde S.; Garcia A. E.; Pratt L. R. (2000) New perspectives on hydrophobic effects. Chem. Phys. 258, 349–370. 10.1016/S0301-0104(00)00115-4. [DOI] [Google Scholar]
- Richards F. M. (1991) The Protein Folding Problem. Sci. Am. 264, 54. 10.1038/scientificamerican0191-54. [DOI] [PubMed] [Google Scholar]
- Cukalevski R.; Yang X.; Meisl G.; Weininger U.; Bernfur K.; Frohm B.; Knowles T. P. J.; Linse S. (2015) The A beta 40 and A beta 42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chemical Science 6, 4215–4233. 10.1039/C4SC02517B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iesmantavicius V.; Dogan J.; Jemth P.; Teilum K.; Kjaergaard M. (2014) Helical Propensity in an Intrinsically Disordered Protein Accelerates Ligand Binding. Angew. Chem., Int. Ed. 53, 1548–1551. 10.1002/anie.201307712. [DOI] [PubMed] [Google Scholar]
- Ozenne V.; Noel J. K.; Heidarsson P. O.; Brander S.; Poulsen F. M.; Jensen M. R.; Kragelund B. B.; Blackledge M.; Danielsson J. (2014) Exploring the Minimally Frustrated Energy Landscape of Unfolded ACBP. J. Mol. Biol. 426, 722–734. 10.1016/j.jmb.2013.10.031. [DOI] [PubMed] [Google Scholar]
- Arai M.; Sugase K.; Dyson H. J.; Wright P. E. (2015) Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc. Natl. Acad. Sci. U. S. A. 112, 9614–9619. 10.1073/pnas.1512799112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.