

Abstract
To fully characterize the CoIII–‘nitrene radical’ species that are proposed as intermediates in nitrene transfer reactions mediated by cobalt(II) porphyrins, different combinations of cobalt(II) complexes of porphyrins and nitrene transfer reagents were combined, and the generated species were studied using EPR, UV–vis, IR, VCD, UHR-ESIMS, and XANES/XAFS measurements. Reactions of cobalt-(II) porphyrins 1P1 (P1 = meso-tetraphenylporphyrin (TPP)) and 1P2 (P2 = 3,5-DitBu-ChenPhyrin) with organic azides 2Ns (NsN3), 2Ts (TsN3), and 2Troc (TrocN3) led to the formation of mono-nitrene species 3P1Ns, 3P2Ts, and 3P2Troc, respectively, which are best described as [CoIII(por)(NR″•−)] nitrene radicals (imidyl radicals) resulting from single electron transfer from the cobalt(II) porphyrin to the ‘nitrene’ moiety (Ns: R″ = –SO2-p-C6H5NO2; Ts: R″ = –SO2C6H6; Troc: R″ = –C(O)OCH2CCl3). Remarkably, the reaction of 1P1 with N-nosyl iminoiodane (PhI=NNs) 4Ns led to the formation of a bis-nitrene species 5P1Ns. This species is best described as a triple-radical complex [(por•−)CoIII(NR″•−)2] containing three ligand-centered unpaired electrons: two nitrene radicals (NR″•−) and one oxidized porphyrin radical (por•−). Thus, the formation of the second nitrene radical involves another intramolecular one-electron transfer to the “nitrene” moiety, but now from the porphyrin ring instead of the metal center. Interestingly, this bis-nitrene species is observed only on reacting 4Ns with 1P1. Reaction of the more bulky 1P2 with 4Ns results again in formation of mainly mono-nitrene species 3P2Ns according to EPR and ESI-MS spectroscopic studies. The mono- and bis-nitrene species were initially expected to be five- and six-coordinate species, respectively, but XANES data revealed that both mono- and bis-nitrene species are six-coordinate Oh species. The nature of the sixth ligand bound to cobalt(III) in the mono-nitrene case remains elusive, but some plausible candidates are NH3, NH2−, NsNH−, and OH−; NsNH− being the most plausible. Conversion of mono-nitrene species 3P1Ns into bis-nitrene species 5P1Ns upon reaction with 4Ns was demonstrated. Solutions containing 3P1Ns and 5P1Ns proved to be still active in catalytic aziridination of styrene, consistent with their proposed key involvement in nitrene transfer reactions mediated by cobalt(II) porphyrins.
Graphical Abstract
INTRODUCTION
Catalytic functionalization of C–H bonds is a desirable tool in organic and organometallic chemistry, as it is an atom-, time-, and cost-efficient alternative to traditional hydrocarbon functionalization.1–3 Therefore, C–H insertion of carbenoid and nitrenoid species has emerged as a very promising protocol in the recent years.4,5 The insertion of metal carbenoids into C–H bonds is now a well-established transformation, and many transition-metal catalysts that can catalyze such reactions have been disclosed in the past decade.4,6,7 In addition to catalytic cyclopropanation and C–H insertion, metal-catalyzed carbene transfer has also made it possible to perform insertions into X–H bonds (X = O, N, S, Si) and to convert alkynes to cyclopropenes (including some cycloaddition reactions).4 Given the ubiquity of nitrogen atoms in biologically active compounds,8 nitrene transfer reactions also have very important applications in making molecules of interest. However, the applicability of metal-nitrenoids has so far been limited to alkene aziridination,9,10 C–H amination11,12 and amidation.13
Among several catalysts that have been reported for nitrene transfer reactions, the most successful ones are still based on non-abundant Rh4–6,13–17 and Ru4,6,7,18–20 metals. Among the first row transition metals, there have been reports of Mn,21,22 Cu,23–26 and Fe27 catalyzed systems. Besides being very efficient in carbene transfer reactions,28–31 cobalt(II) porphyrin catalysts have also attracted attention in nitrene transfer reactions. Until the early 2000s, it was quite common to use iminoiodanes32–36 or haloamine-T23,25,26 compounds as nitrene sources. These are, however, not the most benign nitrene sources as they lead to the formation of undesirable side products like phenyl iodide and other halogen-containing compounds. With the efforts of various groups, organic azides37,38 were discovered to be potentially greener resources to transfer nitrenes, as the only byproduct formed during the generation of nitrenes from azides is dinitrogen. The other disadvantage of using iminoiodanes is their poor solubility and the fact that there is not much scope to change the pre-existing functionality on the nitrogen atom.
With the use of differently meso-substituted cobalt(II) porphyrins (1P1–1P4 in Figure 1) a variety of nitrene transfer reagents (Figure 2) have been employed. For example, with the use of reagents like diphenyl phosphoryl azide (DPPA),39,40 it is possible to further modify the substituent, as the nitrogen–phosphorus bond in the product aziridine is readily hydrolyzed. In the pursuit of finding more nitrene transfer reagents with easily removable groups on the nitrogen atom, trichloroethoxysulfonyl azide41 (TcesN3, Tces = trichloroethoxysulfonyl = CCl3CH2O(SO2)–) was found to be effective. Apart from the nitrene precursors itself, specially tailored cobalt porphyrins, for example, with H-bonding functionalities, have further enhanced the applicability of these systems (see Figure 1; P1 = meso-tetraphenylporphyrin, P2 = 3,5-ditBu-ChenPhyrin; P3 = 2,6- diMeO-ZhuPhyrin; P4 = 3,5-ditBu-IbuPhyrin). With the use of 1P4, organic azides like tosyl azide can also be used as a viable nitrene source. H-bonding interactions between the nitrene moiety and the arms of the catalyst 1P4 facilitate nitrenoid formation42 and lead to an increased catalyst efficiency and lifetime. Enantioselective aziridination of alkenes was reported a few years ago using chiral cobalt(II)-porphyrin 1P3 with TcesN3, and enantioselectivity of 99% could be obtained.41
Figure 1.

Commonly used cobalt(II) porphyrins in nitrene transfer reactions.
Figure 2.

Commonly used nitrene transfer reagents.
The mechanistic aspects of two such catalytic nitrene transfer reactions with cobalt porphyrins have been elucidated by our groups previously (Scheme 1).9,43 The mechanism of C–H bond amination of ethylbenzene, toluene, and 1,2,3,4-tetrahydronaphthalene (tetralin) using a series of different organic azides (N3C(O)OMe, N3SO2Ph, N3C(O)Ph and N3P(O)(OMe)2) as nitrene sources was studied using density functional theory (DFT) and electron paramagnetic resonance (EPR) spectroscopy (Scheme 1a).43 The mechanism of cobalt(II) porphyrin-mediated aziridination of styrene with PhSO2N3 was also studied (Scheme 1b).9 For both amination and aziridination reactions, the DFT calculations revealed a stepwise radical process involving coordination of the azide to the cobalt(II) center, followed by release of dinitrogen to produce an unusual ‘nitrene radical’ intermediate C (Scheme 1). In addition, experimental EPR spectroscopic studies, combined with DFT calculated EPR properties being in good agreement with the experimental data, revealed the formation of a (por)CoIII–N•Y nitrene radical adduct C from the catalyst in the presence of an excess of the azide in benzene. Formation of a nitrene moiety at cobalt(II) effectively leads to electron transfer from the metal to the nitrene, thus reflecting the redox non-innocence of the nitrene ligand. The spin density of this intermediate resides almost entirely on the nitrogen atom of the nitrene moiety. A simplified molecular orbital picture showing the frontier π-interactions and explaining the unusual electronic structure of this intermediate is depicted in Scheme 2.
Scheme 1.

(a) DFT-Calculated Mechanism of Co(II) Porphyrin-Mediated Benzylic C–H Amination of Ethylbenzene by N3C(O)OMe and (b) Co(II) Porphyrin- Mediated Aziridination of Styrene with PhSO2N3
Scheme 2.

Redox Non-Innocent Behaviour of Nitrene Ligands Coordinated to Open-Shell CoII(por) Species (A), a Simplified Bonding Scheme Explaining This Behavior (B), and Alternative Bonding Scheme Involving a Triplet Nitrene (C)
Experimental detection of these radicaloid species led us to investigate the subsequent stepwise C–H amination sequence computationally. According to these investigations, the amination reaction follows a stepwise radical pathway, in which the nitrene radical intermediate C readily abstracts a hydrogen atom from the benzylic position of the organic substrate. This leads to formation of a close-contact pair (D) of the thus-formed organic radical R′• and the cobaltIII–amido complex (por)CoIII–NHR″. Subsequent back-attack of the organic radical onto the amido moiety in D, in a rebound-type mechanism, leads to release of the desired amine products with regeneration of the cobalt(II) catalyst, which proceeds with low activation barriers according to DFT.
Nitrene transfer to styrene also proceeds in a stepwise manner via radical addition of the nitrene radical C to the C= C double bond of styrene to form γ-alkyl radical intermediates D′ (Scheme 1b). The cobalt(III) ion in D′ has an intermediate (S = 1) spin-state with its unpaired electrons antiferromagnetically coupled to the unpaired electron of the γ-alkyl radical, thus explaining its overall doublet (S = 1/2) state. Species D′ easily collapses in an almost barrierless ring closure reaction to form the aziridine, thereby regenerating the cobalt(II) porphyrin catalyst. Additionally, the computed free energy profile well explained the superior performance of the reported CoII(porAmide) system 1P4 with H-bond donor functionalities over the non-functionalized system 1P1 (see Figure 2).42
Thus, the DFT calculations and the X-band EPR data gathered thus far strongly indicate the formation of substratecentered (por)CoIII–N•–R″ nitrene radicals, which are proposed to be key intermediates in the mechanisms of the catalytic C–H amination and alkene aziridination reactions mediated by CoII(por) catalysts and employing organic azides as nitrene sources. These nitrene radicals have thus far only been detected by means of X-band EPR spectroscopy.9,43 Here we disclose their detection and characterization with a variety of spectroscopic and mass spectrometric techniques.
Ligand-centered radical complexes play a pivotal role in a number of bio- and “bioinspired” catalytic transformations, providing a powerful tool to control the reactivity and selectivity of the catalyst.44–46 Such species are well-studied and characterized for transition-metal complexes containing typical polydentate redox non-innocent ligands, such as semi quinone-type radical ligands47–49 and their nitrogen analogs, 47–50 reduced bipyridine and terpyridine ligands,51–53 α-iminopyridine ligands,54–56 and pyridine-2,6-diimine ligands. 57–61 More recently, considerable efforts have been made to understand the (electronic) structure and reactivity of monodentate redox non-innocent substrate-type ligands, such as carbenes,62 which play a key role in catalytic carbene transfer reactions.63,64 So far, only a few examples have been reported in which complexes bearing monodentate nitrogen-based radicaloid ligands were experimentally detected. Thus, well-studied complexes with redox active aminyl ligand radicals are limited to only a few well-characterized examples,65 and related complexes bearing redox non-innocent nitrene ligands are even scarcer.9,43,65–67 Given the importance of these radicaloid nitrene species in metal-catalyzed nitrene transfer and C–H functionalization reactions,10,12,13,39–42,68 we gathered more experimental evidence for the formation of the previously reported mono-nitrene radical complexes upon reaction of cobalt(II) porphyrins with nitrene transfer reagents.9,43 Furthermore, we here reveal the first example of a bis-nitrene species of 1P1 upon reacting the stronger oxidizing nitrene transfer agent N-nosyl iminoiodane 4Ns with 1P1. This bisnitrene species has a markedly different electronic structure than the previously detected mono-nitrene species and also differs from the electronic structure of the previously reported diamagnetic bis-imido RuVI-porphyrin species (por)RuVI((= NR″)2).69–73 The work described herein further bears some similarity with the mono- and bis-nitrene species of non-heme iron complexes disclosed by Che and co-workers,66 but again the electronic structures reported herein are entirely different.
RESULTS AND DISCUSSION
To study the key nitrene intermediates in nitrene transfer reactions mediated by cobalt(II) porphyrins, we chose to react a few of the commonly used nitrene sources with two different types of cobalt(II) porphyrins: the tetraphenyl-substituted porphyrin 1P1 and the bulkier (and chiral) H-bond donor appended porphyrin 1P2 (see Figure 1). Complex 1P2 serves as a model system for all reported cases where CoII porphyrins with H-bonding moieties were proven to give superior catalytic results and where H-bonding stabilizes the formed nitrene intermediates.41,8 In total, four different nitrene sources were employed. As mentioned before, these sources are known in literature to be active in different nitrene transfer reactions like aziridination and amination.8–11 Three of these were organic azides 2Ns, 2Ts, and 2Troc (see Figure 2): Nosyl azide (2Ns; NsN3; Ns = Nosyl = p-NO2–PhSO2–), Tosyl azide (2Ts; TsN3; Ts = Tosyl = p-MePhSO2–), and Troc azide (2Troc; TrocN3; Troc =2,2,2-trichlorethoxycarbonyl = CCl3CH2O-(CO)–). The fourth nitrene transfer reagent investigated was the stronger oxidizing N-nosyl iminoiodane (PhI=NNs) 4Ns. To perform the EPR, ultra high-resolution electron spray ionization mass spectrometry (UHR-ESI), UV–vis, and X-ray absorption spectroscopy (XAS) studies, the following method was employed: To a solution of the catalyst in benzene-d6 we added a 100-fold excess of the nitrene precursor. The catalyst concentrations of these solutions were typically 2.5 mM. The solvent used was benzene-d6 in all cases, and this was chosen to avoid any cases of C–H insertions as is known for these systems in toluene, cyclohexane, and other related solvents.10,11 Unless mentioned otherwise, deuterated benzene was chosen as the solvent to further slow down any C–H activation of the benzene ring itself.
EPR Spectroscopy of the Mono-Nitrene Species
Upon reaction of 1P1 with a 100-fold excess of 2Ns and 1P2 with a 100-fold excess of either 2Ts or 2Troc (Scheme 3), clear gradual changes in the X-band EPR spectra occurred that point to the formation of mono-nitrene radical species, revealing the redox non-innocence of the nitrene moiety. The disappearance of signals characteristic for the cobalt(II) porphyrins is associated with appearance of signals corresponding to mono-nitrene species of the type 3 (for an example, see Figure S5-1; Supporting Information). The room-temperature (r.t.) EPR measurement of the same samples gave spectra characteristic for ligand radical species (Figure 3, top),42 and the simulated spectra fits best to a mono-nitrene species of the type 3.
Scheme 3.

Different Combinations of Cobalt(II) Porphyrins and Organic Azides Used to Study the Formation of Mono-Nitrene Species
Figure 3.

Top: Experimental and simulated (Table 1) X-band EPR spectra of nitrene radical ligand complex 3P1Ns in solution at r.t. Isotropic spectrum in solution recorded at r.t.42 (Freq = 9.38056 GHz; mod. amp. = 1 G; microwave power = 0.2 mW). Bottom: Experimental and simulated (Table 1) high-field EPR spectra of nitrene radical ligand complex 3P1Ns obtained by mixing 1P1 and 2Ns in frozen solution (Freq = 243.76176 GHz, mod. amp. = 1 mT, microwave power = 1 mW, T = 50 K).
The X-band EPR spectrum recorded from a reaction mixture of 1P1 and 2Ns in frozen benzene-d6/o-terphenyl solution at 50 K was quite isotropic, but the g-anisotropy is clearly resolved in high-frequency (244 GHz) EPR measurements (Figure 3, bottom). The experimental g-anisotropy is very small, and smaller than calculated with DFT (Table 1). This may be caused by a small mismatch between the calculated and experimental geometries, as the calculated g-tensor is sensitive to the orientation of the –SO2Ar fragment of the nitrene radical moiety (e.g., optimization with and without dispersion corrections). It should further be noted at this point that complexes of type 3P1Ns need not be five-coordinate. Coordination of an additional ligand to the cobalt(III) center (see also Scheme S5-5; Supporting Information) is perhaps even likely (vide infra), and this will also have some influence on the (calculated) g-tensor and hyperfine coupling parameters of the mono-nitrene complex, albeit very small (see Table 1 for a comparison of the experimental EPR parameters and the DFT calculated values of five-coordinate 3P1Ns and its six-coordinate NsNH− and OH− adducts). Note that a variety of other six-coordinate analogs of 3P1Ns investigated, containing either neutral or anionic ligands bound trans to the nitrene radical moiety, all have very similar calculated g-values and HFIs with cobalt and the nitrogen atom of the spin-bearing nitrene radical ligand (see Table S5-1, Supporting Information).
Table 1.
EPR Parameters of Nitrene Radical Complex 3P1Ns in Frozen Solution at 50 K and in Isotropic Solution at Room Temperature
| 50 K | 298 K | ||||
|---|---|---|---|---|---|
| g11 | g22 | g33 | gav | giso | |
| Exp | 2.0087 | 2.0049 | 2.0005 | 2.0047 | 2.005 |
| DFTa | |||||
| 5-coord | 2.0490 | 2.0154 | 1.9810 | 2.0150 | 2.015 |
| 5-coorde | 2.0287e | 2.0029e | 1.9816e | 2.0044e | 2.004e |
| NsNH−f | 2.0311 | 2.0052 | 1.9966 | 2.0109 | 2.011 |
| OH−g | 2.0387 | 2.0116 | 2.0022 | 2.0175 | 2.018 |
| ACo | ACo11b) | ACo22b) | ACo33b) | ACoavb) | ACoisob) |
| Exp | −50c) | 50c) | 20c) | 7c) | −25 |
| DFTa) | |||||
| 5-coord | −98 | 65 | 13 | −7 | −7 |
| 5-coorde | −18e | −89e | 15e | −42e | −31e |
| NsNH−g | −59 | 30 | 11 | −6 | −6 |
| OH−h) | −62 | 50 | 67 | 18 | 18 |
| AN | AN11b) | AN22b) | AN33b) | ANavb) | ANisob) |
| Exp | n.r.d) | n.r.d) | n.r.d) | n.r.d) | 10 |
| DFTa) | |||||
| 5-coord | −20 | −14 | 81 | 15 | 16 |
| 5-coorde | −23e | −14e | 84e | 16e | 16e |
| NsNH−f | −11 | −9 | 87 | 22 | 22 |
| OH−g | −13 | −10 | 84 | 20 | 20 |
Geometry of 3P1Ns optimized with Turbomole (full atom model, BP86, def2-TZVP) employing Grimme’s D3-dispersion corrections (disp3); EPR parameters calculated with ORCA, (b3-lyp/def2-TZVP).
MHz.
Data from Q-band Davies ENDOR measurements (see Supporting Information).
Not resolved.
Geometry of 3P1Ns (full atom model) optimized with Turbomole at the b3-lyp, def2-TZVP level without dispersion corrections to match the solution data.
Optimized geometry of six-coordinate NsNH− amido adduct of 3P1Ns (full atom model, BP86, def2-TZVP) employing Grimme’s D3-dispersion corrections (disp3).
Optimized geometry of six-coordinate OH− hydroxido adduct of 3P1Ns (full atom model, BP86, def2-TZVP) employing Grimme’s D3-dispersion corrections (disp3).
Additionally, a quite anisotropic cobalt hyperfine interaction (HFI) tensor was revealed by Q-band Davies ENDOR measurements. The Q-band Davies ENDOR experiment (Figure S5-2; Supporting Information) revealed broad features spanning 1–60 MHz. Apart from the characteristic proton signals centered around 52 MHz, strong lines are observed around 22 and 12 MHz. The position of these features is consistent with the 59Co hyperfine parameters estimated from the high-field EPR spectrum (Figure 3), which are summarized in Table 1. Low-frequency contributions (<8 MHz) are attributed to the porphyrin nitrogen atoms. The corresponding DFT calculated values match the experimental ones quite well (Table 1), albeit that the calculated anisotropy of the cobalt hyperfine tensor is a bit larger than in the experimental spectrum. Unfortunately, the anisotropic nitrene nitrogen hyperfine couplings were not resolved for technical reasons (see Supporting Information for discussion).
Interestingly, the mono-nitrene species 3 did not form instantaneously upon mixing the azides and the CoIIporphyrin catalysts at r.t. For example, only 10% of the mono-nitrene species 3P2Troc was formed upon mixing 1P2 and azide 2Troc at r.t. after 15 min. These conversions were determined by comparison of the spin concentrations (double integration) with a sample of TEMPO of the same concentration. It should be mentioned that the relaxation times of TEMPO and the mono-nitrene species 3 are likely markedly different, and hence the spin integrations gave only a rough qualitative comparison of the spin concentrations. However, upon heating the samples, higher intensities were observed immediately; and on letting them heat overnight at 45 °C, the maximum intensities for all of these species were observed. The resulting solutions still contained the (excess) organic azide (as revealed by analysis of crystals recovered upon evaporation of the solvent) and were still catalytically active in aziridination of styrene (as confirmed by 1H NMR and GC-MS analysis of the crude reaction mixture). Among the series of complexes and nitrene precursors studied, the maximum intensities were obtained for the combination of 1P2 and 2Troc (see Figure 4). This can be explained on the basis of better stabilization of the formed nitrene radical intermediate by H-bonding between the amide arms of the porphyrin backbone in 1P2 and the nitrene transfer agent 2Troc.8,41 The concentration of this species was found to be ~80% referenced against a sample of TEMPO with the same concentration as the cobalt precursor. Similar rough spin counting reveals the presence of ~60% of EPR active 3P1Ns upon reaction of 1P1 with excess 2Ns after heating overnight at 45°C.
Figure 4.

EPR spectra of 3P2Troc (r.t.; benzene-d6) showing increasing intensities over time on heating at 45 °C.
EPR Spectroscopy of the Bis-Nitrene Species 5P1Ns
In contrast to the use of organic azides that produced mono-nitrene species of the type 3 upon reaction with the cobalt(II) porphyrin complexes, reaction of an excess of N-nosyl iminoiodane 4Ns with 1P1 at room temperature in benzene-d6 yielded an entirely different species (Scheme 4), as revealed by X-band EPR spectroscopy in solution at r.t. A completely different isotropic spectrum was obtained with 4Ns (Figure 5) than that with nosyl azide 2Ns or tosyl azide 2Ts (Figure 3, top). The multiline spectrum revealed an isotropic g-value close to ge (2.003) with well-resolved hyperfine interactions with the two equivalent Nnitrene atoms (AN-nitrene iso = 10.0 MHz) and the four equivalent Nporphyrin atoms (AN-por iso = 3.5 MHz), again indicating formation of a “ligand radical” complex. The isotropic cobalt HFI in 5P1Ns (ACoiso = 2.0 MHz) was very small, even smaller than in the mono-nitrene case (25 MHz). The spectrum could be satisfactorily simulated based on the DFT calculated EPR parameters (Figure 5 and Table 2).
Scheme 4. Reaction of Cobalt(II) Porphyrin 1P1 and Nitrene Transfer Reagent 4Ns To Give Bis-Nitrene Species 5P1 Nsa.

aPlease note that the formation of a one-electron oxidized porphyrin ligand is indicated with a positive charge on the porphyrin ring, using the “π-cation radical” convention commonly used in cytochrome P450 chemistry,74 but this is in fact a mono-anionic por•− ligand.
Figure 5.

Top: Overlay of the isotropic X-band EPR spectra of unlabeled 5P1Ns (red) and labeled (15N)2-5P1Ns (blue). Bottom: Experimental and simulated isotropic EPR spectra of species (15N)2-5P1Ns in benzene-d6. Freq = 9.38126 GHz, mod. ampl. = 1 G, microwave power = 0.2 mW, T = 298 K.
Table 2.
| Equiv. Nuclei | Expa) | DFTb) | |
|---|---|---|---|
| giso | 2.003 | 2.008 | |
| ACoisoc) | 1(I = 7/2) | −2.0 | −2.5 |
| AN-nitreneiso14Nc) | 2(I=1) | 10.0 | 16.0d) |
| AN-nitreneiso14Nc) | 2(I = ½) | 14.0 | 22.4 |
| AN-porisoc) | 4(I=1) | 3.5 | −1.5e) |
Derived from spectral simulations shown in Figure 5.
Geometry optimized with Turbomole (b3-lyp/def2-TZVP) using a simplified model of 5P1Ns without porphyrin Ph substituents without dispersion corrections to match the r.t. solution EPR data. EPR parameters calculated with ORCA (b3-lyp/def2-TZVP).
MHz.
Average of two AN-nitrene hyperfines.
Average of four AN-por hyperfines.
To prove that the largest detected nitrogen hyperfine couplings stem from the two equivalent nitrene moieties, we further prepared the 15N-labeled iminoiodane 15N-4Ns (PhI=15NNs) and recorded the r.t. EPR spectrum of (15N)2-5P1Ns generated in a mixture of 1P1 and 15N-4Ns. This led to clear differences in the hyperfine coupling pattern compared to the non-labeled analogue 5P1Ns (Figure 5, top).
EPR simulation using an expected 1.4 times larger AN-nitreneiso 15N (I = 1/2) hyperfine coupling (14 MHz) compared to the unlabeled 14N AN-nitrene iso constant of 10 MHz (I = 1) in 5P1Ns and otherwise identical spectral parameters provided an excellent fit of the experimental spectrum (Figure 5, bottom; Table 2). Hence, the largest detected nitrogen hyperfine couplings indeed stem from the two equivalent nitrene moieties.
In case of the bis-nitrene species 5P1Ns the intensity of the signal was not as strong as the mono-nitrene species, for example, 3P1Ns. On heating the sample shortly, the intensity increased only very slightly and was at least 10 times lower than that of the mono-nitrene species (spin counting amounts to ~8%, using a reference sample containing TEMPO at the same concentration as 1P1). The poor solubility of the N-nosyl iminoiodane substrate in benzene-d6 could well be a reason for the low intensities obtained for species 5P1Ns. It is also worth mentioning that the experimentally detected signal of species 5P1Ns stems from a (net) doublet spin state (S = 1/2), leading to the characteristic EPR data shown in Figure 5 and Table 2, but in DFT (b3-lyp) the doublet (S = 1/2) and quartet (S = 3/2; ΔG = +0.2 kcal mol−1) spin states of 5P1Ns are predicted to be of nearly equal energy.75 As only the doublet state is detectable with EPR at r.t. in solution, the potential existence of a (slow) thermal equilibrium between the two spin states might also explain the relatively low EPR intensity of the signal, which stems from the S = 1/2 state of bis-nitrene species only. Furthermore, a word about the higher reactivity (intrinsic instability) of these bis-nitrene species is worth mentioning. In contrast to the mono-nitrene species 3P1Ns, which increased in time upon heating the solution to 45 °C, species 5P1Ns decomposed at this temperature, causing the EPR signal of the bis-nitrene species to disappear completely overnight (i.e., in absence of other substrates). Catalyst decomposition may be less important in the presence of suitable substrates, as heating the solution in the presence of styrene did reveal the expected aziridination activity (as confirmed by 1H NMR and GC-MS analysis of the crude reaction mixture).
Steric and Electronic Influence of the Catalyst on Formation of the Nitrene Intermediates
The steric and electronic influence of the catalyst on the species obtained, even when using the more oxidizing N-nosyl iminoiodane 4Ns, is quite dramatic. For example, on using the bulkier porphyrin complex 1P2 in combination with the oxidizing N-nosyl iminoiodane 4Ns, the bis-nitrene species was detected only in minor amounts. Instead, a major EPR signal corresponding to the mono-nitrene species was detected. Thus, it may be concluded that the bulky side groups on the porphyrin backbone of 1P2 make it difficult for the second molecule of N-nosyl iminoiodane 4Ns to react.
In contrast to the decomposition observed for species 5P1Ns that was obtained from a mixture of 1P1 and an excess of 4Ns, mono-nitrene species 3P2Ns obtained from a mixture of 1P2 and excess 4Ns proved to be much more stable. On heating the latter sample to 45 °C, the signal intensity decreased, with the complete disappearance of the initially detected small amount of bis-nitrene species 5P2Ns. However, a significant amount of mono-nitrene species 5P2Ns remained present in solution, even after heating for a period of 18 h (Figure 6). This was also observed with ESI-MS measurements (Figure S4-20; Supporting Information). These results clearly show that the bis-nitrene species 5 is more reactive than the mono-nitrene species 3 and that complex 1P2 is more stable than complex 1P1 when combined with N-nosyl iminoiodane 4Ns• The latter can perhaps be attributed to stabilizing H-bonding interactions between the nitrene radical moiety and the H-bond-donating amido functionalities incorporated in the porphyrin backbone of 1P2, as was previously observed in aziridination reactions and studied previously using DFT.8,41 At the same time, it also suggests that N-nosyl iminoiodane 4Ns is not a very benign nitrene transfer reagent for these reactions as it seems to undergoes subsequent side reactions with the catalyst (especially for the less bulky catalysts and in the absence of another substrate).
Figure 6.
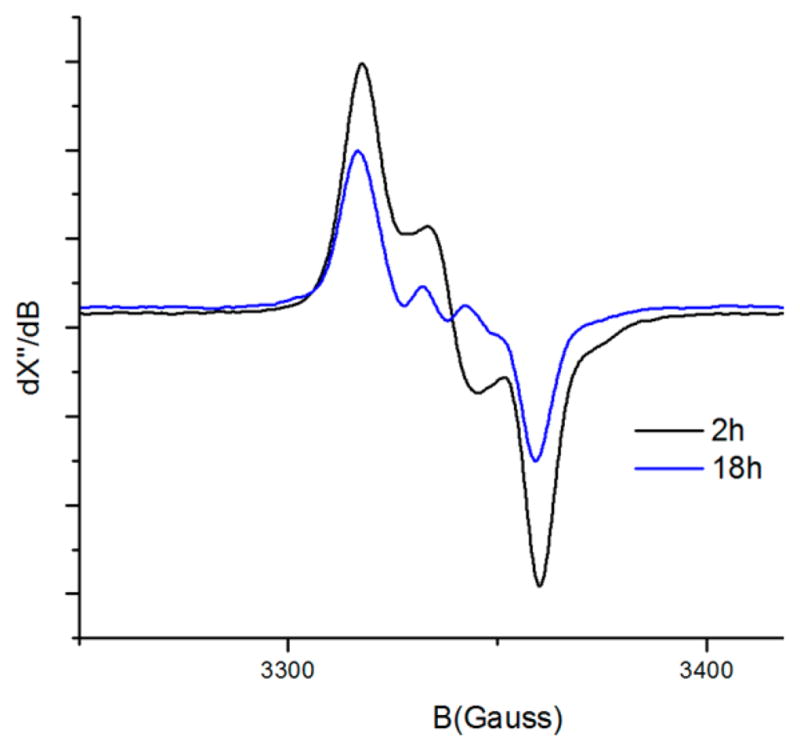
EPR spectra of 1P2 with 4Ns• Sample obtained by heating the reaction mixture to 45 °C; spectra recorded at r.t. in benzene-d6. Note the decrease in the intensity of the signal over time.
UHR-ESI-MS Spectrometry
To further corroborate the above observations in the EPR measurements, the reactions were also investigated with mass spectrometry. We investigated the formation of both the mono- and the bis-nitrene species. High-resolution ESI-MS mass spectrometry proved to be a suitable method to detect the formation of both mono- and bisnitrene species. However, since these species are neutral (or perhaps anionic, vide infra), only signals resulting from protonation, (one-electron) oxidation, and formation of Na+ adducts can be expected in positive mode UHR-ESI-MS.
A reaction mixture of 1P1 with nosyl azide 2Ns in C6D6/ MeCN produced clear ESI-MS signals around m/z 872 characteristic for formation of the mono-nitrene species 3P1Ns (Scheme 3). The isotope distribution pattern and exact masses determined by ESI-MS match the theoretical values (Δm/z < 0.004 Da) for the species in its protonated form [3P1Ns + H]+ (Figure 7a). The relatively low intensity of the detected nitrene radical species derived from reaction of complex 1P1 with azide 2Ns is likely a result of incomplete conversion to 3P1Ns, as the measurements were performed within 30 min of sample preparation. At the same time, subsequent reactions with the cosolvent acetonitrile cannot be neglected. While 3P1Ns is detected as a five-coordinate species in its protonated form with ESI-MS, this does not exclude 3P1Ns from being six-coordinate (see Scheme S5-5; Supporting Information), as the sixth ligand might easily dissociate in the ionization chamber of the ESI-MS spectrometer. We were not able to detect 3P1Ns in negative mode ESI-MS measurements (cold-spray ionization). However, interestingly, in the positive mode ESI-MS measurements (cold-spray ionization), a Na+ adduct of NO2PhSO2NHD was detected from a mixture of 1P1 and 2Ns• Note that formation of NO2PhSO2ND• radicals, detected with EPR, has been reported earlier.8 On this basis, the most likely candidates for the sixth ligand coordinated to the mono-nitrene species are either the NO2PhSO2NDH amine ligand or the NO2PhSO2ND− amido ligand (in both cases detected as the Na+ adduct of the amine in the ESI-MS measurements, in the first case directly and in the second case after HAT).
Figure 7.

Observed (top) and simulated (bottom) ESI-MS spectrum with isotope distribution (a) [3P1Ns + H+] (870–876 au) and (b) [5P1Ns + H• + H+] (1070–1080 au).
ESI-MS spectra recorded from a mixture of 1P1 and N-nosyl iminoiodane 4Ns revealed signals pointing to the presence of bis-nitrene species 5P1Ns (Scheme 4). This species was observed as the protonated form of its hydrogen atom abstraction (HAA) reaction product ([5P1Ns + H• + H+]), thus pointing to rapid HAA from the cosolvent MeCN (Figure 7b). Substantially weaker signals corresponding to the mono-nitrene species 3P1Ns were still detected ([3P1Ns + H]+), which likely resulted from incomplete conversion of 1P1 to 5P1Ns and/or fragmentation of 5P1Ns to 3P1Ns in the ionization chamber of the ESI-MS spectrometer.
Electronic Structures and UV–vis Spectra of Monoand Bis-Nitrene Species
To gain additional insight into the electronic structures of the generated nitrene species, we performed DFT geometry optimizations at the b3-lyp/def2- TZVP level using the simplified models 3P0Ns and 5P0Ns without meso-phenyl substituents on the porphyrin ring P0 (Figure 8). According to these calculations, mono-nitrene species 3P1Ns formed from 1P1 and 2Ns is best described as a CoIII species with one 1e reduced nitrene moiety and a normal, non-oxidized (por2−) porphyrin ligand. Note again the experimental complex 3P1Ns needs not be five-coordinate. Coordination of an additional ligand to the cobalt(III) center cannot be excluded (Scheme S5-5; Supporting Information) and even seems likely (vide infra). However, note that all six-coordinate analogs of 3P0Ns considered computationally have almost identical spin density distributions as the one shown for five-coordinate mono-nitrene complex 3P0Ns in Figure 8. In these calculations full atom models were optimized at the BP86/def2-TZVP/ disp3 level (Turbomole), followed by single point calculations at the b3-lyp/def2-TZVP level (ORCA), considering several different (neutral and anionic) ligands bound trans to the nitrene radical moiety (see also Table S5-1; Supporting Information).
Figure 8.

Spin density plots (b3-lyp/def2-TZVP) of mono-nitrene species 3P0Ns (left) and bis-nitrene species 5P0Ns (right).
The bis-nitrene species 5P0Ns, on the other hand, is a tripleradical containing two 1e-reduced nitrene-moieties and a 1eoxidized porphyrin ring (from por2− to the por•− radical monoanion). As is clear from the positive and negative spin density distribution (Figure 8, right), two of the three unpaired electrons in 5P0Ns are antiferromagnetically coupled, thus effectively leading to a (net) doublet state (Stotal = 1/2). Both 3P1Ns and 5P0Ns were found to contain a low-spin d6 (SCo = 0) CoIII center. Hence, formation of 5 involves one-electron oxidation of CoII to CoIII upon formation of the first nitreneradical (3) and a second intramolecular electron transfer from the porphyrin ring to the second nitrene moiety in the second step.
The DFT calculated electronic structures are qualitatively in agreement with UV–vis measurements, revealing the presence of a non-oxidized porphyrin ring in 3P1Ns and a “porphyrin radical” ligand in 5P1Ns (Figures 9 and 10).
Figure 9.

UV–vis spectra of a mixture of 1P1 and 2Ns followed in time (after 40 min of sample preparation). Clear shifts in both the Q- and Soret-bands were observed corresponding to azide ligation to the cobalt(II) center.
Figure 10.

UV–vis spectral changes upon reaction of (left) complex 1P2 with azide 2Troc to give mono-nitrene species 3P2Troc formed after heating the solution overnight, and (right) reaction of complex 1P1 with iminoiodane 4Ns to give bis-nitrene species 5P1Ns within 1 h.
Metal- (not porphyrin-) centered oxidation of CoII(por) to CoIII(por) is known to produce characteristic red-shifts of both the Soret- and Q-bands.76,77 The same is also true for binding donor ligands, such as pyridine or bipyridyl ligands, to the central metal ion of cobalt(II) porphyrins.78,79 Such shifts were indeed observed upon measuring UV–vis spectra of a mixture of 1P1 and 2Ns immediately upon mixing. In Figure 9, the UV– vis spectrum of a mixture of 1P1 and 2Ns was followed in time (after 40 min of sample preparation). In these spectra, the observed shifts in the Q- and Soret-bands are due to formation of a simple azide adduct (mostly likely a monoazide adduct). Indeed, measuring EPR spectra of these same mixtures (again 40 min after mixing the reagents) without any additional heating revealed extremely weak EPR signals in the region around g = 2.0, corresponding to formation of only a tiny amount of nitrene radical species 3P1Ns•
On measuring the UV–vis spectra of these same solutions after heating them overnight at 45 °C (i.e., solutions that gave strong EPR signals characteristic for 3P1Ns) clearly revealed the presence of a new species in the UV–vis spectra (Figure S6-2, Supporting Information), concomitant with much higher EPR intensities corresponding to nitrene radical species 3P1Ns• Illustrative spectra comparing 1P2 and species 3P2Troc formed upon heating a solution of complex 1P2 in the presence of an excess of 2Troc are shown in Figure 10 (left).
Reaction of complex 1P2 with azide 2Troc resulted in red-shifts of the Soret band at 438 nm and shifting of the Q-band to 648 nm. In addition, a new peak at ~753 nm was also observed (Figure 10, left). The shift in the Soret and the Q-band is indicative of oxidation of the metal from cobalt(II) to cobalt(III) and (or) ligation to the sixth coordination site, leaving the porphyrin ligand intact without oxidation upon formation of mono-nitrene radical species 3P2Troc. The additional band at ~753 nm is likely a charge-transfer band (e.g., a MLCT or LMCT band involving the nitrene radical and the cobalt(III) center).
Formation of porphyrin-based ligand radicals is known to cause drastic changes in the UV–vis spectrum with appearance of red-shifted bands, disappearance of the Q-band and obvious shape changes of the Soret band compared to unmodified (i.e., non-reduced/nonoxidized) dianionic porphyrinato ligands (por2−).80,81 This is what was observed upon formation of bis-nitrene species 5P1Ns in the reaction of complex 1P1 with Nnosyl iminoiodane 4Ns (Figure 10, right). As mentioned before, when using iminoiodane 4Ns, formation of the bis-nitrene species is comparatively faster than formation of the mononitrenes when using azide 2Ns• Accordingly, in the case on measuring the UV–vis spectra of a mixture of 1P1 and 4Ns, the disappearance and shape changes of the Q- and Soret-bands was observed within an hour of making these samples (Figure 10, right). As shown in Figure 10, the species exhibited characteristic, strongly red-shifted bands at 863 and 977 nm. Similar spectral changes have been reported for CoIII complexes with a 1e-oxidized octaethylporphyrin ligand.82,83 Hence, we assign these spectral changes to formation of the bis-nitrene species 5P1Ns• This is also in correspondence with the EPR measurements. Both the EPR and UV–vis signals characteristic for the nitrene radical species 5P1Ns were completely lost upon heating the mixture overnight at 45 °C.
In accordance with the EPR spectroscopic studies, once again the mono-nitrene species 3P2Troc was found to be more stable than bis-nitrene species 5P1Ns• UV–vis spectroscopy clearly indicated that solutions of 3P2Troc obtained from complex 1P2 and 4Troc remained stable for even 2 days, while a significant amount of decomposition already occurred for 5P1Ns obtained from complex 1P1 and 4Ns within 2 h. This points to a higher intrinsic reactivity of bis-nitrene species 5P1Ns compared to mono-nitrene species 3P1Ns• Indeed, DFT calculations (using 3′ and 5′, which are simplified models of 3P1Ns and 5P1Ns without phenyl substituents on the porphyrin ring and having a NSO2Ph base nitrene instead of N-Ns) predict a ca. 1.3 kcal mol−1 lower activation energy for HAA from ethylbenzene for 5′ (ΔG‡ = +30.4 kcal mol−1) than for 3′ (ΔG‡ = +31.7 kcal mol−1).
X-ray Absorption Spectroscopic (XAS) Studies
To gain additional evidence for the coordination mode and the oxidation states of the two intermediates, i.e., the mono- and the bis-nitrene species, XAS measurements were performed. Xray absorption near edge spectroscopic (XANES) studies were carried out at Co K-edge in order to directly probe the metal oxidation states in 3P1Ns, 3P2Troc, and 5P1Ns (Figure 11A). The starting cobalt(II)-meso-tetraphenylporphyrin (TPP) complex 1P1 exhibited an edge inflection energy of ca. 7720.7 eV. A shoulder feature along the rising edge at 7715.4 eV corresponds to a 1s to 4p + LMCT shakedown transition, in accord with the observed four-coordinate square-planar structure of this complex. This transition is strongest in four-coordinate square-planar Co complexes, but is also observed in fivecoordinate square-pyramidal geometries (it is not present in either Td or Oh geometries). Finally, a broad 1s to 3d pre-edge peak was observed at 7708.9 eV, with preliminary peak fitting analysis indicating a peak area of approximately 8.1 units. The energies of the edge and 1s → 3d pre-edge transitions are in accord with the reported values for other cobalt(II) complexes.80,81 XANES of 5P1Ns showed a +1.9 eV blue-shift of the edge inflection energy to 7722.56 eV, relative to complex 1P1, supporting metal-centered oxidation from cobalt(II) to cobalt(III). The 1s → 3d pre-edge transition was also blueshifted by ~1.0 to 7710.2 eV and the pre-edge increased in intensity (peak-area of 12.0 units). Most notably, 5P1Ns lacks the diagnostic 1s → 4p shakedown transition shoulder on the rising edge, providing strong evidence for a six-coordinate Oh cobalt site. Finally, the XANES data on 3P1Ns revealed the existence of edge and 1s → 3d pre-edge transitions at nearly identical positions (7722.8 and 7710.2 eV, respectively) relative to 5P1Ns, thereby indicating that both mono-nitrene complex 3P1Ns and bis-nitrene complex 5P1Ns are cobalt(III) species.
Figure 11.

(A) XANES of 1P1 (red), 3P1Ns (green), and 5P1Ns (blue). Spectra are referenced to the first inflection point of a cobalt reference foil set to 7709.0 eV. (B) Fourier transform EXAFS spectra of 5P1Ns (dotted line) and the best fit (red line); the inset shows the EXAFS data on a wave vector scale weighted by k3 with respective representation. (C) Fourier transform EXAFS spectra of 3P1Ns (dotted line) and the best fit (red line); the inset shows the EXAFS data on a wave vector scale weighted by k3 with respective representation. For further details see Supporting Infomation.
Importantly, similar to the observations for complex 5P1Ns, the absence of the 1s → 4p shakedown transition in the XAS data of 3P1Ns also points to the presence of a six-coordinate Oh cobalt(III) center for this complex. XANES data were also obtained for complex 3P2Troc and compared with that for 3P1Ns and 5P1Ns• Careful examination of the pre-edge of the XANES of different catalyst and oxidant combinations shows no distinct differences between the different generated intermediates (see Figure S3-1; Supporting Information). This indicates that sixcoordinate Oh species were formed in all cases. Extended X-ray absorption fine structure (EXAFS) analysis revealed further structural details (Figures 11B–C and S3-2, S3-3 and Tables 3, S3-1, S3-2, and S3-3). For complex 5P1Ns, the first coordination sphere could be satisfactorily fitted by considering six N/O scatterers at a distance of 1.92 Å (Table 3). Although the additional outer-shell features could be satisfactorily accounted for by considering single scattering paths involving 8 carbons at 2.94 Å, 12 carbons at 3.35 Å, and 4 oxygen donors at 3.57 Å distance from cobalt, the fit could be significantly improved by introducing multiple-scattering pathways. The best fit for 5P1Ns is represented by fit 12 in Table S3-1 and Figure 11B. It is important to note that efforts were also made to include the effect of the sulfur scatterers originating from the two –NNs units. However, all fits (fits 9–11 in Table S3-1) including any kind of S-shell showed negative Debye–Waller factors in the fit parameters; and hence they were not considered in the fitting procedure.
Table 3.
A Comparison of the EXAFS Determined Metrical Parameters (for the first three shells) of 3P1Ns and 5P1Ns with That of the DFT Calculated Values for 3P1Ns(Ns-NH−) and 5P1Nsa
| complex | Co–N/C
|
Co···C/N
|
Co···C/N
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| n | r | σ2 | n | r | σ2 | n | r | σ2 | |
| 3P1Ns (EXAFS) | 6 | 1.95 | 3.7 | 8 | 2.99 | 6.6 | 12 | 3.39 | 14.5 |
| 3P1Ns(Ns-NH−) (DFT) | 6 | 1.94 | 8 | 2.98 | 12 | 3.92 | |||
| 5P1Ns (EXAFS) | 6 | 1.92 | 3.0 | 8 | 2.94 | 4.5 | 12 | 3.35 | 8.7 |
| 5P1Ns (DFT) | 6 | 1.92 | 8 | 2.97 | 12 | 3.91 | |||
Details of the EXAFS simulation and DFT structures can be found in the Supporting Information.
Interestingly, consistent with the XANES data, our attempts to do a set of fits for a back transformation range limited to the first shell (r = 0.8 – 2.1 Å) for 3P1Ns also pointed to a six-coordinate geometry (fits 1–3; Table S3-2). This contrasts with previous assumptions considering mono-nitrene species 3P1Ns to be five-coordinate.8,42 The best fit to the data of the mononitrene species requires six N/O scatterers at ~1.95 Å distance from the cobalt(III) center, which is slightly longer (although almost identical within the error of measurements) than the Co–N distance of 1.92 Å obtained from the EXAFS data of bisnitrene species 5P1Ns•
With these results in hand, we set out to consider plausible ligands that could occupy the sixth coordination site for the mono-nitrene species 3P1Ns• Initially, we considered coordination of a neutral unreacted nosyl azide ligand under the conditions of the EXAFS measurements. However, DFT geometry optimizations and calculation of the Co K-edge features of the optimized structures with DFT methods revealed that such nosyl azide species have too long Co–N distances to explain the experimental XAS data (Figures S8-2; Supporting Information). The same holds for the aqua adduct of 3P1Ns• The Ns-NH2 amine and ammonia (NH3) adducts of 3P1Ns have shorter Co–N bond distances, which substantially decreases the calculated intensity of the pre-edge transitions in the Co K-edge region. However, the best agreement between the calculated and experimental pre-edge intensities is obtained for anionic ligand adducts of mono-nitrene 3P1Ns• The optimized geometries of NsNH−, NH2−, and OH− adducts of 3P1Ns species have comparable Co–N and Co–X distances for the nitrene radical ligand and the sixth ligand X (Figure 12), respectively. Hence, similar pre-edge intensities are also computed as obtained for bis-nitrene species 5P1Ns (Figure S8-3; Supporting Information). Considering the detection of the Na+ adduct of NO2PhSO2NHD as mentioned earlier in the ESI-MS studies in combination with the above-mentioned calculated pre-edge intensities, the NsNH− amido ligand is perhaps the most probable candidate occupying the sixth coordination site of the mono-nitrene species. Notably, the DFT calculated geometrical parameters for 3P1Ns(NsNH−) is in reasonable agreement with the EXAFS data (Table 3). This would mean, however, that 3P1Ns is anionic (perhaps containing an NsNH3 + counterion). Formation of such a charged species in benzene is unexpected, and as such we cannot exclude the sixth ligand being the neutral NsNH2 amine donor (despite a better agreement between the calculated and experimental XAS pre-edge intensities for the NsNH− amido ligand). At this point, we cannot fully exclude formation of dinuclear complexes with ligand X being for example a bridging amido ligand. However, we consider this possibility less likely in view of the fact that similar XAS data were gathered for 3P2Troc, which is based on the bulky porphyrin P2. No peak corresponding to a Co scatterer was observed in the EXAFS spectrum, which would be expected for a homodinuclear Co··· Co species. It is further worth mentioning that the sixth ligand X (Figure 12) does not prevent conversion of mono-nitrene species 3P1Ns to bis-nitrene species 5P1Ns upon treatment with the strongly oxidizing N-nosyl iminoiodane 4Ns• On adding an excess of 4Ns to a solution of previously formed mono-nitrene species 3P1Ns, clear EPR signals corresponding to bis-nitrene species 5P1Ns appeared, once again displaying the strongly oxidizing nature of N-nosyl iminoiodane. Apparently, the ligand X can be replaced or converted to a nosyl nitrene radical moiety upon reaction of mono-nitrene 3P1Ns with iminoiodane 4Ns•
Figure 12.

Left: The six-coordinate mono-nitrene species of the type 3P1Ns obtained from 1P1 and organic azide 2Ns• Right: The sixcoordinate bis-nitrene species 5P1Ns obtained from 1P1 and N-nosyliminoiodane 4Ns•
CONCLUSIONS
In conclusion, we have demonstrated that activation of both organic azides and iminoiodanes by cobalt(II) porphyrin complexes leads to formation of ‘cobalt(III)-nitrene radical’ complexes. Both species bear substantial spin density at the ‘nitrene-moiety’ and are key intermediates in cobalt-catalyzed nitrene transfer reactions. Notably, for the less bulky porphyrin complexes, the obtained species are markedly different. While organic azides generate predominantly mono-nitrene species, hypervalent iodine reagents such as iminoiodanes produce bisnitrene species such as 5P1Ns• This is the first report to demonstrate the formation of a bis-nitrene species of a cobalt porphyrin complex. The stronger oxidizing nature of the nitrene precursor 4Ns is considered important to generate such species. The bis-nitrene species 5P1Ns has a markedly different electronic structure from the mono-nitrene species 3P1Ns, 3P2Ts, and 3P2Troc and also differs markedly from the electronic structure of the previously reported diamagnetic bis-imido RuIV-porphyrin species (por)RuVI((=NR″)2). Electronically, the mono- and the bis-nitrene species are markedly different; but XANES data for these species suggest that both are sixcoordinate (Oh) complexes with similar averaged bond distances between cobalt(III) and the six N/O scatterers (3P1Ns: ~1.95 Å; 5P1Ns: ~1.92 Å). The exact nature of the ligand X coordinated at the sixth available coordination site of the [CoIII(por)(N•R″)] nitrene radical species 3P1Ns, 3P2Ts, and 3P2Troc remains speculative. For 3P1Ns, the most probable candidates (based on a comparison between the experimental and DFT calculated Co K-edge XAS pre-edge intensities, complementary mass data, and the previously detected NsND• radical) seem to be either the neutral NsNHD amine ligand or the anionic NsND− amido ligand.
The revelation of the presence of a sixth ligand even in the mono-nitrene species implies that the use of additives in such catalytic nitrene transfer reactions should not have a positive effect on the catalytic reactions. This is indeed seen to be the case in the earlier reports from the Zhang group, where additives have only a detrimental effect on catalysis if any (except for ee enhancement in one case but in lower yields).39,40
The two different types of catalytically relevant species detected are represented in Figure 12. Overall, these results signify the importance of the nature of nitrene sources in metalcatalyzed nitrene transfer processes. Depending on the nature of the nitrene precursor, the intermediates can be a result of a single oxidation of the catalyst at the metal center or a double oxidation where the second oxidation step occurs at the porphyrin ring. In addition, the bis-nitrene species 5P1Ns, though formed faster, lives shorter and is more reactive than the mono-nitrene species 3P1Ns• Degradation of bis-nitrene species 5P1Ns occurs within hours after initial formation while decomposition of mono-nitrene species 3P1Ns is much slower (solutions of 3P1Ns in benzene-d6 were stable for >24 h according to EPR and UV–vis). In addition, the studies described here clearly demonstrate the better performance of specially tailored cobalt(II) porphyrins like complex 1P2, which not only give higher conversions to the nitrene-radical species but also help in preserving them for longer periods by the stabilizing effects of H-bonding between the catalyst amide arms and the nitrene moiety. The steric influence of the bulk in complex 1P2 also has an implication on the type of intermediates formed, as was shown for the reactions of 1P1 and 1P2 with N-nosyl iminoiodane 4Ns• Detailed studies of the differences in reactivity between the mono- and bis-nitrene and implications in catalysis between these ‘cobalt(III)-nitrene radical’ species are a topic of current investigation and should shed new light on the catalytic mechanisms of nitrene transfer processes catalyzed by cobalt(II) porphyrin complexes.
Supplementary Material
Acknowledgments
We thank Ed Zuidinga (HIMS, UvA) for additional CSI-MS measurements, Dr. Erik R. Farquhar for help with XAS data collection, and Dr. Moniek Tromp for helpful discussions. The work was financially supported by the European Research Council (grant agreement 202886; BdB), The Netherlands Organization for Scientific Research (NWO-CW VICI grant 016.122.613, BdB), CMST COST Action CM1305 (ECOSTBIO), COST action STSM (COST-STSM-CM1305–21241), the University of Amsterdam (UvA), the National Science Foundation (CHE-1152767; X.P.Z.), the National Institutes of Health (R01-GM098777; X.P.Z.), and the “Solar Technologies Go Hybrid” initiative by the State of Bavaria (I.I.-B. and O.T.). K.R. acknowledges financial support from the Cluster of Excellence “Unifying Concepts in Catalysis” (EXC 314/1), Berlin. XAS data were obtained on NSLS beamline X3B (Brookhaven National Laboratory), with support from NIH grant P30-EB-009998 and the U.S. Department of Energy.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Experimental details, synthetic procedures, copies of NMR, MS, IR, VCD, EPR spectra, XANES/XAFS data, energies and geometries of stationary points (DFT), and supporting discussions. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
X. Peter Zhang, Email: xpzhang@usf.edu.
Serena DeBeer, Email: b.debruin@uva.nl.
References
- 1.Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]
- 2.Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- 3.Shilov AE, Shul GB. Chem Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- 4.Doyle MP, Forbes DC. Chem Rev. 1998;98:911–935. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]
- 5.Lu HJ, Zhang XP. Chem Soc Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]
- 6.Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies HML. Angew Chem, Int Ed. 2006;45:6422–6425. doi: 10.1002/anie.200601814. [DOI] [PubMed] [Google Scholar]
- 8.Hili R, Yudin AK. Nat Chem Biol. 2006;2:284–287. doi: 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- 9.Suarez AIO, Jiang H, Zhang XP, de Bruin B. Dalton Trans. 2011;40:5697–5705. doi: 10.1039/c1dt10027k. [DOI] [PubMed] [Google Scholar]
- 10.Jin LM, Xu X, Lu H, Cui X, Wojtas L, Zhang XP. Angew Chem, Int Ed. 2013;52:5309–5313. doi: 10.1002/anie.201209599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu H, Subbarayan V, Tao J, Zhang XP. Organometallics. 2010;29:389–393. [Google Scholar]
- 12.Lu H, Hu Y, Jiang H, Wojtas L, Zhang XP. Org Lett. 2012;14:5158–5161. doi: 10.1021/ol302511f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin L, Lu H, Cui Y, Lizardi CL, Arzua TN, Wojtas L, Cui X, Zhang XP. Chem Sci. 2014;5:2422–2427. doi: 10.1039/C4SC00697F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catino AJ, Nichols JM, Forslund RE, Doyle MP. Org Lett. 2005;7:2787–2790. doi: 10.1021/ol0510973. [DOI] [PubMed] [Google Scholar]
- 15.Shen M, Leslie BE, Driver TG. Angew Chem, Int Ed. 2008;47:5056–5059. doi: 10.1002/anie.200800689. [DOI] [PubMed] [Google Scholar]
- 16.Stokes BJ, Dong H, Leslie BE, Pumphrey AL, Driver TG. J Am Chem Soc. 2007;129:7500–7501. doi: 10.1021/ja072219k. [DOI] [PubMed] [Google Scholar]
- 17.Espino CG, Wehn PM, Chow J, Du Bois J, VSU, April RV, HC J Am Chem Soc. 2001;123:6935–6936. [Google Scholar]
- 18.Shou WG, Li J, Guo T, Lin Z, Jia G. Organometallics. 2009;28:6847–6854. [Google Scholar]
- 19.Liang J, Yuan S, Huang J, Che CM. J Org Chem. 2004;69:3610–3619. doi: 10.1021/jo0358877. [DOI] [PubMed] [Google Scholar]
- 20.Fantauzzi S, Gallo E, Caselli A, Ragaini F, Casati N, Macchi P, Cenini S. Chem Commun. 2009:3952–3954. doi: 10.1039/b903238j. [DOI] [PubMed] [Google Scholar]
- 21.Abu-Omar MM. Dalton Trans. 2011;40:3435–3444. doi: 10.1039/c0dt01341b. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Hong Chan PW, Che C-M. Tetrahedron Lett. 2005;46:5403–5408. [Google Scholar]
- 23.Albone DP, Aujla PS, Taylor PC. J Org Chem. 1998;63:9569–9571. [Google Scholar]
- 24.Clark JS, Roche C. Chem Commun. 2005:5175–5177. doi: 10.1039/b509678b. [DOI] [PubMed] [Google Scholar]
- 25.Fructos MR, Trofimenko S, Diaz-Requejo MM, Perez PJ. J Am Chem Soc. 2006;128:11784–11791. doi: 10.1021/ja0627850. [DOI] [PubMed] [Google Scholar]
- 26.Albone DP, Challenger S, Derrick AM, Fillery SM, Irwin JL, Parsons CM, Takada H, Taylor C, Wilson DJ. J Org Biomol Chem. 2005:107–111. doi: 10.1039/b410883c. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Che CM. Chem - A Eur J. 2010;16:10494–10501. [Google Scholar]
- 28.Chen Y, Ruppel JV, Zhang XP. J Am Chem Soc. 2007;129:12074–12075. doi: 10.1021/ja074613o. [DOI] [PubMed] [Google Scholar]
- 29.Zhu S, Xu X, Perman JA, Zhang XP. J Am Chem Soc. 2010;132:12796–12799. doi: 10.1021/ja1056246. [DOI] [PubMed] [Google Scholar]
- 30.Zhu S, Ruppel JV, Lu H, Wojtas L, Zhang XP. J Am Chem Soc. 2008;130:5042–5043. doi: 10.1021/ja7106838. [DOI] [PubMed] [Google Scholar]
- 31.Zhu S, Perman Ja, Zhang XP. Angew Chem, Int Ed Engl. 2008;47:8460–8463. doi: 10.1002/anie.200803857. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Capretto DA, Rahaman R, He C. Angew Chem, Int Ed. 2007;46:5184–5186. doi: 10.1002/anie.200700760. [DOI] [PubMed] [Google Scholar]
- 33.Chang JWW, Chan PWH. Angew Chem, Int Ed. 2008;47:1138–1140. doi: 10.1002/anie.200704695. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Weinberg R, Breslow R. Chem Commun. 2000:531–532. [Google Scholar]
- 35.Chan J, Baucom KD, Murry JA. J Am Chem Soc. 2007;129:14106–14107. doi: 10.1021/ja073872a. [DOI] [PubMed] [Google Scholar]
- 36.Liang C, Collet F, Robert-Peillard F, Müller P, Dodd RH, Dauban P. J Am Chem Soc. 2008;130:343–350. doi: 10.1021/ja076519d. [DOI] [PubMed] [Google Scholar]
- 37.Driver TG. Org Biomol Chem. 2010;8:3831–3846. doi: 10.1039/c005219c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Intrieri D, Zardi P, Caselli A, Gallo E. Chem Commun. 2014;50:11440–11453. doi: 10.1039/c4cc03016h. [DOI] [PubMed] [Google Scholar]
- 39.Jones JE, Ruppel JV, Gao G, Moore TM, Zhang XP. J Org Chem. 2008;73:7260–7265. doi: 10.1021/jo801151x. [DOI] [PubMed] [Google Scholar]
- 40.Gao G, Jones JE, Vyas R, Harden JD, Zhang XP. J Org Chem. 2006;71:6655–6658. doi: 10.1021/jo0609226. [DOI] [PubMed] [Google Scholar]
- 41.Subbarayan V, Ruppel JV, Zhu S, Perman JA, Zhang XP. Chem Commun. 2009:4266–4268. doi: 10.1039/b905727g. [DOI] [PubMed] [Google Scholar]
- 42.Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP. Org Lett. 2008;10:1995–1998. doi: 10.1021/ol800588p. [DOI] [PubMed] [Google Scholar]
- 43.Lyaskovskyy V, Suarez AIO, Lu H, Jiang H, Zhang XP, de Bruin B. J Am Chem Soc. 2011:12264–12273. doi: 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]
- 44.Luca OR, Crabtree RH. Chem Soc Rev. 2013;42:1440– 1459. doi: 10.1039/c2cs35228a. [DOI] [PubMed] [Google Scholar]
- 45.Lyaskovskyy V, de Bruin B. ACS Catal. 2012;2:270–279. [Google Scholar]
- 46.Van der Vlugt JI. Eur J Inorg Chem. 2012;2012:363–375. [Google Scholar]
- 47.Pierpont CG, Buchanan RM. Coord Chem Rev. 1981;38:45–87. [Google Scholar]
- 48.Kaim W, Schwederski B. Coord Chem Rev. 2010;254:1580– 1588. [Google Scholar]
- 49.Boyer JL, Rochford J, Tsai MK, Muckerman JT, Fujita E. Coord Chem Rev. 2010;254:309–330. [Google Scholar]
- 50.Lever ABP. Coord Chem Rev. 2010;254:1397–1405. [Google Scholar]
- 51.Jones GD, Martin JL, Mcfarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Konovalova T, Desrochers PJ, Pulay P, Vicic DA. J Am Chem Soc. 2006;128:13175–13183. doi: 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]
- 52.Ciszewski JT, Mikhaylov DY, Holin KV, Kadirov MK, Budnikova YH, Sinyashin O, Vicic DA. Inorg Chem. 2011;50:8630–8635. doi: 10.1021/ic201184x. [DOI] [PubMed] [Google Scholar]
- 53.Scarborough CC, Lancaster KM, DeBeer S, Weyhermüller T, Sproules S, Wieghardt K. Inorg Chem. 2012;51:3718–3732. doi: 10.1021/ic2027219. [DOI] [PubMed] [Google Scholar]
- 54.Tejel C, Ciriano MA, del Rio MP, van den Bruele FJ, Hetterscheid DGH, Tsichlis i Spitas N, de Bruin B. J Am Chem Soc. 2008;130:5844–5845. doi: 10.1021/ja711495v. [DOI] [PubMed] [Google Scholar]
- 55.Tejel C, Asensio L, del Río MP, de Bruin B, López JA, Ciriano MA. Angew Chem, Int Ed. 2011;50:8839–8843. doi: 10.1002/anie.201104045. [DOI] [PubMed] [Google Scholar]
- 56.Lu CC, Bill E, Weyhermüller T, Bothe E, Wieghardt K. J Am Chem Soc. 2008;130:3181–3197. doi: 10.1021/ja710663n. [DOI] [PubMed] [Google Scholar]
- 57.de Bruin B, Bill E, Bothe E, Weyhermüller T, Wieghardt K. Inorg Chem. 2000;39:2936–2947. doi: 10.1021/ic000113j. [DOI] [PubMed] [Google Scholar]
- 58.Chirik PJ, Wieghardt K. Science. 2010;327:794–795. doi: 10.1126/science.1183281. [DOI] [PubMed] [Google Scholar]
- 59.Tondreau AM, Milsmann C, Patrick AD, Hoyt HM, Lobkovsky E, Wieghardt K, Chirik PJ. J Am Chem Soc. 2010;132:15046–15059. doi: 10.1021/ja106575b. [DOI] [PubMed] [Google Scholar]
- 60.Intermolecular I, Russell SK, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2011;133:8858–8861. doi: 10.1021/ja202992p. [DOI] [PubMed] [Google Scholar]
- 61.Bouwkamp MW, Bowman AC, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2006;128:13340–13341. doi: 10.1021/ja064711u. [DOI] [PubMed] [Google Scholar]
- 62.Dzik WI, Zhang XP, de Bruin B. Inorg Chem. 2011;50:9896–9903. doi: 10.1021/ic200043a. [DOI] [PubMed] [Google Scholar]
- 63.Dzik WI, Xu X, Zhang XP, Reek JNH, de Bruin B. J Am Chem Soc. 2010;132:10891–10902. doi: 10.1021/ja103768r. [DOI] [PubMed] [Google Scholar]
- 64.Lu H, Dzik WI, Xu X, Wojtas L, de Bruin B, Zhang XP. J Am Chem Soc. 2011;133:8518–8521. doi: 10.1021/ja203434c. [DOI] [PubMed] [Google Scholar]
- 65.Suarez AIO, Lyaskovskyy V, Reek JNH, van der Vlugt JI, de Bruin B. Angew Chem, Int Ed. 2013;52:12510–12529. doi: 10.1002/anie.201301487. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y, Guan X, Wong EL, Liu P, Huang J, Che C. J Am Chem Soc. 2013;135:7194–7204. doi: 10.1021/ja3122526. [DOI] [PubMed] [Google Scholar]
- 67.Kundu S, Miceli E, Farquhar E, Pfaff FF, Kuhlmann U, Hildebrandt P, Braun B, Greco C, Ray K. J Am Chem Soc. 2012;134:14710–14713. doi: 10.1021/ja306674h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu H, Tao J, Jones JE, Wojtas L, Zhang XP. Org Lett. 2010;12:1248–1251. doi: 10.1021/ol100110z. [DOI] [PubMed] [Google Scholar]
- 69.Au S, Huang J, Yu W, Fung W, Che CM. J Am Chem Soc. 1999;121:9120–9132. [Google Scholar]
- 70.Leung SK, Tsui W, Huang J, Che C, Liang J, Zhu N. J Am Chem Soc. 2005;127:16629–16640. doi: 10.1021/ja0542789. [DOI] [PubMed] [Google Scholar]
- 71.Guo Z, Guan X, Huang J-S, Tsui W-M, Lin Z, Che C-M. Chem— Eur J. 2013;19:11320–11331. doi: 10.1002/chem.201300021. [DOI] [PubMed] [Google Scholar]
- 72.Law SM, Chen D, Chan SLF, Guan X, Tsui WM, Huang JS, Zhu N, Che CM. Chem—Eur J. 2014;20:11035– 11047. doi: 10.1002/chem.201305084. [DOI] [PubMed] [Google Scholar]
- 73.Manca G, Gallo E, Intrieri D, Mealli C. ACS Catal. 2014;4:823–832. [Google Scholar]
- 74.See for example: Nam W. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. and references therein.
- 75.The quartet-doublet gap might be underestimated, as hybrid functionals often overestimate the stability of higher-spin states: Swart, M. J Chem Theory Comput. 2008;4:2057–2066. doi: 10.1021/ct800277a. [DOI] [PubMed] [Google Scholar]
- 76.Campo Dall’ Orto V, Carballo R, Hurst JA, Rezzano I. Spectrochim Acta, Part A. 2005;61:2089–2093. doi: 10.1016/j.saa.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 77.Mu XH, Kadish KM. Inorg Chem. 1989;28:3743–3747. [Google Scholar]
- 78.Lin XQ, Kadish KM. Inorg Chem. 1986;25:3242–3248. [Google Scholar]
- 79.Dey S, Ikbal SA, Rath SP. New J Chem. 2014;38:1458– 1470. [Google Scholar]
- 80.Gasyna Z, Stillman MJ. Inorg Chem. 1990;29:5101–5109. [Google Scholar]
- 81.Gasyna Z, Browett WR, Stillman MJ. Inorg Chem. 1988;27:4619–4622. [Google Scholar]
- 82.Yu RP, Darmon JM, Milsmann C, Margulieux GW, Stieber SCE, DeBeer S, Chirik PJ. J Am Chem Soc. 2013;135:13168–13184. doi: 10.1021/ja406608u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tomson NC, Crimmin MR, Petrenko T, Rosebrugh LE, Sproules S, Boyd WC, Bergman RG, DeBeer S, Toste FD, Wieghardt K. J Am Chem Soc. 2011;133:18785–18801. doi: 10.1021/ja206042k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.