

Summary
Vα24 invariant natural killer T (iNKT) cells are a subset of T lymphocytes implicated in the regulation of broad immune responses. They recognize lipid antigens presented by CD1d on antigen-presenting cells and induce both innate and adaptive immune responses, which enhance effective immunity against cancer. Conversely, reduced iNKT cell numbers and function have been observed in many patients with cancer. To recover these numbers, we reprogrammed human iNKT cells to pluripotency and then re-differentiated them into regenerated iNKT cells in vitro through an IL-7/IL-15-based optimized cytokine combination. The re-differentiated iNKT cells showed proliferation and IFN-γ production in response to α-galactosylceramide, induced dendritic cell maturation and downstream activation of both cytotoxic T lymphocytes and NK cells, and exhibited NKG2D- and DNAM-1-mediated NK cell-like cytotoxicity against cancer cell lines. The immunological features of re-differentiated iNKT cells and their unlimited availability from induced pluripotent stem cells offer a potentially effective immunotherapy against cancer.
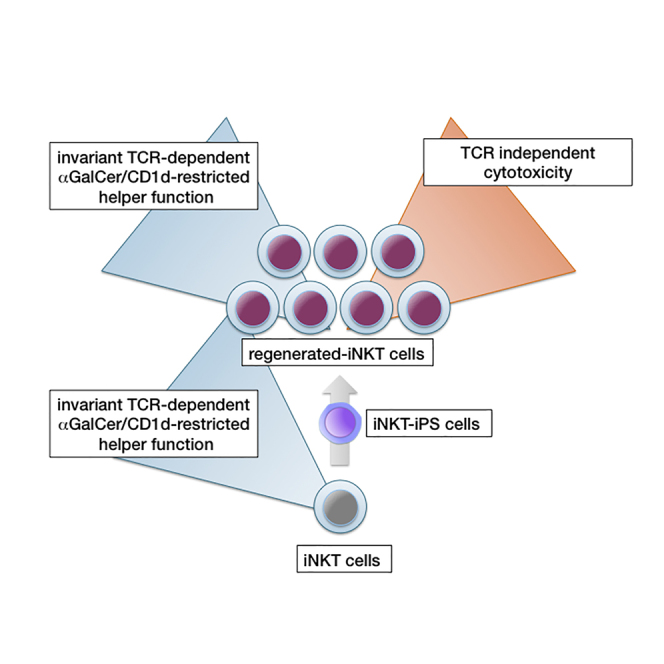
Graphical Abstract
Highlights
-
•
Human iNKT cell-derived iPSCs have differentiated into Vα24 iNKT-like cells in vitro
-
•
Re-differentiated iNKT-like (Re-iNKT) cells have functionally recovered properties
-
•
Re-iNKT cells function as an adjuvant to activate antigen-specific CTLs and NK cells
-
•
Re-iNKT cells exert cytotoxic activity via NKG2D- and DNAM-1-dependent mechanism
Kaneko, Uemura, and colleagues examined functional Vα24+ human iNKT cells induced from iPS cells in vitro. The re-differentiated iNKT cells demonstrated immune-adjuvant properties to induce cancer antigen-specific cytotoxic T cells via dendritic cell stimulation and direct cytotoxicity to cancer cells by induced NKG2D expression and increased DNAM-1 activity. These features indicate that re-differentiated iNKT cells are a potential source for new immunotherapies against cancer.
Introduction
Cytotoxic T lymphocytes (CTLs) play a crucial role in the eradication of cancer cells by precisely recognizing them via tumor antigen-specific T cell receptors (TCRs) in a peptide-dependent, human leukocyte antigen (HLA)-restricted manner (Maus et al., 2014). Sometimes, however, cancer cells can proliferate due to absent or dysfunctional CTLs, thus creating demand for immunotherapies. We and another group recently reported the unlimited production of target antigen-specific human CD8+ T lymphocytes from induced pluripotent stem cells (iPSCs) (Nishimura et al., 2013, Vizcardo et al., 2013). This technology has the potential to overcome two important problems currently facing T cell immunotherapies: a shortage of tumor antigen-specific T cells and their exhaustion induced by continuous TCR stimulation and overproliferation (Schietinger and Greenberg, 2014). However, other problems in T cell immunotherapies must also be overcome. One example is the emergence of tumor escape from antigen-specific monoclonal CTLs due to tumor immune-editing involving tumor antigen mutagenesis or HLA depression (Schreiber et al., 2011). Another problem is local immunosuppression in the tumor microenvironment by instigated immune cells, which supports tumor growth and inhibits CTL activities (Mittal et al., 2014, Motz and Coukos, 2013, Noy and Pollard, 2014). A good approach to overcome these problems would be combination therapy using a cellular adjuvant, i.e., invariant natural killer T (iNKT) cells, as iNKT cells exert helper functions to induce antigen-specific polyclonal CTLs (Cerundolo et al., 2009), improve the immunosuppressive milieu (De Santo et al., 2010), and maintain memory CD8+ T cells (Hong et al., 2009).
iNKT cells are a unique subset of T cells that express a canonical invariant TCR α chain (Vα24-Jα18 in humans) and TCR β chains that use limited Vβ segments (Vβ11 in humans), and also play a key role in the regulation of innate and adaptive immunity (Berzins et al., 2011, Brennan et al., 2013). In contrast to conventional αβ T cells, iNKT cells recognize a limited number of lipid antigens presented by the MHC class I-like molecule CD1d. Stimulation of iNKT cells by α-galactosylceramide (α-GalCer), a synthetic glycosphingolipid, results in the rapid production of Th1 and Th2 cytokines (e.g., interleukin-γ [IFN-γ] and interleukin-4 [IL-4]) and increased expression of CD40 ligand (CD40L), which induces dendritic cell (DC) maturation and production of IL-12p70 (Liu et al., 2008, McEwen-Smith et al., 2015, Uemura et al., 2009). These events ultimately lead to downstream activation of critical effectors of antitumor immunity, including NK cells, CTLs, and Th cells (Hong et al., 2009, Salio et al., 2014). Because CD1d is non-polymorphic, the modification of DC function by iNKT cells is independent of HLA restriction, making this process attractive for broad clinical application.
The antitumor potential of iNKT cells has been demonstrated in several clinical trials (Chang et al., 2005, McEwen-Smith et al., 2015, Motohashi et al., 2006, Motohashi et al., 2009, Nicol et al., 2011, Richter et al., 2013, Song et al., 2009, Uchida et al., 2008, Yamasaki et al., 2011). Infiltration of iNKT cells into tumor tissue is a favorable prognostic factor and is associated with improved survival, while low levels of circulating iNKT cells predict a poor clinical outcome (Molling et al., 2007). Although human iNKT cells are present wherever conventional T cells are found, their frequency relative to other T cells is less than 0.1%. In addition, a deficiency of iNKT cells and/or defects in their function has been reported in patients with many types of cancer (Berzins et al., 2011, Molling et al., 2005). Consequently, acquiring sufficient numbers of iNKT cells from patients to induce effective antitumor immune responses is currently an obstacle to iNKT cell-based immunotherapy.
A previous study has shown that iNKT cell TCR-harboring mouse iPSCs can differentiate into mature iNKT cells in vivo (Watarai et al., 2010). It remains unclear, however, whether human iNKT cell-derived iPSCs can differentiate into functional iNKT cells ex vivo. Here, we demonstrate that reprogramming human iNKT cells to pluripotency and subsequent re-differentiation of functional iNKT-like cells are possible ex vivo. These regenerated iNKT-like cells are functionally recovered and available in an unlimited supply from iPSCs. Moreover, they show both the expected adjuvant function of inducing leukemic antigen-specific polyclonal cytotoxic T cells via DC activation as well as TCR-independent direct killing of leukemic cell lines. This second feature is controlled by NKG2D signaling and, unexpectedly, DNAM-1 signaling, which is conceivably enhanced by the lack of TIGIT expression in the re-differentiated iNKT-like cells (re-iNKT cells). The adjuvant property and newly identified cytotoxic features of re-iNKT cells, which we demonstrate here against cancer cell lines, may have wide application in the field of immunotherapy.
Results
iNKT Cell-Derived Human iPSCs Are Preferentially Generated from a CD4+ Subset
Human iNKT cells can be classified into three phenotypically distinct subsets: CD4-positive (CD4+), CD8-positive (CD8+: CD8αβ or CD8αα), and CD4/8 double-negative (DN) (Brennan et al., 2013). The CD4+ subset has the potential to produce a large amount of Th2 cytokines, while the DN and CD8+ subsets have a Th1-biased profile. In addition, the CD8+ subset produces more IFN-γ and more cytotoxicity than the CD4+ or DN subsets. In this study, functionally distinct CD4+ and DN iNKT cell lines were established from the peripheral blood of healthy donors and reprogrammed into iPSCs. We could not reprogram CD8+ iNKT cells because of an insufficient number of cells obtained from donor peripheral blood. However, both CD4+ and DN iNKT cells were efficiently transduced with defective Sendai virus vectors harboring four reprogramming factors, OCT3/4, KLF4, SOX2, and C-MYC (SeVdp(KOSM302L)) (Nishimura et al., 2011) and SV40 T antigen (SeV18 + SV40T/TS15ΔF) (Nishimura et al., 2013), and then formed into embryonic stem cell (ESC)-like colonies. However, most of the DN iNKT cell-derived colonies could not be maintained under the culture conditions used for human pluripotent stem cells, and those that were maintained showed no differentiation potential toward hematopoietic cells (Figures S1A and S1B). In addition, the rarely established ESC-like colonies derived from DN iNKT cells were positive for residual Sendai virus vector. In contrast, all established clones from CD4+ iNKT cells were negative for residual transgenes (Figure S1C), showed pluripotency characterized by the expression of pluripotency-related genes (Figure S1D) and teratoma formation in immunodeficient mice (Figure S1E), and were confirmed to have a normal karyotype (Figure S1F). Although from different donors, all established clones showed the same TCRα chain with an invariant Vα24-Jα18 (TRAV10∗01-TRAJ18∗01) junctional sequence, which is a distinctive feature of human iNKT cells (Table 1).
Table 1.
Junction Sequences of TCR Genes in Reprogrammed and Redifferentiated Cells
| Clone | Productivity | TRAV | TRAJ | 3′ Va | P(N) | 5′ Ja | Frequency |
|---|---|---|---|---|---|---|---|
| V-J Junction Sequence of Rearranged TRA Genes in the Genome of CD4+iNKT-iPSC | |||||||
| 1 | productive | 10∗1 | 18∗01 | TGTGTGGTGAGCG | ACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 5/8 productive | |
| productive | 30∗01 | 10∗01 | TG | TGGCACAGAGAGG | ACGGGAGGAGGAAACAAACTCACCTTT | 3/8 productive | |
| 2 | productive | 10∗01 | 18∗01 | TGTGTGGTGAGC | GACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 1/1 productive | |
| 4 | productive | 10∗01 | 18∗01 | TGTGTGGTGAGC | GACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 1/1 productive | |
| 8 |
productive | 10∗01 | 18∗01 | TGTGTGGTGAGCG | ACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 5/10 productive | |
| productive | 30∗01 | 10∗01 | TG | TGGCACAGAGAGG | ACGGGAGGAGGAAACAAACTCACCTTT | 5/10 productive | |
| V-J Junction Sequence of Rearranged TRA mRNAs of Re-iNKT Cells | |||||||
| 1 | productive | 10∗1 | 18∗01 | TGTGTGGTGAGCG | ACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 9/13 productive | |
| productive | 30∗01 | 10∗01 | TG | TGGCACAGAGAGG | ACGGGAGGAGGAAACAAACTCACCTTT | 4/13 productive | |
| 2 | productive | 10∗01 | 18∗01 | TGTGTGGTGAGC | GACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 5/5 productive | |
| 4 | productive | 10∗01 | 18∗01 | TGTGTGGTGAGC | GACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 2/2 productive | |
| 8 | productive | 10∗01 | 18∗01 | TGTGTGGTGAGCG | ACAGAGGCTCAACCCTGGGGAGGCTATACTTT | 6/12 productive | |
| productive | 30∗01 | 10∗01 | TG | TGGCACAGAGAGG | ACGGGAGGAGGAAACAAACTCACCTTT | 6/12 productive | |
| Clone | Productivity | TRBV | TRBD | TRBJ | 3′ Vb | N1-P-Db-N2 | 5′ Jb | Frequency |
|---|---|---|---|---|---|---|---|---|
| V-D-J Junction Sequence of Rearranged TRB Genes in Genome of CD4+iNKT-iPSC | ||||||||
| 1 | productive | 25-1∗01 | 1∗01 | 1-5∗01 | TGTGCCAGCAGTGAA | CTAGGGGAGAATTTG | CAGCCCCAGCATTTT | 6/6 productive |
| unproductive | – | 1∗01 | 1-6∗01 | – | GGGTGCCGATGGAAT | TTCACCCCTCCACTTT | ||
| 2 | productive | 25-1∗01 | 1∗01 | 2-3∗01 | TGTGCCAGCAGTG | GCAGGAGCCTAAA | CACAGATACGCAGTATTTT | 2/2 productive |
| unproductive | germline | 1∗01 | 2-7∗01 | TACAAAGCTGTAACATTGTG | GGGACAGGGGGCCGC | TCCTACGAGCAGTACTTCGGGCCGG | ||
| 4 | productive | 25-1∗01 | 1∗01 | 2-3∗01 | TGTGCCAGCAG | GGGATCCGGGACAGGGGCC | GATACGCAGTATTTT | 4/4 productive |
| unproductive | 11-1∗01 | – | 2-6∗01 | GGCAG | – | CAGG | ||
| 8 |
productive | 25-1∗01 | 1∗01 | 1-5∗01 | TGTGCCAGCAGTGAA | CTAGGGGAGAATTTG | CAGCCCCAGCATTTT | 6/6 productive |
| unproductive | – | 1∗01 | 1-6∗01 | – | GGGTGCCGATGGAAT | TTCACCCCTCCACTTT | ||
| V-D-J Junction Sequence of Rearranged TRB mRNAs of Re-iNKT Cells | ||||||||
| 1 | productive | 25-1∗01 | 1∗01 | 1-5∗01 | TGTGCCAGCAGTGAA | CTAGGGGAGAATTTG | CAGCCCCAGCATTTT | 5/5 productive |
| 2 | productive | 25-1∗01 | 1∗01 | 2-3∗01 | TGTGCCAGCAGTG | GCAGGAGCCTAAA | CACAGATACGCAGTATTTT | 5/6 productive |
| productive | 12-3∗01 | 2∗01 | 2-3∗01 | TGTGCCAGCAGTT | CTTCATCCACTCCCGTC | GATACGCAGTATTTT | 1/6 productive | |
| 4 | productive | 25-1∗01 | 1∗01 | 2-3∗01 | TGTGCCAGCAG | CGGATCCGGGACAGGGGCC | GATACGCAGTATTTT | 5/5 productive |
| unproductive | 7-9∗04 | 1∗01 | 2-1∗01 | AGAGA | CACAGGCAGGGAA | GGGCC | ||
| unproductive | 20-1∗03 | – | 2-2P∗01 | GATGG | – | AGAGG | ||
| 8 | productive | 25-1∗01 | 1∗01 | 1-5∗01 | TGTGCCAGCAGTGAA | CTAGGGGAGAATTTG | CAGCCCCAGCATTTT | 6/7 productive |
| productive | 20-1∗01 | 2∗01 | 2-7∗01 | TGCAGTGCTAGA | TCCTACTTGGGGGGAGAA | GAGCAGTACTTC | 1/7 productive | |
Vα24-Jα18/Vβ11 TCR-Expressing T Cells Can Be Re-differentiated in the Presence of IL-15 and Expanded by TCR Stimulation
In contrast to conventional αβT cells, whose differentiation is controlled via TCR ligation by specific peptide antigens presented by polymorphic HLA molecules on thymic epithelial cells (Shah and Zuniga-Pflucker, 2014), iNKT cells differentiate in a unique fashion within the thymus (Godfrey et al., 2010). Once early T cell progenitors obtain the Vα24 invariant TCR at the CD4+CD8+ double-positive (DP) stage, they become sensitive to stimulation by specific glycolipid antigens presented by the non-polymorphic molecule CD1d. In addition, they become sensitive to self-stimulation by CD150, also known as SLAM (signaling lymphocytic activation molecule) (Veillette et al., 2007). Cytokine dependency during differentiation differs slightly between αβT and iNKT cells, as IL-15 is crucial for iNKT cell differentiation (Gordy et al., 2011). When iNKT-iPSCs were differentiated into T-lineage cells without IL-15, they stayed at the CD4−CD8− DN stage but were partially positive for iNKT cell-related molecules, including CD56, CD69, CD161 (NKRP1; NK cell-related protein 1), CD314 (cellular stress-ligand receptor NKG2D), and invariant TCR (stained by 6B11, a monoclonal antibody (mAb) specific for the invariant Vα24-Jα18 CDR3 loop), and expressed low levels of CD150 and CD122 (IL-2/-15 receptor β chain) at the third week of culture on OP9/DL1 (Figure 1A). Although they expressed invariant TCR, iPSC-derived differentiating cells showed minimal expansion upon stimulation by α-GalCer-loaded peripheral blood mononuclear cells (PBMCs) (data not shown). This prompted us to speculate whether other signals, such as IL-15 and/or SLAM-SLAM interaction, were necessary for further maturation. In light of the positive feedback effect of IL-15 signaling on CD122 expression (Castillo et al., 2010), which may induce further maturation, we modified the culture medium, adding 10 ng/ml IL-15 from the second or third week of culture on OP9/DL1. Improving the culture conditions led to greater numbers of invariant TCR-positive cells, and a fraction of these cells showed further enhancement of CD56, CD69, CD122, CD150, CD161, and CD314 expression (Figure 1A). It is generally difficult to exactly identify control/check points (the expressions of TCR or NK markers) or immature stages in the differentiation process (Godfrey and Berzins, 2007), since the differentiation of TCR pre-rearranged iPSC-derived hematopoietic cells into T-lineage cells on OP9/DL1 deviates from normal T-lymphopoiesis (Nishimura et al., 2013, Vizcardo et al., 2013). We were able to confirm, however, that additional IL-15 induced further maturation past control/check point 2 to differentiating cells (Figures S2A and S2B). By adding IL-2 to the culture, we increased cell numbers 7.5-fold over that seen with IL-15 alone (n ≥ 3). In subsequent experiments, therefore, we cultured re-differentiated T cells with a combination of Flt3L (FMS-like tyrosine kinase 3 ligand), IL-7, IL-2, and IL-15 (Figure 1B).
Figure 1.

Re-differentiation of iNKT Cells from iNKT-iPSCs
(A) Surface antigen profiles of re-differentiating T cells from CD4+ iNKT-iPSC (clone 2) on OP9/DL1 feeder cells on day 21 in the presence of the indicated cytokines. The results shown are representative of four independent experiments examining the surface protein expression patterns of the cells gated by CD45, CD3, and 6B11.
(B) Schematic illustration showing the culture protocol for iNKT cell re-differentiation from CD4+ iNKT cell-derived iPSCs. EB differentiation medium: Iscove's modified Dulbecco's medium supplemented with 15% fetal bovine serum and a cocktail of 10 μg/ml human insulin, 5.5 μg/ml human transferrin, 5 ng/ml sodium selenite, 2 mM L-glutamine, 0.45 mM monothioglycerol, and 50 μg/ml ascorbic acid. OP9 medium: α-MEM supplemented with 15% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
(C) Fold expansion of parental CD4+ iNKT cells and newly induced re-iNKT cells around 2 weeks after stimulation with α-GalCer-pulsed PBMCs. The result obtained with each indicated cell in three independent experiments is shown as a filled circle. Experiment 1: clone 2, parental clone and subclone of donor A; experiment 2: clone 1, clone 8, and donor A; experiment 3: clone 1, clone 2, clone 8, and parental clones of donor A and B. Horizontal lines indicate medians. NS, not significant.
(D) Co-expression profile of PD-1 and TIGIT on re-iNKT cells and parental iNKT cells after TCR stimulation (gated by CD45, CD3, and 6B11). The results shown are representative of two independent experiments. The numbers in the quadrants indicate percentages of cells.
(E) Telomere length in parental CD4+ iNKT cells and induced re-iNKT cells normalized to the tetraploid leukemic cell line 1301. Datasets from three independent experiments that include data from three different re-iNKT cell clones (1, 2, and 8), and two parental iNKT cell clones (donor A and donor B) are shown as filled diamonds, squares, and triangles, respectively. t Tests for paired data were used to compare results from the same donor; ∗p < 0.05.
Vα24-Jα18/Vβ11 TCR-expressing re-differentiated T cells were stained with α-GalCer/CD1d tetramer to the same degree as the parental iNKT cells (Figure S2C) and were responsive to stimulation with α-GalCer-pulsed PBMCs. Although there were some differences in the responsiveness to stimulation among iNKT-iPSC clones, the final yield after around 2 weeks was nearly the same as that of the parental iNKT cells (Figure 1C). In addition, repeated stimulation with α-GalCer-pulsed PBMCs or the mitogen phytohemagglutinin (PHA-P) in vitro resulted in expansion of the initial cell number by about 103- to 104-fold (Figure S2D). Because of their phenotypic and functional similarity to iNKT cells, the re-differentiated T cells expressing Vα24-Jα18/Vβ11 TCR are hereafter referred to as “re-iNKT cells.”
Generation of iNKT-like Cells through Reprogramming
Based on previous reports (Johnston et al., 2014, Postow et al., 2015) and our previous observations of CD8+ T cell rejuvenation through reprogramming (Nishimura et al., 2013), we estimated reprogramming-mediated functional recovery by comparing the expression of the immunological exhaustion-related checkpoint proteins PD-1 (programmed cell death protein 1) and TIGIT (T cell immunoreceptor with immunoglobulin and ITIM domains) and cell exhaustion-related telomere length in expanded re-iNKT cells with those in parental CD4+ and DN iNKT cells. Middle to high expression of PD-1 and TIGIT was observed in both CD4+ and DN iNKT cells, whereas re-iNKT cells expressed minimal levels of PD-1 and almost no TIGIT, even after several cycles of expansion induced by TCR stimulation (Figure 1D). The re-iNKT cells also showed a recovery of telomere length as previously reported (Nishimura et al., 2013) (Figure 1E).
Gene and Protein Expression Profiles Predict Re-iNKT Cells to Have the Potential of CD4+ iNKT Cells but Are Biased toward the Th1-like Phenotype
All expanded re-iNKT cells showed invariant TCR expression (Figure S2B and Table 1) and expressed CD122 and CD150 at similar levels. In addition, the activation- and cytotoxicity-associated molecules CD56, CD69, CD161, and CD314 were detected in most of the expanded re-iNKT cells (Figure 2A). Re-iNKT cells also changed their expression dominance from CD45RA to CD45RO during α-GalCer-based stimulation, consistent with the effector memory phenotype (Figure S2E). Although re-iNKT cells were originally generated from CD4+ iNKT cells, their surface phenotype was more similar to DN iNKT cells. We therefore analyzed the global gene expression profiles of re-iNKT cells and other primary lymphoid cell subsets. Hierarchical clustering of transcriptomes enabled by mRNA sequencing indicated that re-iNKT cells are closest to freshly isolated iNKT cells and next closest to expanded iNKT cells after TCR stimulation (Figure 2B). This relationship was confirmed by the hierarchical clustering of selected genes related to NK/T cells, including 30 transcription factors, 13 cytokines, and ten cytokine receptors (Figures 2C and Table S1). Several genes known to characterize iNKT cells, including the innate lymphoid cell-related transcription factor ZBTB16 and the Th1 type pro-inflammatory cytokine and receptor genes IFNG, IFNGR1, and IL-12RB1, were preferentially expressed in re-iNKT cells before TCR stimulation (Figure 2C, blue box). On the other hand, the Th2 helper T cell-related transcription factor GATA3, the Notch signal-related transcription factor RBPJ, and the NK/T cell-related cytokine and receptor genes TNF, IL-5, IL-13, CSF1, and IL2RA were preferentially expressed in activated re-iNKT cells and expanded primary iNKT cells (Figure 2C, red boxes). Taken together, these findings indicate that although re-iNKT cells show a DN iNKT cell-like surface phenotype, their expression profile covers CD4+ iNKT cell-related Th1 and Th2 type genes.
Figure 2.

Gene and Molecular Expression Profile of Re-iNKT Cells
(A) Flow cytometric analysis of T cells re-differentiating from CD4+ iNKT-iPSCs on day 10 after TCR stimulation. iPSC-derived re-differentiating cells were stimulated with α-GalCer-pulsed (10 ng/ml) and irradiated PBMCs from an HLA-A24-negative donor. The results shown are the surface protein expression patterns of re-differentiating cells gated by HLA-A24, CD45, CD3, and 6B11, and are representative of four experiments. The blue and red populations in the right two panels respectively indicate unstained and stained samples for the indicated molecules.
(B and C) Global gene expression profiles (B) and two-way clustering of 53 selected gene expression profiles (C) related to NK/T cell differentiation and function for re-iNKT cells (clones 2 and 4; both from donor B) and primary immune cells. Levels of mRNA expression in re-iNKT cells and primary immune cells were determined using RNA sequencing. Heatmaps show the differential expression of genes among samples. Red and blue indicate increased and decreased expressions, respectively. Blue and red boxes in (C) indicate gene clusters preferentially up-regulated in resting and activated re-iNKT cells, respectively.
Microarray-based DNA methylation analysis was performed to evaluate the epigenetic status of re-iNKT cells. Correlation plots indicated that the methylation level of re-iNKT cells was generally higher than that of original CD4+ or DN iNKT cells, which were similar (Figure S3A). A detailed profile of each gene revealed that master transcriptional factors of iNKT cells, such as ZBTB16, TBX21, and GATA3, were similarly methylated among the three cell populations. Th1-related cytokine genes such as tumor necrosis factor α (TNF-α) and IFN-γ were also similarly methylated among the populations, but Th2-related cytokine genes such as IL-4 and IL-5 were preferentially methylated in re-iNKT cells. The hypermethylation of CD4 gene could contribute to the absence of CD4 expression in CD4+ iNKT-iPSC-derived re-iNKT cells (Figure S3B).
When DCs were used as antigen-presenting cells (APCs), re-iNKT cells produced IFN-γ and negligible amounts of IL-4 (Figure 3A), and the IFN-γ production was inhibited by the addition of a blocking mAbs specific for CD1d (Figure 3B). CD1d restriction was also confirmed by using C1R-CD1d as the APC (Figure 3C). When anti-CD3 mAb was used for TCR stimulation, re-iNKT cells produced both Th1 and Th2 cytokines, including IL-2, IL-4, IL-5, IL-13, GM-CSF (granulocyte macrophage colony-stimulating factor), IFN-γ, and TNF-α, just as parental CD4+ iNKT cells do (Figures 3D and S4). The IFN-γ/IL-4 ratio in re-iNKT cells stimulated with anti-CD3 mAb was higher than that in parental iNKT cells, indicating that re-iNKT cells had a more Th1-biased profile than the parental cells (Figures 3A, 3D, and S4).
Figure 3.

CD1d-Restricted, α-GalCer-Specific Response and Cytokine Profile of Re-iNKT Cells
(A) IFN-γ and IL-4 production. Re-iNKT cells or parental iNKT cells were co-cultured with DCs in the presence of vehicle or 100 ng/ml α-GalCer.
(B) Re-iNKT cells were cultured with α-GalCer DCs in the presence of the indicated blocking Abs. Blockade was assessed based on IFN-γ production.
(C) α-GalCer-dependency and CD1d-restriction. Re-iNKT cells were cultured with C1R cells expressing mock or the indicated molecules in the presence of vehicle or 100 ng/ml α-GalCer.
(D) Cytokine profiles. Re-iNKT cells or parental iNKT cells were stimulated for 24 hr with plate-bound control immunoglobulin G (IgG) or anti-CD3 mAb (10 μg/ml), after which levels of the indicated cytokines in the culture supernatants were evaluated.
Data were run in triplicate, and the experiment was repeated three times. The results of one representative experiment are shown. Error bars depict mean ± SD.
Activation of Re-iNKT Cells Induces DC Maturation and Downstream Activation of Both Cancer Antigen-Specific CTLs and NK Cells
iNKT cells significantly influence the efficacy of immune responses by modulating DC function. CD40L molecules expressed by activated iNKT cells stimulate DCs through CD40 ligation, which induces DC maturation characterized by CD80 (B7-1) and CD86 (B7-2) elevation following IL-12p70 production (Brennan et al., 2013). IL-12p70 is a strong NK cell activator and a crucial inducer of functional antigen-specific Th1 cells and CTL responses (Trinchieri, 2003). We initially confirmed that the stimulation of TCRs on re-iNKT cells using immobilized anti-CD3 mAb activated the cells and enhanced surface CD40L expression (Figure 4A). Thereafter, vehicle DCs or α-GalCer DCs were co-cultured with re-iNKT cells, and the expression of the co-stimulatory molecule CD86 along with IL-12p70 production in the differentially conditioned DCs was evaluated (Figures 4B and 4C). α-GalCer DCs conditioned by re-iNKT cells (re-iNKT/α-GalCer DCs) expressed higher levels of CD86 than re-iNKT/vehicle DCs, and re-iNKT/α-GalCer DCs produced IL-12p70, although at a level lower than that produced by α-GalCer DCs conditioned by the parental iNKT cells (iNKT/α-GalCer DCs) (Figure 4C). In addition, the IL-12p70 production could be inhibited using a blocking anti-CD1d or anti-CD40L mAb (Figure S5A). These findings indicate that activated re-iNKT cells are able to induce DC maturation and IL-12p70 production via CD40 ligation.
Figure 4.

Induction of Tumor Antigen-Specific CTLs via Re-iNKT Cell-DC Interaction
(A) Re-iNKT cells or parental iNKT cells were stimulated with plate-bound anti-CD3 mAb, after which CD40L expression was evaluated. The mean fluorescence intensity (MFI) of CD40L is shown.
(B) Vehicle- or α-GalCer DCs were cultured with re-iNKT cells at a DC/re-iNKT cell ratio of 10:1, after which CD86 expression on DCs was evaluated. The MFI of CD86 is shown. Medium DCs and OK432 DCs served as references.
(C) IL-12p70 production by DCs stimulated using the indicated conditions.
(D) Schematic representation of the WT1235–243-specific CTL priming assay.
(E and F) Vehicle- or α-GalCer DCs were cultured with or without re-iNKT cells (clone 2). Differentially conditioned DCs were irradiated and cultured with autologous CD8+ T cells ± WT1235–243 peptide. (E) Proliferative responses were measured as [3H]thymidine incorporation. (F) Increased frequencies of WT1235–243-specific CTLs after repeated stimulation were determined using WT1 tetramer. HIV tetramer served as a negative control.
(G) Cytotoxic activities of pWT1-primed CD8+ T cells toward C1R-A∗24:02 cells ± WT1 peptide were determined using 51Cr-release assays at the indicated E/T ratios.
(H) Proliferative responses of NK cells (1.0 × 105) cultured for 48 hr in the presence of 25% cell-free supernatants that were from iNKT-DC co-culture. Medium control and IL-2 (300 IU/ml) plus IL-12 (20 ng/ml) control served as references.
In (A), (B), (F), and (G), one representative result from at least two independent experiments is shown. In (C), (E), and (H) data were run in triplicate, and experiments were repeated at least twice; the results of one representative experiment are shown. Error bars depict mean ± SD.
Studies have shown that iNKT cells act via DCs to provide help in priming diverse, tumor antigen-specific CD8+ T cells (Brennan et al., 2013). Functional re-iNKT cells would be extremely useful for preventing tumor escape from antigen-specific monoclonal CTLs through tumor antigen mutagenesis if functionality could be confirmed. We therefore performed CTL priming experiments using a representative cancer antigen, Wilms' tumor 1 (WT1) (Figure 4D) (Cheever et al., 2009). Initially, re-iNKT cells were co-cultured with vehicle DCs or α-GalCer DCs to induce DC maturation. The DCs were then loaded with an HLA-A∗24:02-restricted, modified 9-mer peptide derived from WT1 (WT1235–243), irradiated, and cultured with autologous CD8+ T cells for antigen-specific priming of CTLs. After 6 days of culture, we observed markedly greater proliferation of WT1235–243-tetramer-positive CD8+ T cells in cultures stimulated by WT1235–243 peptide-loaded α-GalCer DCs than in cultures stimulated by WT1235–243 peptide-loaded vehicle DCs (Figures 4E, 4F, and S5B). Moreover, the CTLs induced by WT1235–243 peptide-loaded α-GalCer DCs exhibited cytotoxic activity toward WT1235–243 peptide-loaded HLA-A24+ C1R cells, but not toward vehicle-loaded HLA-A24+ C1R cells (Figure 4G). These data indicate that re-iNKT cells activated by α-GalCer exert cellular adjuvant effects that boost tumor antigen-specific polyclonal CTL responses via DCs and could be useful for developing re-iNKT cell-based adjuvant therapies. Another important function of iNKT cells is that they have the potential to induce downstream activation of NK cells via interaction with α-GalCer-loaded DCs. As indicated in Figures 4H and S5C, NK cells up-regulated the activation marker CD69 and showed effective expansion in the presence of supernatant from re-iNKT cell/α-GalCer-loaded DC interaction.
Re-iNKT Cells Show NK Cell-like Cytotoxic Activity through a Mechanism Dependent on NKG2D and Enhanced DNAM-1 Pathways
Earlier studies indicate that iNKT cells exhibit perforin- or TRAIL-mediated cytotoxicity toward some hematopoietic malignancies and DCs (Metelitsa et al., 2003, Nicol et al., 2000, Nieda et al., 2001, Takahashi et al., 2000). In addition, certain NKG2D-expressing DN iNKT cell subsets kill K562 NK-sensitive cells, which lack CD1d and HLA-I but are rich in the NKG2D ligands MICA and MICB, and also express ULBP2 and ULBP4 (Kuylenstierna et al., 2011). Because the major re-iNKT cell population expresses NKG2D as well as CD56 and CD161, two other NK markers, we evaluated the cytotoxic potential of re-iNKT cells in vitro using 51Cr-release assays. Like the parental iNKT cells, re-iNKT cells displayed minimal cytotoxic activity toward vehicle-pulsed DCs (vehicle DCs) and only weak cytotoxicity toward α-GalCer-pulsed DCs (α-GalCer DCs) (Figure 5A). However, in contrast to the parental iNKT cells, which exhibited little cytotoxicity toward K562 or U937 cells, re-iNKT cells showed remarkable cytotoxic activity, even at low effector/target (E/T) ratios (Figure 5B). On the other hand, re-iNKT cells showed only weak cytotoxicity toward Daudi cells, which lack NKG2D ligands such as MICA/B, ULBP1, ULBP2, and ULBP3, and DNAM-1 ligands such as poliovirus receptor (PVR; CD155) or nectin-2 (PVRL; CD112) (Figures S6A and S6B). Both re-iNKT cells and parental iNKT cells expressed intracellular perforin and granzyme B as well as surface DNAM-1 (CD226) (Figure 5C). However, the expression of Fas ligand (FasL; CD178), natural cytotoxicity receptors (NCRs; NKp44 and NKp46), and NKG2D was restricted to re-iNKT cells and was absent from parental CD4+ NKT cells (Figure 5C). Blocking experiments revealed that re-iNKT cells mediated perforin-, NKG2D-, and DNAM-1-dependent K562 cell lysis, while U937 cell lysis induced by re-iNKT cells was mediated mainly by perforin and DNAM-1 (Figure 5D). These data suggest that re-iNKT cells can kill cancer cells via NKG2D- and DNAM-1-dependent mechanisms.
Figure 5.

NKG2D- and DNAM-1 Mediated TCR-Independent Cytotoxicity of re-iNKT Cells
Parental CD4+ iNKT cells and re-iNKT cells were stimulated with irradiated PBMCs pulsed with α-GalCer in the presence of rhIL-2 and rhIL-15 and then expanded and used for the assays.
(A) Cytotoxicity of re-iNKT cells and parental CD4+ iNKT cells toward autologous vehicle DCs and α-GalCer DCs at the indicated E/T ratios.
(B) Cytotoxicity of re-iNKT cells and parental CD4+ iNKT cells toward K562 and U937 cells at the indicated E/T ratios.
(C) Expression of cytotoxicity-related molecules in IL-2-pretreated re-iNKT cells or parental iNKT cells. Expression of the indicated molecules (open) and isotype-matched controls (filled) is shown. The MFI of each molecule is shown in the upper right corner.
(D) Cytotoxicity toward the indicated tumor cell lines at an E/T ratio of 2.5:1 was tested in the presence of the indicated Abs blocking receptor/ligand interactions or after pretreating re-iNKT cells with 10 nM concanamycin A (CMA) to block perforin.
(E) CD96 and TIGIT expression. Re-iNKT cells or parental iNKT cells were stimulated with plate-bound control IgG or anti-CD3 mAb (10 μg/ml). Expression of the indicated molecules (open) and isotype-matched controls (filled) is shown. The MFI of each molecule is shown in the upper right corner.
In (C) and (E), one representative result from two to three independent experiments is shown. In (D), data were run in triplicate, and experiments were repeated twice; the results of one representative experiment are shown. Error bars depict mean ± SD.
Because DNAM-1 is expressed in both re-iNKT cells and parental iNKT cells (Figure 5C), we asked why DNAM-1-mediated cytotoxicity was restricted to re-iNKT cells. The ligands for DNAM-1, PVR, and nectin-2 share with CD96 and TIGIT an inhibitory receptor important for the modulation of NK cell or CTL function. Both CD96 and TIGIT attenuate effector responses by competitively inhibiting or interfering with DNAM-1-mediated co-stimulation of CTLs (Chan et al., 2014, Johnston et al., 2014). Flow cytometric analysis revealed that TIGIT is strongly expressed on parental iNKT cells but never on re-iNKT cells, whereas CD96 is expressed on both (Figures 1D and 5E). These findings suggest that weak expression of TIGIT on re-iNKT cells may be crucial for enhanced DNAM-1-mediated target cell killing and that re-iNKT cells exert cytotoxicity through this mechanism, which is unlike parental iNKT cells.
Discussion
In this study, we demonstrated the regeneration and expansion of human iNKT cells via iPSCs. Re-iNKT cells were originally induced from CD4+ iNKT cells, although re-iNKT cells were CD4−CD8− DN and exhibited Th1-biased properties and cytotoxicity. One possible explanation for this phenotype is that it reflects the effect of IL-15, which is reportedly indispensable for the proper development and terminal maturation of iNKT cells (Gordy et al., 2011). Indeed, our established protocol, in which exogenous IL-15 was added, achieved differentiation and functional maturation of re-iNKT cells through the up-regulation of T-bet, which accelerates IL-2/-15Rβ expression, leading to differentiation of Th1-biased re-iNKT cells via a positive feedback effect of IL-15 signaling. The re-differentiation may depend in part on the aberrant differentiation of progenitor T cells into mature re-iNKT cells. Specifically, iNKT cells derived from iPSCs never exhibit a CD4+CD8+ DP stage during in vitro differentiation, which suggests re-iNKT cells skip that physiological step of iNKT cell differentiation. It is known that some lymphoid cells with innate-like immune functionality are directly derived at the CD4−CD8− DN progenitor T cell stage under the control of the transcriptional repressor ID2. These innate lymphoid cells, which include NK cells, have been characterized by their effector functionality, and some exhibit CD4+ T helper cell-like functions that are also observed in iNKT cell subsets (Brennan et al., 2013, Eberl et al., 2015). At the moment, we do not know whether the normal counterpart of re-iNKT cells is within the innate lymphoid subsets. Further investigation to clarify the mechanism of re-iNKT cell differentiation will improve the protocol to generate re-iNKT cells that are completely equivalent to natural iNKT cells.
It is often pointed out that tumor cells can escape immune cells by changing their antigenicity through the modification of targeted peptide sequences or reduction of HLA-molecule expression (Schreiber et al., 2011). This is usually the case when immune therapies with antigen-specific monoclonal CTLs fail. Given their functional similarity to natural iNKT cells, re-iNKT cells, which would be available at an unlimited supply, would be expected to exert cellular adjuvant effects via DCs following the activation of tumor antigen-specific polyclonal CTLs and NK cells, although the level of cytokine production induced by α-GalCer DCs was lower in re-iNKT than in parental cells and there was less cytokine variety. In addition, the NK cell-like cytotoxicity of re-iNKT cells could assist with TCR-independent antitumor responses via NKG2D- and DNAM-1-mediated direct killing of tumor cells. TIGIT inhibits DNAM-1 function via the same ligands not only by competitively inhibiting ligand binding but also through direct interaction with DNAM-1, which physically prevents DNAM-1 homodimerization (Johnston et al., 2014, Stanietsky et al., 2009, Yu et al., 2009). The balance between DNAM-1 and TIGIT expression may control iNKT cell cytotoxicity; that is, re-iNKT and parental iNKT cells express equal levels of DNAM-1, but re-iNKT cells do not express TIGIT, which may enable stronger cytotoxic signaling than primary iNKT cells. In addition, TIGIT was recently identified as a new exhaustion marker expressed in tumor-infiltrating lymphocytes and is thought to be an attractive candidate for blockade, along with PD-1, to reverse CTL dysfunction in cancer (Johnston et al., 2014). That re-iNKT cells scarcely express TIGIT and PD-1 is a property that would be beneficial for cells used in cancer immunotherapy. In addition, iNKT cells reportedly control the tumor microenvironment by killing tumor-associated macrophages (Song et al., 2009) and converting myeloid-derived suppressor cells into APCs (De Santo et al., 2010). The general mechanism of the direct killing activity in re-iNKT cells is unknown. The low expression and hypermethylation of BCL11B may be partly responsible (Figures 2C and S3B), as the loss of the Bcl11b expression induces lineage reprogramming from murine T cells to NK cells (Li et al., 2010). Thus, Th1-biased cytokine production and direct TCR-independent killing may enable re-iNKT cells to exert therapeutic effects against the tumor microenvironment. Nevertheless, it will be important to gather additional therapeutic evidence in vitro and in vivo using appropriate animal models, as the characteristics and responsiveness of the re-iNKT cells were not completely identical to those of parental iNKT cells.
iNKT cells have been implicated in the regulation of immune responses associated with a broad range of diseases, including autoimmunity, infectious diseases, and cancer, and their contribution to cell therapy has been much anticipated. Although many technical challenges lie ahead, re-iNKT cells are expected to open new avenues toward broadly applicable iNKT cell-based immunotherapies.
Experimental Procedures
Preparation of Vα24 iNKT Cells and Generation of iNKT-iPSCs
The preparation of human iNKT cells was as described previously (Liu et al., 2008). In brief, PBMCs from three healthy donors were cultured for 15 days in the presence of 10 ng/ml α-GalCer, after which Vα24+6B11+CD4−CD8β− or Vα24+6B11+CD4+CD8β− cells were sorted using a flow cytometer. The sorted cells were then re-stimulated with irradiated PBMCs pulsed with α-GalCer in the presence of recombinant human IL-2 (rhIL-2) and rhIL-15. Establishment and characterization of iPSCs from the iNKT cells were performed as described previously (Nishimura et al., 2013). In brief, iNKT cells were stimulated with irradiated PBMCs pulsed with α-GalCer and transduced with both reprogramming factors (OCT3/4, KLF4, SOX2, and C-MYC) using SeVdp (KOSM) 302L and SV40 large T antigen using SeV18 + SV40LTA/TS15ΔF. Finally, the cells were cultured for several weeks after removal of the SeV vectors from the cytoplasm (Nishimura et al., 2011). The entire study was conducted in accordance with the Declaration of Helsinki and with the approval of institutional ethics boards.
iNKT Cell Differentiation from iNKT-iPSCs
To differentiate iNKT-iPSCs into iNKT cells, we used a previously described T cell differentiation method with slight modification (Nishimura et al., 2013). In brief, clumps of iPSCs were transferred onto C3H10T1/2 feeder cells and cultured in EB medium containing rhVEGF (vascular endothelial growth factor). On day 7, rhSCF and rhFlt3L were added to the culture. On day 14, hematopoietic progenitor cells were collected and transferred onto OP9-DL1 cells and co-cultured in OP9 medium in the presence of rhIL-7 and rhFlt3L. On day 29, rhIL-15 and rhIL-2 were added to the culture (Figure 1B).
Flow Cytometry-Based Assays
Information about the mAbs used for flow cytometry and functional assays are listed in Table S2. Stained cell samples were analyzed using a FACSCalibur or FACSAriaII flow cytometer (BD Biosciences), and the data were processed using FlowJo (Tree Star). CD1d-tetramer (Proimmune) and HLA-A∗24:02/WT1235–243 tetramer were used to detect iNKT cells and WT1 peptide-specific CTLs, respectively, with HLA-A∗24:02/HIV Env584–592 tetramer serving as a negative control. Relative telomere length was measured with a telomere PNA kit/fluorescein isothiocyanate (DAKO). Levels of human cytokines in the culture supernatants were evaluated using a bead-based multiplex immunoassay (FlowCytomix; Affymetrix eBiosciences and BD Cytometric Beads Array; BD Biosciences) or ELISA (Affymetrix eBiosciences). For the multiplex assay, data were acquired on a FACSCanto flow cytometer (BD Biosciences).
Cell Proliferation
Cell proliferation was evaluated by direct enumeration of cells and by using [3H]thymidine incorporation assays as described by Liu et al. (2008).
CTL Priming and Cytotoxicity Assays
Both parental iNKT cells and re-differentiated iNKT cells were stimulated with irradiated PBMCs pulsed with α-GalCer in the presence of rhIL-2 and rhIL-15. Vehicle- or α-GalCer-pulsed autologous DCs were cultured with the cells for 12 hr. The differentially conditioned DCs were then irradiated and cultured with CD8+ T cells in the presence of WT1235–243 peptide. After 6 days of culture, the proliferative responses of the cells were measured by [3H]thymidine incorporation assay. After 10 days of culture, the frequencies of WT1 peptide-specific CTLs were determined using HLA-A∗24:02/WT1235–243 tetramer, and the cells were re-stimulated using irradiated autologous PBMCs pre-pulsed with WT1 peptide in the presence of rhIL-2. After an additional 9 days of culture, the expansion of WT1 peptide-specific CTLs was determined. The cytotoxicity of the CTLs and re-iNKT cells was measured using standard 51Cr-release assays.
RNA Sequencing of Re-iNKT Cells
We extracted total RNA using an RNeasy micro kit (Qiagen) as instructed by the manufacturer. cDNA was synthesized using a SMARTer Ultra Low Input RNA for Illumina Sequencing -HV kit (Clontech), after which the Illumina library was prepared using a Low Input Library Prep kit (Clontech). The libraries were sequenced using HiSeq 2500 in 101 cycle Single-Read mode. All sequence reads were extracted in FASTQ format using BCL2FASTQ Conversion Software 1.8.4 in the CASAVA 1.8.2 pipeline. The sequence reads were mapped to hg19 reference genes, downloaded on December 10, 2012 using TopHat v2.0.8b, and quantified using RPKMforGenes. The data have been deposited in NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO: GSE76371 for gene expression microarray and GEO: GSE76372 for genome-wide DNA methylation analysis.
Statistics
PRISM (GraphPad Software) was used for all statistical analyses. t Tests for paired or unpaired data were used to assess the significance of differences between the means of two experimental groups.
Author Contributions
S. Kitayama and R.Z. performed iPSC establishment and differentiation experiments, and were involved in all analyses; T.Y.L., N.U., S.I., Y.K., Y.Y., M.T., N.H., and T.I. performed parts of the cellular and molecular analyses; A.W. and Y.M. performed gene expression and DNA methylation analysis; M.N. and K.K. provided materials critical for the study; and Y.U. and S. Kaneko were responsible for the overall design of the study and wrote the manuscript.
Acknowledgments
The authors thank Drs. Shinya Yamanaka, Yasuhiro Yamada, Hiroshi Kawamoto, Yuta Mishima (Kyoto University), and Tetuya Nakatsura (NCC) for helpful discussion; Dr. Ken Nishimura (University of Tsukuba), Ms. Manami Ohtaka (AIST), Kaho Hiramatsu (ACCRI), and Yukiko Kobayashi (Kyoto University) for technical assistance; Dr. Peter Karagiannis (Kyoto University) for editing the manuscript; and Dr. Hiromitsu Nakauchi (The University of Tokyo) for providing the cell lines. This work was supported in part the Japanese Ministry of Education, Culture, Sports, Science and Technology, 23592022 (Y.U.), 26462078 (Y.U.), 25861253 (R.Z.), 26670578 (S. Kaneko), 25114707 “Carcinogenic Spiral”(S. Kaneko), and Core Center for iPS Cell Research of Research Center Network for Realization of Regenerative Medicine (S. Kaneko) by the Japanese Ministry of Education, Culture, Sports, Science and Technology; the Aichi Cancer Research Foundation (Y.U.); Takeda Science Foundation (K.K.), The National Cancer Center Research and Development Fund (25-A-7, Y.U.); Daiwa Securities Health Foundation (Y.U.); Pancreas Research Foundation of Japan (Y.U.); and SENSHIN Medical Research Foundation (Y.U.). S. Kaneko is a founder and shareholder at AsTlym Co., Ltd, and Thyas Co., Ltd. M.N. is a founder and CTO of TOKIWA-Bio Inc.
Published: February 9, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.01.005.
Contributor Information
Yasushi Uemura, Email: yuemura@east.ncc.go.jp.
Shin Kaneko, Email: kaneko.shin@cira.kyoto-u.ac.jp.
Supplemental Information
References
- Berzins S.P., Smyth M.J., Baxter A.G. Presumed guilty: natural killer T cell defects and human disease. Nat. Rev. Immunol. 2011;11:131–142. doi: 10.1038/nri2904. [DOI] [PubMed] [Google Scholar]
- Brennan P.J., Brigl M., Brenner M.B. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013;13:101–117. doi: 10.1038/nri3369. [DOI] [PubMed] [Google Scholar]
- Castillo E.F., Acero L.F., Stonier S.W., Zhou D., Schluns K.S. Thymic and peripheral microenvironments differentially mediate development and maturation of iNKT cells by IL-15 transpresentation. Blood. 2010;116:2494–2503. doi: 10.1182/blood-2010-03-277103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerundolo V., Silk J.D., Masri S.H., Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat. Rev. Immunol. 2009;9:28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- Chan C.J., Martinet L., Gilfillan S., Souza-Fonseca-Guimaraes F., Chow M.T., Town L., Ritchie D.S., Colonna M., Andrews D.M., Smyth M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014;15:431–438. doi: 10.1038/ni.2850. [DOI] [PubMed] [Google Scholar]
- Chang D.H., Osman K., Connolly J., Kukreja A., Krasovsky J., Pack M., Hutchinson A., Geller M., Liu N., Annable R. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J. Exp. Med. 2005;201:1503–1517. doi: 10.1084/jem.20042592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever M.A., Allison J.P., Ferris A.S., Finn O.J., Hastings B.M., Hecht T.T., Mellman I., Prindiville S.A., Viner J.L., Weiner L.M. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santo C., Arscott R., Booth S., Karydis I., Jones M., Asher R., Salio M., Middleton M., Cerundolo V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat. Immunol. 2010;11:1039–1046. doi: 10.1038/ni.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G., Di Santo J.P., Vivier E. The brave new world of innate lymphoid cells. Nat. Immunol. 2015;16:1–5. doi: 10.1038/ni.3059. [DOI] [PubMed] [Google Scholar]
- Godfrey D.I., Berzins S.P. Control points in NKT-cell development. Nat. Rev. Immunol. 2007;7:505–518. doi: 10.1038/nri2116. [DOI] [PubMed] [Google Scholar]
- Godfrey D.I., Stankovic S., Baxter A.G. Raising the NKT cell family. Nat. Immunol. 2010;11:197–206. doi: 10.1038/ni.1841. [DOI] [PubMed] [Google Scholar]
- Gordy L.E., Bezbradica J.S., Flyak A.I., Spencer C.T., Dunkle A., Sun J., Stanic A.K., Boothby M.R., He Y.W., Zhao Z. IL-15 regulates homeostasis and terminal maturation of NKT cells. J. Immunol. 2011;187:6335–6345. doi: 10.4049/jimmunol.1003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C., Lee H., Park Y.K., Shin J., Jung S., Kim H., Hong S., Park S.H. Regulation of secondary antigen-specific CD8(+) T-cell responses by natural killer T cells. Cancer Res. 2009;69:4301–4308. doi: 10.1158/0008-5472.CAN-08-1721. [DOI] [PubMed] [Google Scholar]
- Johnston R.J., Comps-Agrar L., Hackney J., Yu X., Huseni M., Yang Y., Park S., Javinal V., Chiu H., Irving B. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26:923–937. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Kuylenstierna C., Bjorkstrom N.K., Andersson S.K., Sahlstrom P., Bosnjak L., Paquin-Proulx D., Malmberg K.J., Ljunggren H.G., Moll M., Sandberg J.K. NKG2D performs two functions in invariant NKT cells: direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur. J. Immunol. 2011;41:1913–1923. doi: 10.1002/eji.200940278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Burke S., Wang J., Chen X., Ortiz M., Lee S.C., Lu D., Campos L., Goulding D., Ng B.L. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science. 2010;329:85–89. doi: 10.1126/science.1188063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.Y., Uemura Y., Suzuki M., Narita Y., Hirata S., Ohyama H., Ishihara O., Matsushita S. Distinct subsets of human invariant NKT cells differentially regulate T helper responses via dendritic cells. Eur. J. Immunol. 2008;38:1012–1023. doi: 10.1002/eji.200737838. [DOI] [PubMed] [Google Scholar]
- Maus M.V., Fraietta J.A., Levine B.L., Kalos M., Zhao Y., June C.H. Adoptive immunotherapy for cancer or viruses. Annu. Rev. Immunol. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen-Smith R.M., Salio M., Cerundolo V. The Regulatory role of invariant NKT cells in tumor immunity. Cancer Immunol. Res. 2015;3:425–435. doi: 10.1158/2326-6066.CIR-15-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metelitsa L.S., Weinberg K.I., Emanuel P.D., Seeger R.C. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia. 2003;17:1068–1077. doi: 10.1038/sj.leu.2402943. [DOI] [PubMed] [Google Scholar]
- Mittal D., Gubin M.M., Schreiber R.D., Smyth M.J. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr. Opin. Immunol. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molling J.W., Kolgen W., van der Vliet H.J., Boomsma M.F., Kruizenga H., Smorenburg C.H., Molenkamp B.G., Langendijk J.A., Leemans C.R., von Blomberg B.M. Peripheral blood IFN-gamma-secreting Valpha24+Vbeta11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int. J. Cancer. 2005;116:87–93. doi: 10.1002/ijc.20998. [DOI] [PubMed] [Google Scholar]
- Molling J.W., Langius J.A., Langendijk J.A., Leemans C.R., Bontkes H.J., van der Vliet H.J., von Blomberg B.M., Scheper R.J., van den Eertwegh A.J. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J. Clin. Oncol. 2007;25:862–868. doi: 10.1200/JCO.2006.08.5787. [DOI] [PubMed] [Google Scholar]
- Motohashi S., Ishikawa A., Ishikawa E., Otsuji M., Iizasa T., Hanaoka H., Shimizu N., Horiguchi S., Okamoto Y., Fujii S. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. 2006;12:6079–6086. doi: 10.1158/1078-0432.CCR-06-0114. [DOI] [PubMed] [Google Scholar]
- Motohashi S., Nagato K., Kunii N., Yamamoto H., Yamasaki K., Okita K., Hanaoka H., Shimizu N., Suzuki M., Yoshino I. A phase I-II study of alpha-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J. Immunol. 2009;182:2492–2501. doi: 10.4049/jimmunol.0800126. [DOI] [PubMed] [Google Scholar]
- Motz G.T., Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol A., Nieda M., Koezuka Y., Porcelli S., Suzuki K., Tadokoro K., Durrant S., Juji T. Human invariant valpha24+ natural killer T cells activated by alpha-galactosylceramide (KRN7000) have cytotoxic anti-tumour activity through mechanisms distinct from T cells and natural killer cells. Immunology. 2000;99:229–234. doi: 10.1046/j.1365-2567.2000.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol A.J., Tazbirkova A., Nieda M. Comparison of clinical and immunological effects of intravenous and intradermal administration of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells. Clin. Cancer Res. 2011;17:5140–5151. doi: 10.1158/1078-0432.CCR-10-3105. [DOI] [PubMed] [Google Scholar]
- Nieda M., Nicol A., Koezuka Y., Kikuchi A., Lapteva N., Tanaka Y., Tokunaga K., Suzuki K., Kayagaki N., Yagita H. TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood. 2001;97:2067–2074. doi: 10.1182/blood.v97.7.2067. [DOI] [PubMed] [Google Scholar]
- Nishimura K., Sano M., Ohtaka M., Furuta B., Umemura Y., Nakajima Y., Ikehara Y., Kobayashi T., Segawa H., Takayasu S. Development of defective and persistent Sendai virus vector: a unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011;286:4760–4771. doi: 10.1074/jbc.M110.183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Kaneko S., Kawana-Tachikawa A., Tajima Y., Goto H., Zhu D., Nakayama-Hosoya K., Iriguchi S., Uemura Y., Shimizu T. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Noy R., Pollard J.W. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow M.A., Callahan M.K., Wolchok J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J., Neparidze N., Zhang L., Nair S., Monesmith T., Sundaram R., Miesowicz F., Dhodapkar K.M., Dhodapkar M.V. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood. 2013;121:423–430. doi: 10.1182/blood-2012-06-435503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salio M., Silk J.D., Jones E.Y., Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu. Rev. Immunol. 2014;32:323–366. doi: 10.1146/annurev-immunol-032713-120243. [DOI] [PubMed] [Google Scholar]
- Schietinger A., Greenberg P.D. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35:51–60. doi: 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Shah D.K., Zuniga-Pflucker J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014;192:4017–4023. doi: 10.4049/jimmunol.1302259. [DOI] [PubMed] [Google Scholar]
- Song L., Asgharzadeh S., Salo J., Engell K., Wu H.W., Sposto R., Ara T., Silverman A.M., DeClerck Y.A., Seeger R.C. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J. Clin. Invest. 2009;119:1524–1536. doi: 10.1172/JCI37869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanietsky N., Simic H., Arapovic J., Toporik A., Levy O., Novik A., Levine Z., Beiman M., Dassa L., Achdout H. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA. 2009;106:17858–17863. doi: 10.1073/pnas.0903474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Nieda M., Koezuka Y., Nicol A., Porcelli S.A., Ishikawa Y., Tadokoro K., Hirai H., Juji T. Analysis of human V alpha 24+ CD4+ NKT cells activated by alpha-glycosylceramide-pulsed monocyte-derived dendritic cells. J. Immunol. 2000;164:4458–4464. doi: 10.4049/jimmunol.164.9.4458. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Uchida T., Horiguchi S., Tanaka Y., Yamamoto H., Kunii N., Motohashi S., Taniguchi M., Nakayama T., Okamoto Y. Phase I study of alpha-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer. Cancer Immunol. Immunother. 2008;57:337–345. doi: 10.1007/s00262-007-0373-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura Y., Liu T.Y., Narita Y., Suzuki M., Nakatsuka R., Araki T., Matsumoto M., Iwai L.K., Hirosawa N., Matsuoka Y. Cytokine-dependent modification of IL-12p70 and IL-23 balance in dendritic cells by ligand activation of Valpha24 invariant NKT cells. J. Immunol. 2009;183:201–208. doi: 10.4049/jimmunol.0900873. [DOI] [PubMed] [Google Scholar]
- Veillette A., Dong Z., Latour S. Consequence of the SLAM-SAP signaling pathway in innate-like and conventional lymphocytes. Immunity. 2007;27:698–710. doi: 10.1016/j.immuni.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Vizcardo R., Masuda K., Yamada D., Ikawa T., Shimizu K., Fujii S., Koseki H., Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Watarai H., Fujii S., Yamada D., Rybouchkin A., Sakata S., Nagata Y., Iida-Kobayashi M., Sekine-Kondo E., Shimizu K., Shozaki Y. Murine induced pluripotent stem cells can be derived from and differentiate into natural killer T cells. J. Clin. Invest. 2010;120:2610–2618. doi: 10.1172/JCI42027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K., Horiguchi S., Kurosaki M., Kunii N., Nagato K., Hanaoka H., Shimizu N., Ueno N., Yamamoto S., Taniguchi M. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol. 2011;138:255–265. doi: 10.1016/j.clim.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Yu X., Harden K., Gonzalez L.C., Francesco M., Chiang E., Irving B., Tom I., Ivelja S., Refino C.J., Clark H. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.