

Abstract
Failure of lens fiber cell denucleation (LFCD) is associated with congenital cataracts, but the pathobiology awaits elucidation. Recent work has suggested that mechanisms that direct the unidirectional process of LFCD are analogous to the cyclic processes associated with mitosis. We found that lens-specific mutations that elicit an unfolded-protein response (UPR) in vivo accumulate p27(Cdkn1b), show cyclin-dependent kinase (Cdk)-1 inhibition, retain their LFC nuclei, and are cataractous. Although a UPR was not detected in lenses expressing K6W-Ub, they also accumulated p27 and showed failed LFCD. Induction of a UPR in human lens epithelial cells (HLECs) also induced accumulation of p27 associated with decreased levels of S-phase kinase-associated protein (Skp)-2, a ubiquitin ligase that regulates mitosis. These cells also showed decreased lamin A/C phosphorylation and metaphase arrest. The suppression of lamin A/C phosphorylation and metaphase transition induced by the UPR was rescued by knockdown of p27. Taken together, these data indicate that accumulation of p27, whether related to the UPR or not, prevents the phosphorylation of lamin A/C and LFCD in maturing LFCs in vivo, as well as in dividing HLECs. The former leads to cataract and the latter to metaphase arrest. These results suggest that accumulation of p27 is a common mechanism underlying retention of LFC nuclei.—Lei, L., Whitcomb, E. A., Jiang, S., Chang, M.-L., Gu, Y., Duncan, M. K., Cvekl, A., Wang, W.-L., Limi, S., Reneker, L. W., Shang, F., Du, L., Taylor, A. Unfolded protein response–associated stabilization of p27(Cdkn1b) interferes with lens fiber cell denucleation, leading to cataract.
Keywords: ubiquitin, cell cycle, endoplasmic reticulum stress, nuclear disassembly
Opacification of the eye lens, or cataract, is probably the most common malady of the elderly and still blinds 20 million people worldwide in 2010 (1). In addition to age-related cataracts, there are congenital cataracts (0.4% of all births) that present early and result from the expression of abnormal proteins (2). The lens is formed by epithelial cells. During differentiation, lens epithelial cells undergo extreme morphologic and biochemical changes as they become fiber cells. This process includes induction of expression of a large quantity of proteins called crystallins and the removal of virtually all organelles from the central fiber cells. Removal of the cell nuclei, mitochondria, lysosomes, endoplasmic reticulum (ER), and Golgi apparatus forms an organelle-free zone (OFZ) in the center of the lens. The process continues throughout life, and, although cell nuclei are destroyed, there is no detectable lens fiber cell (LFC) turnover (3–6). The formation of an OFZ is essential for the transparency and function of the lens. LFC denucleation (LFCD) appears to be initiated by cyclin-dependent kinase (Cdk)-1–induced phosphorylation of lamin A/C, with subsequent disassembly of the nuclear envelope. Failure to remove lens fiber cell nuclei is associated with various congenital cataracts (7–14). Despite significant progress in lens development research, the mechanisms underlying the LFCD process remain largely unknown.
The nuclear lamina is a meshwork of intermediate filament proteins that closely associates with several inner nuclear membrane proteins and interacts with portions of the chromatin (15–18). Initiation of the nuclear envelope disassembly process during mitosis is regulated by phosphorylation of lamins, particularly by Cdk1 (19–21). We recently demonstrated that Cdk1 is critical for nuclear envelope disassembly in the eye lens in vivo as well (22).
Ubiquitination is best known for targeting proteins for degradation via the proteasome (23). Studies have shown that expression of mutant ubiquitin (Ub) in the lens in vivo results in the accumulation of p27 (12), a Cdk inhibitor that plays important roles in regulating the cell cycle (24–27). The stability of p27 is determined by its phosphorylation status. When p27 is phosphorylated on Thr187, it is degraded by the Ub proteasome system (UPS) via SCFskp2-mediated ubiquitination (28–30). Several lines of evidence suggest that p27 is a common upstream regulator of the Cdk1-driven cyclic process of mitosis and the unidirectional LFCD process: 1) overexpression of mutant Ub in human lens epithelial cells (HLECs) also causes the accumulation of p27, concurrent with G2/M phase arrest (12, 31); 2) a few reports indicate that p27 has roles in directing the G2/M transition during the cell cycle via activation of Cdk1 (32–34); and 3) Cdk1 is necessary in directing LFCD (19–22).
In eukaryotic cells, if the influx of secretory and transmembrane proteins into the ER exceeds the folding or processing capacity or mutant proteins enter and cannot be folded properly, ER stress is induced (35). In response to ER stress, cells activate a set of adaptive intracellular signal-transduction pathways, known as the unfolded-protein response (UPR) (35–37). The UPR is reportedly activated in the lens during embryogenesis, and moderate levels of ER stress are thought to play a physiologic role during lens development (14). A UPR is induced in the lenses of transgenic mice that express mutant collagen IV (Col IV), dominant negative fibroblast growth factor receptor (dnFGFR), and other mutant proteins (7, 8, 13). These mutant mice have cataracts and retain their LFC nuclei in the OFZ. The retention of LFC nuclei is similar to that observed in the lenses of mutant Ub transgenic mice (12) in which p27 accumulates. Recent reports have indicated that the UPR can stabilize p27 through the suppression of the expression of the E3 ligase S-phase kinase-associated protein (Skp)-2 (38, 39). In this study, we hypothesized that UPR-induced accumulation of p27 is a mechanism that links UPR to defects in the LFCD process.
To test this hypothesis, we used several in vivo and in vitro models of the UPR in transgenic mice [mutant Col IV, dnFGFR, and dominant negative nuclear receptor coactivator 6 (dnNCOA6)] (7, 8, 40) and HLECs, respectively. The UPR in both the cultured cells and lenses in vivo resulted in the accumulation of p27. We compared the findings to systems in which p27 accumulated but did not show a UPR. In all cases, accumulation of p27 was associated with inhibition of the phosphorylation of lamin A/C. In contrast, knockdown of p27 (p27KD) partially rescued the inhibition of lamin A/C phosphorylation and cell cycle arrest at the G2/M phase in HLECs. The data suggest that the accumulation of p27 induced by the UPR mechanistically links inhibition of nuclear envelope disassembly in both the cell cycle and LFCD.
MATERIALS AND METHODS
Antibodies
The following primary antibodies were used: p27kip1 (610241, 1:2,000; BD Biosciences, San Jose, CA, USA); BiP (ab21685, 1:5,000), phospho-lamin A/C (ab58528, 1:5,000 for Western blot analysis and 1:200 for immunohistochemistry), and Lamp1 (ab24170, 1:100) (Abcam, Cambridge MA, USA); phospho-serine CDKs substrate (P-S2-100, 1:1,000; Cell Signaling, Danvers, MA, USA); Skp2 (sc-7164, 1:1,000), DDB1 (sc-136180, 1:500), Kpc1 (sc-101122, 1:500), cyclin B1 (sc-245, 1:1,000), securin (sc-5839, 1:1,000), and cyclin A (sc-751, 1:1,000) (Santa Cruz Biotechnology, Dallas, TX, USA); Pirh2 (A300-357A, 1:1,000; Bethyl Laboratories, Montgomery, TX, USA); GAPDH (G9545, 1:10,000) and β-actin (A5441, 1:10,000) (Sigma-Aldrich, St. Louis, MO, USA); phospho-T2055- nuclear mitotic apparatus protein (NuMA; 1:1,000; a gift from Dr. P. Gönczy, Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland); and DNAse IIβ (1:100; a gift from S. Nagata, Kyoto University, Toshida, Kyoto, Japan). Primary antibodies were detected with species-specific secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) conjugated to IgG-horseradish peroxidase (HRP; 1:2000) for Western blot analysis and FITC or Cy3 (1:250) for immunohistochemistry.
Animals
The transgenic mouse line 520, which overexpresses Col IVα3 under the regulation of a modified αA crystallin promoter, was maintained on the FVB/N genetic background in the University of Delaware Animal Facility as has been described (7). The Cryaa-dnNcoa6 transgenic line was generated on the FVB/N background at the Albert Einstein College of Medicine (New York, NY, USA) and analyzed as has been described (40). The dnFGFR mouse was constructed as according to a published method (41). Transgenic mice expressing truncated FGFR1 (or tR1) in the ocular lens, driven by the αA-crystallin promoter, were maintained on the FVB/N genetic background in the animal facility of the University of Missouri Animal Science Research Center (ASRC). The homozygous tR1 female mice were mated to C57BL6/J male mice and the eyes were enucleated from newborn pups for experiments. The K6W-Ub mouse was maintained in the Human Nutrition Research Center on Aging (Tufts University, Boston, MA, USA) (12). All procedures used in the study were approved by the Institutional Animal Care and Use Committee at Tufts University and were conducted in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Histology and immunohistochemistry
Lenses dissected from wild-type (WT) and transgenic mice (7, 8, 40) were fixed with 4% paraformaldehyde for 2 h, embedded in optimal cutting temperature compound (Sakura Finetek, Torrance, CA, USA) frozen, and subsequently sectioned according to standard protocols. HLECs were grown on 4-well chamber slides (Thermo Scientific, Waltham, MA, USA) and fixed with 4% paraformaldehyde for 15 min. Before they were stained, the lens cryosections (10 μm) and cells grown on slides were permeabilized with 0.05% Triton X100 for 5 min at room temperature. The slides were blocked with 5% normal donkey or goat serum (Jackson ImmunoResearch) in 5% bovine serum albumin (BSA)/PBS for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C and secondary antibodies for 45 min at room temperature. Nuclear DNA was stained with 1 μg/ml DAPI. Fluorescent images were obtained with an inverted fluorescence microscope (Axiovert 200 and AxioVision V 4.5 software; Zeiss, Jena, Germany). The detection and localization of p27, p-lamin A/C, and DNase IIβ, as well as their respective semiquantitations, were repeated in more than 2 lenses, each examining multiple sections.
Cell culture and synchronization
HLECs (cell line SRA 01/04) were grown in DMEM (Thermo Scientific) and supplemented with fetal bovine serum (10% v/v; Thermo Scientific), penicillin (100 U/ml) and streptomycin (100 μg/ml) (Thermo Scientific).
For synchronization, the HLECs were grown to 60% confluence and synchronized in medium containing 2 mM hydroxyurea (HU; Sigma-Aldrich). After 21 h, the cells were washed twice with PBS and fresh medium was added to allow the G1/S phase–synchronized cells to enter the cell cycle.
UPR induction in cultured HLECs
HLECs grown to 70% confluence were incubated with 1 μg/ml tunicamycin (TM), 1 μM thapsigargin (Thg), or 5 mM homocysteine (HC) (all from Sigma-Aldrich) for times indicated in the figure legends.
Generation of p27KD HLECs
Short hairpin (sh)RNAs (TRCN0000039930 and TRCN0000295863) and a nontarget shRNA control virus (SHC016-1EA) were purchased from Sigma-Aldrich. HLECs were cultured in 6-well plates to 80% confluence. p27 shRNA lentiviruses [104 titer units (TU)] or nontarget control (NC) virus was added in the presence of 8 μg/ml hexadimethrine bromide (Sigma-Aldrich). The medium containing the lentiviral particles was removed 24 h later, and the cells were grown in fresh medium overnight. Stable cell lines were selected with 2 μg/ml of puromycin (Santa Cruz Biotechnology).
MTS assay
NC and p27KD cell lines were plated in 96-well plates at 5000 cells/well. Cell counts were assessed with the MTS assay kit (Promega, Madison, WI, USA), as soon as they adhered to the plate, or after 24 and 48 h of growth, according to the manufacturer’s instructions. Absorbance at 495 nm was read with a microplate reader (ELx808; BioTek China, Beijing, China).
Quantitative real time-PCR
Total RNA was extracted from cells with the RNeasy Mini Kit (Qiagen, Redwood City, CA, USA), and cDNA was synthesized from 500 ng of RNA with SuperScript III First-Strand Synthesis System for RT-PCR (Thermo Scientific). Quantitative real-time PCR reactions were performed in duplicate. Each reaction contained 1.5 μl cDNA, target-gene–specific primers, and SYBR Green (GoTaq qPCR Master Mix; Promega), according to the manufacturer’s instructions. Forty cycles of qPCR were performed with the 7300 Real Time PCR System (Thermo Scientific). The sequences of primers used were the following: p27, forward, gagtggcaagaggtggagaa, and reverse, gtccgacggatcagtctttg; GAPDH, forward, acccagaagactgtggatgg and reverse, tttctagacggcaggtcagg. The results were analyzed by the comparative Ct (δδCt) method. The quantified values for p27 were normalized by GAPDH.
Western blot analysis
Cells were washed with cold PBS and lysed in SDS-PAGE loading buffer at various time points. Lenses from WT and transgenic mice were also homogenized directly in loading buffer. Equal quantities of samples were separated by SDS-PAGE. Proteins were transferred by electroblotting onto nitrocellulose membranes (0.2 μm; Bio-Rad, Hercules, CA, USA), after which the membranes were probed with primary antibodies overnight at 4°C and HRP-conjugated secondary antibodies for 45 min at room temperature. Signals were detected by chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific).
p27 turnover assay
HLECs were grown to 70% confluence and incubated with 1 μg/ml TM (Sigma-Aldrich) or with the same amount of DMSO for 24 h and then treated with 25 μg/ml cycloheximide (CHX) (Sigma-Aldrich). The cells were collected at various times after the addition of CHX, for immunoblot analysis of p27.
Flow cytometry analysis
Synchronized HLECs treated with 1 μg/ml TM or DMSO were harvested as single cells with 0.25% trypsin/EDTA and fixed in 70% ethanol overnight at 4°C. The cells were then washed with PBS and treated with 0.5 μg/ml RNase A (Sigma-Aldrich) for 1 h at room temperature. Propidium iodide (10 μg/ml; Sigma-Aldrich) was added for DNA staining. The cells were analyzed on an Accuri C6 Cytometer (BD Biosciences). The fluorescence activated cell sorting (FACS) data regarding cell cycle distribution were analyzed with FlowJo v7.6.1 (FlowJo, LLC, Ashland, OR, USA).
Statistical analysis
All data from at least 3 independent experiments are presented as averages ± se. Statistical analysis and comparisons were performed with Student’s t test. Western blot images were plotted and analyzed with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
RESULTS
The UPR is associated with stabilization of p27, failed LFCD, and cataracts in vivo
To investigate the mechanism underlying the relationship between the UPR and failure of LFCD, we first examined several transgenic mouse lines that were reported to induce a UPR and showed failed LFCD. Elevated levels of the ER chaperone BiP in mutant Col IV lenses, one of the hallmarks of the UPR (42), indicated that a UPR was induced, as has been reported (7) (Fig. 1A). We then asked whether p27 was increased in Col IV-expressing lenses by UPR induction. As shown in Fig. 1A and Supplemental Fig. S1B, lenses that expressed mutant Col IV contained ∼3-fold higher p27, compared with WT lenses. Immunohistochemical analyses showed a gradual decrease in nuclear p27 (red) from the bow region to the center of the WT lenses , but retention of higher levels of nuclear p27 toward the center of the Col IV lenses (Fig. 1B). Semiquantitative analysis indicated that 3.5-fold more (P = 6 × 10−5) nuclei retained p27 in the Col IV lens than in the WT lens. To further explore the etiologic relationship of the UPR with accumulation of p27 and aberrant LFCD, we examined 2 additional in vivo models that exhibit lens fiber nuclei retention and cataract. dnFGFR transgenic mice, a confirmed UPR model (8), and the dnNCOA6 model (40) elicited a UPR, as evidenced by the increase in BiP (Fig. 1C). They also expressed (>5-fold higher levels of p27, compared with their WT littermates (Fig. 1C; Supplemental Fig. S1B).The expression of K6W-Ub in the lens also elicited >4-fold p27 accumulation and a denucleation defect (12), but did not induce the UPR (Supplemental Fig. S1). These findings imply that the accumulation of p27 is a common mechanism for impaired denucleation.
Figure 1.
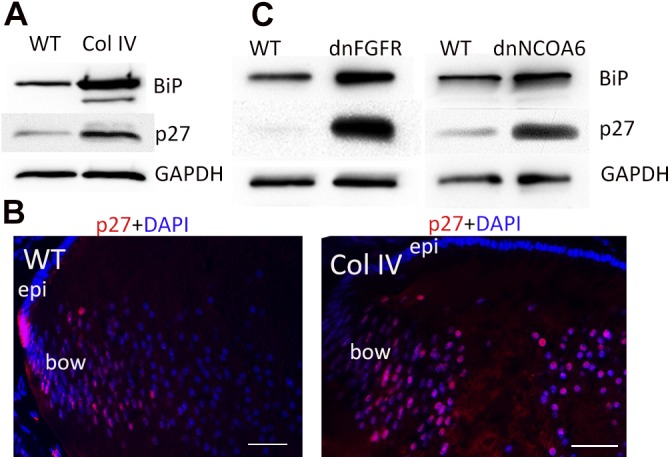
Expression of cataract-inducing proteins induces a UPR and sustained p27 expression. A) Lenses from Col IV transgenic mice (P1) were lysed with SDS-gel loading buffer. Levels of BiP, p27, and GAPDH were determined by Western blot analysis. GAPDH was used as a loading control. B) P1 lenses from WT (left) and mutant Col IV (right) transgenic mice were cryosectioned and immunostained with anti-p27 (red) and counterstained with DAPI (blue). Note that p27 staining diminishes more peripherally in the WT than in the Col IV lens. Bow, bow region; epi, epithelium. C) Lens lysates from dn-FGFR (P0) and dnNCOA6 (P1) mice were also examined for levels of BiP and p27 by Western blot analysis, as described in (A). Scale bars, 50 μm.
Phosphorylation of lamin A/C by Cdk1 drives LFCD and establishment of a clear lens (12, 22). In our K6W-Ub mutant, the stabilization of p27 was associated with inhibition of Cdk1activation, delayed phosphorylation of lamin A/C, and delayed entry of DNAse IIβ into the nucleus. To determine whether the accumulation of p27 in UPR-inducing transgenic lenses is also associated with diminished Cdk1 activation in vivo, we monitored the phosphorylation of lamin A/C. In WT mice, the level of phosphorylated lamin A/C at the bow region was very low (Fig. 2A, left), but increased dramatically at the nuclear envelope of the fiber cells (appearing as green rings) that are scheduled to denucleate to form the OFZ (Fig. 2A). In contrast, phosphorylated lamin A/C was barely detectable surrounding the nuclei in the Col IV lenses (Fig. 2B, compare detail, right), and the fiber cells in these lenses retained their nuclei at a rate that was more than 70% higher (P = 0.0002) than in WT lenses (Fig. 2A, B, compare enlargements of dashed boxes, right). We also did not observe phosphorylation of lamin A/C in dnFGFR lenses (Supplemental Fig. S2A). These data suggest that disassembly of the LFC nuclear envelope in vivo is interrupted by the UPR-elicited accumulation of p27 and the associated inhibition of Cdk1 activation.
Figure 2.

Phosphorylation of lamin A/C in lens fiber cells is inhibited by the UPR. P1 lenses from WT (A) and mutant Col IV transgenic mice (B) were cryosectioned and immunostained with anti-phospho-lamin A/C (green) and counterstained with DAPI (blue). Right: higher magnifications of boxed areas. Scale bars, 50 μm.
DNAse IIβ or DLAD, generally a lysosomal enzyme, is a lens-specific acidic DNAse that enters the nucleoplasm to degrade chromatin at the conclusion of LFC differentiation (11). Because elevated p27 correlates with impaired nuclear envelope disassembly and decreased entry of DNAse IIβ into the nuclei in the K6W transgenic lenses, we predicted that elevated p27 in the UPR-inducing transgenic mice would also lead to failed disassembly of the nuclear membrane and exclusion of DNAse IIβ from the nucleus. In WT lenses, DNAse IIβ entered into the nuclei in the fiber cells in the denucleating region (Fig. 3A, red circles surrounding nuclei or red within nuclei) (9, 12). However, in lenses that expressed mutant Col IV, more DNAse IIβ was observed in the cytoplasm and less translocation into nuclei was noted (Fig. 3B). Moreover, DNase IIβ in Col IV transgenic lens was found more peripherally in the bow region and colocalized with lysosomes (Lamp1) that were also retained in what should be an OFZ (Fig. 3B, enlargement, right). Semiquantitative analysis suggested that the proportion of Col IV lens fiber cell nuclei that contained DNase IIβ was 6 times less (P = 0.0007) than that in the WT. In addition, because of the poor retention of structure during processing of these tissues, there was more nonspecific staining in Col IV lens, which was likely to have caused an overestimate of the DNase IIβ in the Col IV lenses. The results suggest that the proportion of Col IV fiber cell nuclei in which DNase IIβ has been delivered is significantly lower. The distribution of DNase IIβ was similar in dnFGFR lenses, with no staining observed in the inner cell nuclei (Supplemental Fig. S2B). These data are consistent with the hypothesis that UPR-associated elevation of p27 inhibits disassembly of the nuclear membrane that is associated with entry of DNase IIβ.
Figure 3.
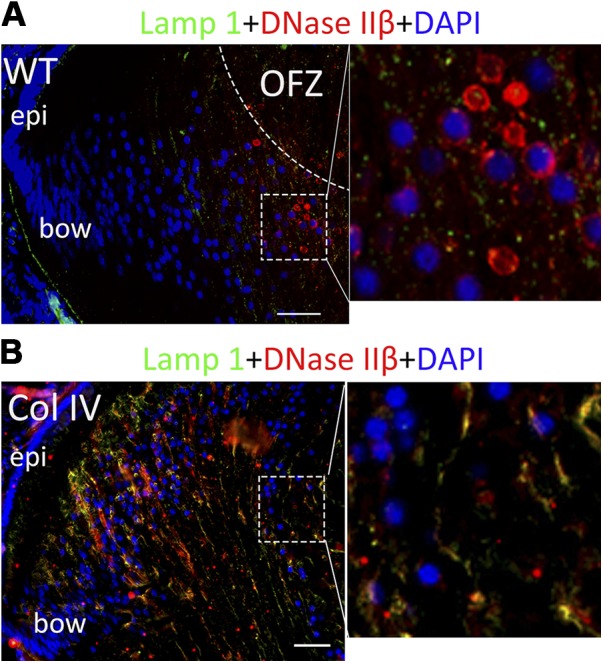
UPR blocks the nuclear translocation of DNase IIβ in lens fiber cells. P1 lenses from WT (A) and mutant Col IV transgenic mice (B) were cryosectioned and immunostained with anti- DNAse IIβ (red) and counterstained with DAPI (blue). Lysosomes are stained with anti-Lamp1 (green). Right: higher magnification of boxed areas. Scale bars, 50 μm.
The UPR inhibits lamin A/C phosphorylation and leads to G2/M arrest in proliferating cells through a p27-dependent mechanism
LFCs do not progress though mitosis in vivo. However, during denucleation, they appear to appropriate some of the mitotic machinery to terminally remove their nuclei, an obligatory process for establishing a clear lens (12, 43). Thus, to further explore the role of UPR-induced p27 expression in nuclear envelope breakdown, we asked whether induction of a UPR causes p27 accumulation in cultured HLECs. If a UPR elicits stabilization of p27, then that should also cause a delay in cell cycle progression and events associated with mitosis. Exposure of HLECs to TM, an established ER stressor that inhibits protein glycosylation, elicited a UPR, as indicated by a dramatic increase in levels of BiP (Fig. 4A). Exposure to TM also resulted in a 2-fold increase in p27 (Fig. 4A; Supplemental Fig. S3A) (38, 39).
Figure 4.

Induction of a UPR in HLECs increases p27 through reduced degradation and increased mRNA expression. A) Cells were treated with 1 μg/ml TM and collected at 16, 20, and 24 h. Levels of BiP and p27 were determined by Western blot. β-Actin was used as a loading control. B) Cells were treated with TM or left untreated for 16 h, and CHX was then added to stop protein synthesis. The p27 level was determined by Western blot. MG132 was used to inhibit proteasome-dependent degradation. C) The effect of UPR induction on the levels of Ub ligases, Skp2, DDB1, Kpc1, and Pirh2, which are related to p27 stability, was determined by Western blot analysis. Cells were treated with 1 μg/ml TM and collected at 16, 20, and 24 h. D) mRNA levels of p27 and BiP in the presence or absence of TM were determined by real-time quantitative PCR, with GAPDH as the control. *P < 0.001, vs. 0 h after TM treatment.
Levels of p27 are known to be controlled by the UPS (28–30); thus, we determined the effects of UPR induction on proteasomal degradation of p27. In the absence of TM (DMSO control), the level of p27 decreased rapidly such that it was barely detectable by 2 h (Fig 4B). In contrast, the level of p27 in cells treated with TM decreased more slowly: ∼70% of p27 protein remained 2 h after protein synthesis was arrested by addition of CHX in TM-treated cells. A similar delay in p27 degradation was observed in the presence of the bona fide proteasome inhibitor, MG132 (Fig. 4B;Supplemental Fig. S3B).
We then asked whether induction of the UPR affected the levels of E3 ligases known to be involved in p27 degradation (44–50). We found that the level of Skp2, the substrate recognition subunit of the E3 that is usually associated with p27 degradation, SCFskp2 (27, 39), was significantly reduced upon treatment with TM, whereas the levels of DDB1, KPC1, and Pirh1, other E3s known to target p27 for proteasomal degradation, were not affected (Fig. 4C).
Quantitative RT-PCR analysis showed that TM treatment significantly increased both BiP and p27 mRNA in HLECs (Fig. 4D). At the 24-h time point, TM induced 3-fold and >25-fold elevations in the levels of p27 and BiP mRNA, respectively. These data indicate that reduced UPS-dependent degradation, likely via a Skp2-mediated pathway, and increased p27 synthesis contribute to the UPR-induced stabilization of p27.
Next, we determined whether decreases in p27 are necessary to set in motion a sequence of events involving activation of Cdk1, phosphorylation of lamin A/C, and disassembly of the nuclear envelope. These processes are required for both LFCD and transition through the M phase in cycling cells (22, 51, 52). To investigate the effect of UPR-associated accumulation of p27 in mitosis, HLECs were synchronized at the G1/S phase boundary using HU (Fig. 5A). HU itself does not induce a UPR or p27 accumulation (data not shown). When the majority of the cells (80%) were in the S phase (3 h after the removal of HU), a UPR was induced with TM. p27 stabilization (Supplemental Fig. S4) was similar to that observed in the nonsynchronized cells (Fig. 4A). Consistent with the reduced level of phosphorylated lamin C in TM-treated HLECs (Supplemental Fig. S4A, middle right), the percentage of phosphorylated lamin A/C+ cells in the TM-treated group was significantly less than those in the control group (Fig. 5B, red arrows), indicating that a UPR is associated with decreased lamin A/C phosphorylation in HLECs.
Figure 5.

UPR induction prevents nuclear lamin phosphorylation and leads to cell cycle arrest at the G2/M phase. A) Experimental scheme. Cells were synchronized by HU treatment. TM was added when most of the cells were in the S phase. B) Cellular localization of phosphorylated lamin A/C was determined with immunostaining after 8 h of TM treatment. The bar graph shows the percentage of cells with phosphorylated lamin. C) Cell cycle profiles were determined with propidium iodide staining and FACS analysis. D) The level of multiple Cdk substrates indicated as phospho-Ser substrates (top) and the level of specific Cdk1 product phospho-NuMA (T2055) (middle) were blotted after 24 h, in the presence or absence of TM, as the indicator of Cdk1 activation. *P < 0.01; ** P < 0.001 vs. the control cells at the specified time points.
To further explore the role of the UPR and accumulation of p27 in cycling cells, progression through the cell cycle was monitored. The majority of the cells (∼70%) were at G1 phase after treatment with HU (t = −3 h; Fig. 5C, top, blue bars). Eighty percent of the cells were in S phase 3 h after the removal of HU (middle, t = 0, blue bars). In the absence of TM, most of the cells progressed from S phase to G2/M phase within 8 h (bottom, blue bars), and the cells started to transition from G2/M phase to the next G1 phase within 12 h (Fig. 5C, top, blue bars). TM slowed, but did not block, the transition from the S phase to the G2/M phase (Fig. 5C, bottom, 8 h). However, TM treatment significantly inhibited the transition beyond the G2/M phase into the next G1 phase (Fig 5C, top, red bars). By 16 to 24 h after TM treatment, there were ∼20% more cells at the G2/M phase in the presence of TM than in its absence. Because TM treatment increases p27 levels and p27 inhibits Cdk1, we hypothesized that the activity of Cdk1 would in turn be decreased. Consistent with this notion, total phosphoserine Cdk1 substrates decreased during TM treatment (Fig. 5D, bar graph, Supplemental Fig. S4B). We also examined the level of phosphorylated (NuMA), an established Cdk1 substrate (53). The decreased levels of phosphorylated NuMA after 24 h of TM treatment also indicate that Cdk1 activity is suppressed during UPR (Fig. 5D).
To determine whether the UPR-associated G2/M arrest requires p27, we stably knocked down p27. Western blots indicate that ∼80–90% of the p27 was depleted in the p27 shRNA-infected cells (p27KD) as compared to the NC shRNA-infected cells (Fig. 6A, lane 9 vs. 3). Diminishing p27 did not affect the TM-induced UPR, as the level of BiP increased to an extent similar to that in the control cells (lanes 10–12 vs. 4–6). Consistent with the role of p27 as a cell cycle inhibitor, p27KD was associated with an increase in cell proliferation (Fig. 6B).
Figure 6.

p27KD abolishes the inhibition of phospho-lamin A/C and G2/M arrest caused by the UPR. A) p27 was knocked down in HLECs using lentivirus-mediated shRNA. The NC and p27KD HLECs were synchronized by HU and treated with TM (Fig. 5A). The levels of p27 and BiP, in the absence or presence of TM, were detected at the indicated time points with Western Blot. B) The growth rates of NC and p27KD cells were determined by the MTS assay, as described in experimental procedures. C) The percentages of phospho-lamin A/C+ cells in the synchronized NC, and p27KD cells were detected by immunostaining 8 h after TM treatment. D) The synchronized NC and p27KD cells were treated with TM or left untreated (Fig. 5A). The cell cycle progressions were determined by FACS analysis. *P < 0.001 vs. TM-treated NC cells at the indicated time points.
We then asked whether p27KD could reverse the UPR-induced decrease in lamin A/C phosphorylation and delay progression through the G2/M phase. TM treatment significantly reduced the phosphorylation of lamin A/C in NC cells (Fig. 6C). However, in p27KD cells, lamin A/C phosphorylation was not decreased upon TM treatment. Rather, it was more than twice that of TM-treated NC cells. These data are consistent with the hypothesis that accumulation of p27 plays a critical role in UPR-induced mitotic arrest, apparently by the inhibition of Cdk1 and subsequent lamin A/C phosphorylation. We then directly tested whether p27KD has an effect on TM-induced cell cycle arrest. Results from 2 independent shRNA clones indicate that p27KD abolished the G2/M arrest induced by TM (Fig. 6D, Supplemental Fig. S5). This finding is of additional interest, because p27 is usually associated with inhibiting the G1/S transition in cell cycle.
To gain additional corroboration that an increase in p27 and cell cycle arrest is a common feature of UPR induction, we asked whether 2 other inducers of the UPR, Thg, and HC, also cause an increase in p27 and cell cycle arrest (39, 54) (Supplemental Fig. S6B, C). In the control cells, the presence of these UPR inducers significantly delayed progression of the cell cycle at G2/M phase, consistent with the results of the addition of TM (Fig. 5C), whereas depletion of p27 enhanced the rate at which the cells advanced to the G1 phase, compared with treated NC cells (Supplemental Fig. S6C).
We were interested to know at which point in the M phase UPR induction causes arrest. Thus, we examined the levels of mitotic regulators cyclin A, cyclin B1, and securin (55–57). Although these substrates are all degraded in mitosis, cyclin A is degraded in the prometaphase, whereas cyclin B and securin are degraded in the metaphase. We found that cyclin A did not accumulate in the TM-treated cells (Supplemental Fig. S6A, B) whereas cyclin B1 and securin were stabilized in cells exposed to these UPR inducers when p27 was stabilized (Supplemental Fig. S6A, lane 2 vs. 1; Supplemental Fig. S6B, lanes 2–3 vs. lane 1). Together these results suggest that the arrest occurred during the metaphase. TM, Thg, and HC had no effect on cyclin B1 and securin levels in p27-depeleted cells (Supplemental Fig. S6A, lane 4 vs. lane 3; Supplemental Fig. S6B, lanes 5, 6 vs. lane 4). These data suggest that in cultured cells, a UPR induced during the S phase results in the accumulation of p27 and that this accumulation of p27 results in metaphase arrest, via decreased Cdk1 activation and subsequent inhibition of nuclear membrane disassembly.
DISCUSSION
Timely removal of nuclei and other cytoplasmic organelles in maturing lens fibers is crucial for establishing a transparent lens. Failure to remove LFC nuclei is a common phenotype of congenital cataracts caused by various mutations (7, 8, 11–13, 58). Initial reports suggested that, to accomplish denucleation, lens fiber cells adapt a mechanism that is involved in the disassembly of the cell nucleus that occurs during the anaphase of the cell cycle (12, 22). Data from this study also support this hypothesis. Furthermore, our data indicate that reduced p27 levels facilitate Cdk1-dependent LFCD and suggest that p27 accumulation is a common pathobiological mechanism that underlies various LFCD defects.
p27 is a critical Cdk inhibitor and cell cycle regulator. p27 is usually associated with stabilization of the G1/S transition (27, 47, 59), but p27 control of mitotic progression has also been reported (32–34). We have shown that lenses expressing mutant Ub exhibit increased p27 and a denucleation defect (12), but do not show evidence of a UPR (Fig 1C). In this study, we demonstrate that a UPR associated with expression of mutant proteins in lenses or induced by TM treatment in lens cells in culture also causes the accumulation of p27 (Figs. 1, 5), thus mechanistically linking increased p27 to denucleation defects in lenses that do and do not elicit a UPR.
Moreover, we observed that the stabilization of p27 involved enhanced transcription, as well as delayed degradation, the latter apparently occurring via down-regulation of Skp2 (Fig. 4C, D). This finding is consistent with previous observations that the UPR stabilizes p27 by decreased Skp2-mediated degradation and increased transcription (39). In our study, other E3s that are known to catalyze degradation of p27 appeared not to be involved (Fig. 4C).
We used multiple in vivo models of UPR and found corroboration in in vitro systems. In each of the in vivo UPR models—mutant Col IV, dnFGFR, and dnNCOA6 transgenic mice—as in the K6W-Ub transgenic mouse lenses, we found accumulation of p27, retention of nuclei in LFCs, smaller lenses, disorderly lens fiber alignment, and cataracts (Fig. 1) (7, 8, 12, 40). These features are in keeping with delayed synthesis of major lens proteins and impaired replication, thus causing a smaller lens. Taken together, these data suggest that when p27 accumulates, Cdk1 activity is markedly reduced. This decrease results in arrest at the G2/M phase in cells in culture and multiple developmental abnormalities, including delayed disassembly of the nucleus in LFCs in vivo (Fig. 2). Consistent with the hypothesized role of p27 in this process, in our study, p27KD rescued the effects of the UPR on lamin A/C phosphorylation and cell cycle arrest (Fig. 6; Supplemental Fig. S5).
The findings enhance our appreciation of p27 biology by demonstrating in new in vivo systems that p27 is a major regulator of lens development and that a UPR is associated with p27 stabilization. The data also corroborate observations (12, 22) that the spatiotemporal activation of Cdk1 and subsequent phosphorylation of lamin A/C is necessary for LFCD. The down-regulation of p27 in the center of the lens appears to play an important role in Cdk1 activation near the OFZ. Analogies between events associated with LFCD and p27–Cdk1-catalyzed control of the cell cycle are striking. Specifically, we observed that nature exploits a major control node (p27 levels) of the cyclical pathway of cell proliferation for the unidirectional control of LFCD. It appears that mutations or stresses that interfere with the degradation or expression of p27 in the LFCs may impede LFCD and cause cataracts.
Supplementary Material
Acknowledgments
The authors thank Dr. S. Nagata for providing the DNAse IIβ antibody; Dr. P. Gönczy for the gift of the pNUMA antibody; and Dr. S. Rowan, L. Bailey, and Z. Crosser for critical reading and suggestions during the preparation of the manuscript. This work was supported by U.S. Department of Agriculture, Agricultural Research Service Agreement 58-1950-4-003; U.S. National Institutes of Health, National Eye Institute Grants EY13250 and EY11717; and the China Scholarship Council.
Glossary
- Cdk
cyclin-dependent kinase
- CHX
cycloheximide
- Col IV
collagen IV
- dnFGFR
dominant negative fibroblast growth factor receptor
- dnNCOA6
dominant negative nuclear receptor coactivator 6
- ER
endoplasmic reticulum
- FACS
fluorescence-activated cell sorting
- HC
homocysteine
- HLEC
human lens epithelial cells
- HRP
horseradish peroxidase
- HU
hydroxyurea
- LFC
lens fiber cell
- LFCD
LFC denucleation
- NC
nontarget control
- NuMA
nuclear mitotic apparatus protein
- OFZ
organelle-free zone
- p27KD
p27 knockdown
- shRNA
short hairpin RNA
- Skp
S-phase kinase-associated protein
- Thg
thapsigargin
- TM
tunicamycin
- Ub
ubiquitin
- UPR
unfolded-protein response
- UPS
ubiquitin proteasome system
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Pascolini D., Mariotti S. P. (2012) Global estimates of visual impairment: 2010. Br. J. Ophthalmol. 96, 614–618 [DOI] [PubMed] [Google Scholar]
- 2.Francis P. J., Moore A. T. (2004) Genetics of childhood cataract. Curr. Opin. Ophthalmol. 15, 10–15 [DOI] [PubMed] [Google Scholar]
- 3.Danysh B. P., Duncan M. K. (2009) The lens capsule. Exp. Eye Res. 88, 151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloemendal H. (1977) The vertebrate eye lens. Science 197, 127–138 [DOI] [PubMed] [Google Scholar]
- 5.Fukushi S., Spiro R. G. (1969) The lens capsule. Sugar and amino acid composition. J. Biol. Chem. 244, 2041–2048 [PubMed] [Google Scholar]
- 6.Piatigorsky J. (1981) Lens differentiation in vertebrates. A review of cellular and molecular features. Differentiation 19, 134–153 [DOI] [PubMed] [Google Scholar]
- 7.Firtina Z., Danysh B. P., Bai X., Gould D. B., Kobayashi T., Duncan M. K. (2009) Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J. Biol. Chem. 284, 35872–35884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reneker L. W., Chen H., Overbeek P. A. (2011) Activation of unfolded protein response in transgenic mouse lenses. Invest. Ophthalmol. Vis. Sci. 52, 2100–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakahara M., Nagasaka A., Koike M., Uchida K., Kawane K., Uchiyama Y., Nagata S. (2007) Degradation of nuclear DNA by DNase II-like acid DNase in cortical fiber cells of mouse eye lens. FEBS J. 274, 3055–3064 [DOI] [PubMed] [Google Scholar]
- 10.De Maria A., Bassnett S. (2007) DNase IIbeta distribution and activity in the mouse lens. Invest. Ophthalmol. Vis. Sci. 48, 5638–5646 [DOI] [PubMed] [Google Scholar]
- 11.Nishimoto S., Kawane K., Watanabe-Fukunaga R., Fukuyama H., Ohsawa Y., Uchiyama Y., Hashida N., Ohguro N., Tano Y., Morimoto T., Fukuda Y., Nagata S. (2003) Nuclear cataract caused by a lack of DNA degradation in the mouse eye lens. Nature 424, 1071–1074 [DOI] [PubMed] [Google Scholar]
- 12.Caceres A., Shang F., Wawrousek E., Liu Q., Avidan O., Cvekl A., Yang Y., Haririnia A., Storaska A., Fushman D., Kuszak J., Dudek E., Smith D., Taylor A. (2010) Perturbing the ubiquitin pathway reveals how mitosis is hijacked to denucleate and regulate cell proliferation and differentiation in vivo. PLoS One 5, e13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alapure B. V., Stull J. K., Firtina Z., Duncan M. K. (2012) The unfolded protein response is activated in connexin 50 mutant mouse lenses. Exp. Eye Res. 102, 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Firtina Z., Duncan M. K. (2011) Unfolded protein response (UPR) is activated during normal lens development. Gene Expr. Patterns 11, 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davidson P. M., Lammerding J. (2014) Broken nuclei: lamins, nuclear mechanics, and disease. Trends Cell Biol. 24, 247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broers J. L. V., Ramaekers F. C. S., Bonne G., Yaou R. B., Hutchison C. J. (2006) Nuclear lamins: laminopathies and their role in premature ageing. Physiol. Rev. 86, 967–1008 [DOI] [PubMed] [Google Scholar]
- 17.Andrés V., González J. M. (2009) Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 187, 945–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.González J. M., Andrés V. (2011) Synthesis, transport and incorporation into the nuclear envelope of A-type lamins and inner nuclear membrane proteins. Biochem. Soc. Trans. 39, 1758–1763 [DOI] [PubMed] [Google Scholar]
- 19.Ward G. E., Kirschner M. W. (1990) Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell 61, 561–577 [DOI] [PubMed] [Google Scholar]
- 20.Peter M., Nakagawa J., Dorée M., Labbé J. C., Nigg E. A. (1990) In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 61, 591–602 [DOI] [PubMed] [Google Scholar]
- 21.Heald R., McKeon F. (1990) Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61, 579–589 [DOI] [PubMed] [Google Scholar]
- 22.Chaffee, B. R., Shang, F., Chang, M.-L., Clement, T. M., Eddy, E. M., Wagner, B. D., Nakahara, M., Nagata, S., Robison, M. L., and Taylor, A. (2014) Nuclear removal during terminal lens fiber cell differentiation requires CDK1 activity: appropriating mitosis-related nuclear disassembly. Development. 141, 3388–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glickman M. H., Ciechanover A. (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82, 373–428 [DOI] [PubMed] [Google Scholar]
- 24.Polyak K., Lee M. H., Erdjument-Bromage H., Koff A., Roberts J. M., Tempst P., Massagué J. (1994) Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78, 59–66 [DOI] [PubMed] [Google Scholar]
- 25.Slingerland J., Pagano M. (2000) Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 183, 10–17 [DOI] [PubMed] [Google Scholar]
- 26.Coats S., Flanagan W. M., Nourse J., Roberts J. M. (1996) Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 272, 877–880 [DOI] [PubMed] [Google Scholar]
- 27.Lu Z., Hunter T. (2010) Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle 9, 2342–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheaff R. J., Groudine M., Gordon M., Roberts J. M., Clurman B. E. (1997) Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 11, 1464–1478 [DOI] [PubMed] [Google Scholar]
- 29.Vlach J., Hennecke S., Amati B. (1997) Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 16, 5334–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montagnoli A., Fiore F., Eytan E., Carrano A. C., Draetta G. F., Hershko A., Pagano M. (1999) Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 13, 1181–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q., Shang F., Zhang X., Li W., Taylor A. (2006) Expression of K6W-ubiquitin inhibits proliferation of human lens epithelial cells. Mol. Vis. 12, 931–936 [PubMed] [Google Scholar]
- 32.Hu R., Aplin A. E. (2008) Skp2 regulates G2/M progression in a p53-dependent manner. Mol. Biol. Cell 19, 4602–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakayama K., Nagahama H., Minamishima Y. A., Miyake S., Ishida N., Hatakeyama S., Kitagawa M., Iemura S., Natsume T., Nakayama K. I. (2004) Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 6, 661–672 [DOI] [PubMed] [Google Scholar]
- 34.Auld C. A., Fernandes K. M., Morrison R. F. (2007) Skp2-mediated p27(Kip1) degradation during S/G2 phase progression of adipocyte hyperplasia. J. Cell. Physiol. 211, 101–111 [DOI] [PubMed] [Google Scholar]
- 35.Malhotra J. D., Kaufman R. J. (2007) The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18, 716–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ron D., Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 37.Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 38.Han C., Jin L., Mei Y., Wu M. (2013) Endoplasmic reticulum stress inhibits cell cycle progression via induction of p27 in melanoma cells. Cell. Signal. 25, 144–149 [DOI] [PubMed] [Google Scholar]
- 39.Chen M., Gutierrez G. J., Ronai Z. A. (2011) Ubiquitin-recognition protein Ufd1 couples the endoplasmic reticulum (ER) stress response to cell cycle control. Proc. Natl. Acad. Sci. USA 108, 9119–9124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W. L., Li Q., Xu J., Cvekl A. (2010) Lens fiber cell differentiation and denucleation are disrupted through expression of the N-terminal nuclear receptor box of NCOA6 and result in p53-dependent and p53-independent apoptosis. Mol. Biol. Cell 21, 2453–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson M. L., MacMillan-Crow L. A., Thompson J. A., Overbeek P. A. (1995) Expression of a truncated FGF receptor results in defective lens development in transgenic mice. Development 121, 3959–3967 [DOI] [PubMed] [Google Scholar]
- 42.Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 [DOI] [PubMed] [Google Scholar]
- 43.Gao C. Y., Rampalli A. M., Cai H. C., He H. Y., Zelenka P. S. (1999) Changes in cyclin dependent kinase expression and activity accompanying lens fiber cell differentiation. Exp. Eye Res. 69, 695–703 [DOI] [PubMed] [Google Scholar]
- 44.Shiyanov P., Nag A., Raychaudhuri P. (1999) Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem. 274, 35309–35312 [DOI] [PubMed] [Google Scholar]
- 45.Kamura T., Hara T., Matsumoto M., Ishida N., Okumura F., Hatakeyama S., Yoshida M., Nakayama K., Nakayama K. I. (2004) Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat. Cell Biol. 6, 1229–1235 [DOI] [PubMed] [Google Scholar]
- 46.Kotoshiba S., Gopinathan L., Pfeiffenberger E., Rahim A., Vardy L. A., Nakayama K., Nakayama K. I., Kaldis P. (2014) p27 is regulated independently of Skp2 in the absence of Cdk2. Biochim. Biophys. Acta 1843, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsvetkov L. M., Yeh K. H., Lee S. J., Sun H., Zhang H. (1999) p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 9, 661–664 [DOI] [PubMed] [Google Scholar]
- 48.Hattori T., Isobe T., Abe K., Kikuchi H., Kitagawa K., Oda T., Uchida C., Kitagawa M. (2007) Pirh2 promotes ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. Cancer Res. 67, 10789–10795 [DOI] [PubMed] [Google Scholar]
- 49.Hu J., McCall C. M., Ohta T., Xiong Y. (2004) Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 6, 1003–1009 [DOI] [PubMed] [Google Scholar]
- 50.Lu Y., Adegoke O. A., Nepveu A., Nakayama K. I., Bedard N., Cheng D., Peng J., Wing S. S. (2009) USP19 deubiquitinating enzyme supports cell proliferation by stabilizing KPC1, a ubiquitin ligase for p27Kip1. Mol. Cell. Biol. 29, 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adhikari D., Zheng W., Shen Y., Gorre N., Ning Y., Halet G., Kaldis P., Liu K. (2012) Cdk1, but not Cdk2, is the sole Cdk that is essential and sufficient to drive resumption of meiosis in mouse oocytes. Hum. Mol. Genet. 21, 2476–2484 [DOI] [PubMed] [Google Scholar]
- 52.Mall M., Walter T., Gorjánácz M., Davidson I. F., Nga Ly-Hartig T. B., Ellenberg J., Mattaj I. W. (2012) Mitotic lamin disassembly is triggered by lipid-mediated signaling. J. Cell Biol. 198, 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kotak S., Busso C., Gönczy P. (2013) NuMA phosphorylation by CDK1 couples mitotic progression with cortical dynein function. EMBO J. 32, 2517–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikesugi K., Yamamoto R., Mulhern M. L., Shinohara T. (2006) Role of the unfolded protein response (UPR) in cataract formation. Exp. Eye Res. 83, 508–516 [DOI] [PubMed] [Google Scholar]
- 55.Holt L. J., Krutchinsky A. N., Morgan D. O. (2008) Positive feedback sharpens the anaphase switch. Nature 454, 353–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parry D. H., Hickson G. R., O’Farrell P. H. (2003) Cyclin B destruction triggers changes in kinetochore behavior essential for successful anaphase. Curr. Biol. 13, 647–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeffrey P. D., Russo A. A., Polyak K., Gibbs E., Hurwitz J., Massagué J., Pavletich N. P. (1995) Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320 [DOI] [PubMed] [Google Scholar]
- 58.Cui X., Wang L., Zhang J., Du R., Liao S., Li D., Li C., Ke T., Li D. W., Huang H., Yin Z., Tang Z., Liu M. (2013) HSF4 regulates DLAD expression and promotes lens de-nucleation. Biochim. Biophys. Acta 1832, 1167–1172 [DOI] [PubMed] [Google Scholar]
- 59.Tane S., Ikenishi A., Okayama H., Iwamoto N., Nakayama K. I., Takeuchi T. (2014) CDK inhibitors, p21(Cip1) and p27(Kip1), participate in cell cycle exit of mammalian cardiomyocytes. Biochem. Biophys. Res. Commun. 443, 1105–1109 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.