

Abstract
Immune responses to coagulation factors VIII (FVIII) and IX (FIX) represent primary obstacles to hemophilia treatment. Previously, we showed that hematopoietic stem cell (HSC) retroviral gene therapy induces immune nonresponsiveness to FVIII in both naive and preimmunized murine hemophilia A settings. Liver-directed adeno-associated viral (AAV)-FIX vector gene transfer achieved similar results in preclinical hemophilia B models. However, as clinical immune responses to FVIII and FIX differ, we investigated the ability of liver-directed AAV-FVIII gene therapy to affect FVIII immunity in hemophilia A mice. Both FVIII naive and preimmunized mice were administered recombinant AAV8 encoding a liver-directed bioengineered FVIII expression cassette. Naive animals receiving high or mid-doses subsequently achieved near normal FVIII activity levels. However, challenge with adjuvant-free recombinant FVIII induced loss of FVIII activity and anti-FVIII antibodies in mid-dose, but not high-dose AAV or HSC lentiviral (LV) vector gene therapy cohorts. Furthermore, unlike what was shown previously for FIX gene transfer, AAV-FVIII administration to hemophilia A inhibitor mice conferred no effect on anti-FVIII antibody or inhibitory titers. These data suggest that functional differences exist in the immune modulation achieved to FVIII or FIX in hemophilia mice by gene therapy approaches incorporating liver-directed AAV vectors or HSC-directed LV.
Introduction
Hemophilia A and B are X-linked recessive bleeding disorders that result from decreased synthesis or functionality of coagulation factors VIII (FVIII) and IX (FIX), respectively. They are characterized clinically by prolonged and at times spontaneous bleeding into the joints and soft tissues resulting in significant hemophilic arthropathy. Left untreated, severe hemophilia (A or B), as defined by <1% circulating FVIII or FIX activity (respectively), is uniformly lethal. The primary therapeutic option is protein replacement therapy with optimal results being obtained through prophylaxis consisting of two to three injections per week of plasma-derived or recombinant (r) human (h) FVIII or FIX. Although nearly equivalent on a unit activity basis, the mass equivalents differ by ~50-fold (2–5 µg/kg FVIII and 100–250 µg/kg FIX). Additionally, hemophilia A and B differ regarding the immune responses observed against protein replacement products. Pathogenic inhibitors against FVIII develop in up to 33% of persons with severe hemophilia A while anti-FIX inhibitors occur in only 3% of individuals with severe hemophilia B.1 Furthermore, anaphylactoid reactions and nephrotic syndrome are observed in the setting of FIX, but not FVIII inhibitors. Immune tolerance induction has been shown to successfully eradicate inhibitory antibodies in 63–100% of treated hemophilia A patients; however, the patient inclusion criteria for this treatment are limiting and the cost easily can exceed $1,000,000 USD per patient.2 Although the exact mechanism of action of immune tolerance induction is not well understood, preclinical studies suggest administration of high doses of FVIII inhibits the restimulation of FVIII-specific memory B cells and prevents their differentiation into antibody-secreting plasma cells.3 Depletion of FVIII-specific memory B cells may deplete efficient antigen-presenting cells and shift restimulation of effector T cells (Teff) to induction of regulatory T cells (Tregs). T-cell anergy, anti-idiotypic antibodies, and suppressor T cells are other possible mechanisms suggested to play a role in successful immune tolerance induction.4–6 Gene therapy offers not only the potential for a cure of FVIII and FIX deficiencies, but also the opportunity to modulate both naive and primed immune systems through both central and peripheral tolerance mechanisms.7–11 For example, in vivo production of FVIII and FIX achieved through gene transfer approaches could be an efficient and cost effective modality for immune tolerance induction.
Adeno-associated viral (AAV) and lentiviral (LV) vectors have become leading strategies for clinical gene transfer due to their target cell transduction capabilities, limited toxicities, and ability to confer high-level therapeutic transgene expression. Safety and clinical efficacy are being demonstrated in the setting of several blood cell disorders using autologous transplantation of ex vivo LV vector-transduced hematopoietic stem cells (HSC).12,13 HSC-directed gene therapy involves the ex vivo transduction of autologous HSCs followed by transplantation into an HSC depleted (i.e., “conditioned”) recipient. HSCs are efficiently transduced using integrating viruses such as γ-retroviruses and HIV-1-based LV vectors allowing for replication of the proviral genome during cell division. As genetically modified HSCs undergo both self-renewal and differentiation, they create a reservoir of transgene-expressing cells that persist in the bone marrow compartment for the lifetime of the individual and amplify the transgene sequence within the recipient up to 106-fold. Additionally, as the HSC progeny undergo cellular differentiation, the immune system is repopulated with genetically modified cells of the myeloid and lymphoid lineages that now promote recognition of the therapeutic transgene product as a “self” protein within the host proteome. In addition to clinical trials of HSC transplantation (HSCT) gene therapy, multiple trials are underway for in vivo delivery of liver-directed AAV vectors encoding FIX.14,15 AAV gene therapy is less invasive in that it requires a single peripheral infusion of vector into an unconditioned patient. As the packaged viral vector reaches its target tissue, cells undergo in vivo transduction resulting in episomal transgene expression and secretion of transgene product into the periphery. AAV gene transfer is being developed for the treatment of a variety diseases including hemophilia B, human alpha-1 antitrypsin deficiency, lysosomal storage disease, and certain forms of congenital blindness.16–20 Several groups currently are in the late-preclinical stage of both LV- and AAV-vector-based approaches for the treatment of hemophilia A and clinical trials are anticipated.21 As clinical gene therapy trials designed to evaluate the efficacy and toxicity associated with FVIII gene transfer are eminent, understanding the immunological consequences ranging from establishment of tolerance to the development of pathogenic inhibitors is of practical concern.
Previously, we demonstrated correction of the bleeding phenotype in FVIII naive hemophilia A mice using either in vivo liver-directed AAV gene transfer or ex vivo HSC-targeted LV gene transfer incorporating bioengineered high-expression FVIII transgenes.22–25 Furthermore, ex vivo HSC-directed gene therapy was shown to both eradicate anti-FVIII inhibitors and restore circulating FVIII levels to hemophilia A mice with preexisting immunity to FVIII.24,26 However, the stringency of conditioning and the use of gamma retroviruses to achieve adequate levels of transduction preclude the use of HSCT gene therapy as a viable therapeutic for patients with preexisting FVIII inhibitors. Recently, two groups showed that liver-directed expression of FIX, achieved using either LV or AAV vectors, could promote the disappearance of pathological inhibitory antibodies in mouse models of hemophilia B with preexisting immunity to human FIX.10,11 Both IgG and IgE anti-FIX antibodies were eliminated following AAV-FIX gene therapy and this correlated with the establishment of therapeutic levels of circulating FIX antigen. Additionally, treated animals demonstrated therapeutic correction of the bleeding phenotype as well as inhibition and elimination of anti-FIX reactive B cells and plasma cells. Given that the immune responses to FVIII and FIX differ in several aspects, herein, we sought to investigate the durability of the apparent immune tolerance achieved in the FVIII naive gene therapy setting, assess the capability of liver-directed gene therapy for hemophilia A to modulate preexisting immunity to FVIII, and draw comparisons between the outcomes of preclinical hemophilia A and B gene therapy using either liver-directed AAV or HSC-directed LV.
Results
Immune nonresponsiveness to FVIII achieved through LV HSCT gene therapy
Previously, our group showed that retroviral transduction and transplantation of HSCs under both myeloablative and nonmyeloablative conditioning using a high expressing FVIII transgene produced sustained, curative plasma FVIII levels in a murine model of hemophilia A.25–27 Furthermore, HSCT gene therapy recipient animals were shown to be immunologically nonresponsive to serial weekly injections of recombinant FVIII as evidenced by long-term FVIII expression, the absence of anti-FVIII antibody production, and apparent T-cell tolerance in coculture activation assays.26 However, most of these studies were performed using γ-retroviral vectors with intact long terminal repeats driving expression of a porcine FVIII transgene. Due to risk of insertional mutagenesis by γ-retroviruses with intact long terminal repeats and the potential immunogenicity of the porcine FVIII transgene, we modified our gene therapy approach to incorporate a self-inactivating (SIN) HIV-1-based LV vector encoding the bioengineered ET3 FVIII transgene (SIN-LV-ET3) (Figure 1a). ET3 is a human/porcine hybrid FVIII molecule, previously referred to as HP47, designed to have the high expressing sequences of porcine FVIII substituted into the A1, activation peptide, and A3 domains of B-domain-deleted human FVIII.22,28,29 For the current study, lethally irradiated (11 Gy total body irradiation (TBI)) hemophilia A mice were transplanted with genetically modified sca-1+ cells isolated from congenic C57Bl/6 animals. Donor sca-1+ cells were transduced with SIN-LV-ET3 vector containing either a cell type-independent, constitutively active promoter derived from the human elongation factor-1α (EEF1A1) gene locus, designated SIN-LV-EF1α-ET3, or a myeloid-restricted promoter generated from the human CD68 locus, designated SIN-LV-CD68-ET3. One million sca-1+-enriched transduced cells were injected retro-orbitally into lethally irradiated hemophilia A mice to ensure engraftment. Under these transplantation conditions, the recipient mice displayed <10% genetically modified peripheral blood mononuclear cells expressing ET3. In the setting of autologous gene therapy, modeled herein through congenic HSCT, a small number of genetically modified cells (microchimerism) express a single (or unimolecular) neoantigen that distinguishes a fraction of the transplanted cells. We term this state “unimolecular microchimerism” and define it by a state of low-level (<10% of total) engraftment of genetically modified autologous cells expressing a single protein that is absent from the host proteome. In the current study, all transplanted animals displayed <0.1 transgene copy per diploid genome equivalent (N = 6; mean vector copy number (peripheral blood) less than the level of detection; <0.06 proviral genomes per diploid genome equivalent). At these low levels of engraftment, animals transplanted with SIN-LV-CD68-ET3-transduced cells displayed measurable levels of circulating FVIII prior to immunological challenge (mean activity of 0.3 units/ml) while levels of circulating FVIII SIN-LV-EF1α-ET3 transplanted mice were not detectable (Figure 1b). At 10 weeks, weekly immunological challenges consisting of i.v. injection of 1 µg of highly purified recombinant FVIII were initiated. Under these conditions, none of the transplanted animals developed measurable ELISA titers as measured at and beyond 7 weeks postimmunization. Conversely, all of the control mice developed robust immune responses to FVIII that persisted beyond 7 weeks postimmunization (Figure 1c). Despite low to undetectable vector copy number and plasma FVIII activity, transplanted animals displayed an attenuated immune response compared to immunized controls. These results extend our previous studies by showing that even under conditions of unimolecular chimerism, nonresponsiveness to heterologous as well as exogenous FVIII is achieved in hemophilia A mice.
Figure 1.

Immunological challenge following LV-FVIII gene therapy. (a) Schematic of FVIII-LV design. (b) Baseline FVIII activity was determined in LV-CD68-ET3 (closed symbols, each symbol represents an individual mouse) and LV-EF1α-ET3 (open symbols, each symbol represents an individual mouse) hematopoietic stem cell-transplanted (HSCT) mice prior to immunological challenges and subsequently measured after four weekly immunizations (at weeks −3, −2, −1, and 0 indicated by arrows) of 1 µg recombinant ET3. Dashed line indicates limit of detection. (c) Anti-FVIII IgG titers were measured by ELISA and are shown for LV-CD68-ET3 (closed symbols, each symbol represents an individual mouse) and LV-EF1α-ET3 (open symbols, each symbol represents an individual mouse) HSCT-treated mice following the final immunization as well as the immunized controls (×). All mice received four weekly injections of 1 µg of recombinant ET3 posttransplant. The data represent 2, 4, and 7 weeks post final immunization (n = 3 for each experimental cohort, n = 8 for the control cohort). Dashed line indicates limit of detection. A1, A2, A3, C1, and C2, functional domains of a FVIII molecule; BGsPA, bovine growth hormone synthetic polyadenylation tail; CD68, myeloid restricted promoter sequence from the human CD68 locus; CMV, cytomegalovirus promoter; cPPT/CTS, central polypurine tract/central termination sequence; EF1α, elongation factor 1α promoter; L, linker sequence; LV, lentiviral; R, U5, and U3, subcomponents of the 5′ and 3′ long terminal repeats; RRE, Rev protein response element; Ψ (psi), target RNA-site signaling viral packaging.
Partial FVIII nonresponsiveness achieved through AAV-FVIII in the naive hemophilia A setting
In addition to HSC-directed LV gene therapy, liver-directed AAV-FVIII vectors have shown preclinical efficacy in several animal models of hemophilia A.30–32 However, these studies have been limited in sample size (e.g., canine and nonhuman primate studies), duration (<8 weeks), or have utilized immune deficient (e.g., CD4-deficient) animals to obviate immune complications. Therefore, the risk of inhibitor development and the durability of immune tolerance achieved in the absence of inhibitors has yet to be established in any preclinical model. In an earlier study, we demonstrated that clinically relevant doses of AAV8 vector encoding a liver-directed, apolipoprotein E hepatic control region enhancer-human α-1 antitrypsin promoter driving the ET3 transgene conferred correction of the FVIII deficiency and bleeding phenotype in hemophilia A mice despite being oversized, i.e., larger than the estimated packaging capacity of recombinant AAV8.22 However, we also observed that a small fraction of experimental animals developed inhibitory antibodies to FVIII 8–12 weeks after vector administration which correlated with a loss of detectable levels of circulating FVIII. This occurred only in the highest dose cohorts and despite the presence of super physiologic FVIII levels immediately prior to inhibitor development. To investigate this observation further in the current study, naive hemophilia A mice were treated with either a mid-dose (4 × 1012 vp/kg, n = 3) or a high dose (2 × 1013 v/kg, n = 3) of a promoter shortened, liver-directed AAV8-HLP-ET3 vector (Figure 2a). Plasma FVIII activity was monitored for 24 weeks. AAV dose-dependent expression was observed with the mid-dose cohort averaging 70% normal human levels (0.7 IU/ml) and the high-dose cohort averaging 200% normal levels (2 IU/ml) that was sustained for the 5-month observation period (Figure 2b,c). While the high vector doses resulted in super physiological FVIII levels, contrary to previous work, FVIII levels were maintained beyond the 8–12 week windows indicating the lack of inhibitory immune development despite high FVIII levels. Subsequently, these animals also were challenged with a series of intravenous injections of recombinant ET3 (1 µg each) without adjuvant. During this challenge, FVIII activity levels in the mid-dose cohort declined to undetectable levels. Disappearance of plasma FVIII activity corresponded with the appearance of robust anti-ET3 humoral immune responses as detected by anti-ET3 IgG ELISA. Animals treated with high-dose AAV8-ET3 displayed a trend toward a transient decline in FVIII activity during the immunization period. However, their FVIII levels did not decrease to baseline and subsequently increased following termination of the recombinant FVIII infusion challenge protocol. These data suggest that higher doses of AAV8-ET3 are capable of establishing and maintaining active tolerance, while lesser doses may be insufficient.
Figure 2.

Immunological challenge following AAV-FVIII gene therapy. (a) Schematic of FVIII-AAV vector design. (b,c) FVIII activity was measured beginning at 2 weeks post AAV8-HLP-ET3 infusion; mid-dose = 4 × 1012 vp/kg (n = 3) or high dose = 2 × 1013 vp/kg (n = 3), respectively. Arrows indicate weekly immunizations of 1 µg recombinant ET3. (d,e) FVIII activity versus anti-ET3 ELISA titer beginning at the initial immunization for the mid-dose = 4 × 1012 vp/kg (n = 3) or high dose = 2 × 1013 vp/kg (n = 3), respectively. A1, A2, A3, C1, and C2, functional domains of a FVIII molecule; AAV, adeno-associated virus; HLP, hybrid liver-specific promoter; ITR, inverted terminal repeat sequence; L, linker sequence; sPA, synthetic polyadenylation tail.
Efficacy of AAV-FVIII in the setting of preexisting FVIII immunity
Liver-directed expression of FIX following AAV or LV gene transfer has been shown to eliminate preexisting high-titer inhibitory anti-FIX antibodies in a mouse model of hemophilia B.10,11 While two different viral vector systems were utilized, similar results were obtained with both approaches affecting a decrease in anti-FIX ELISA and Bethesda titers after vector administration. Additionally, each group showed that liver-directed expression resulted in the depletion of FIX-specific memory B cells and plasma cells. In an attempt to extend these findings to hemophilia A, we evaluated the therapeutic potential of the AAV8-HLP-ET3 vector in hemophilia A mice with preexisting immunity to FVIII. In the current study, hemophilia A mice were preimmunized with commercially available recombinant hFVIII via four weekly injections of 1 µg FVIII each. Baseline humoral immune responses were characterized by both total anti-hFVIII IgG ELISA titers and anti-hFVIII inhibitory (Bethesda) titers. Upon confirmation of anti-FVIII immunity in 100% of the animals, they were administered AAV8-HLP-ET3 at a dose (4 × 1012 vp/kg) previously shown to restore FVIII to near normal human levels in circulation and similar to doses shown to evade an anticapsid immune response in clinical trials. During a 16-week observation period, no significant changes in anti-hFVIII ELISA or Bethesda titer were observed (Figure 3a,b; Supplementary Figure S2).
Figure 3.

Effect of AAV-ET3 gene therapy on preexisting anti-human FVIII immunity. (a) Individual anti-hFVIII ELISA titers for (closed shapes) the naive hemophilia A mice preimmunized with hFVIII and treated with 4 × 1012 vp/kg of AAV8-HLP-ET3 vector and (open shapes) the preimmunized controls. (b) Anti-hFVIII Bethesda titers in (closed shapes) the naive hemophilia A mice preimmunized with hFVIII and treated with 4 × 1012 vp/kg of AAV8-HLP-ET3 vector and (open shapes) the preimmunized controls. AAV, adeno-associated virus; BU, Bethesda units; HLP, hybrid liver-specific promoter.
A confounding variable of the above experiment was the sequence disparity between the FVIII immunogen and the FVIII transgene product. The immunogen consisted of full-length recombinant hFVIII product possessing B domain sequence of varying length established by heterogeneous intracellular processing by paired basic amino acid cleaving enzyme/furin. Conversely, the transgene product is B domain-deleted ET3 that has ~9% amino acid sequence disparity to B domain-deleted hFVIII. The disparate sequence is present in the A1, activation peptide, and A3 domains of the protein. The immune consequences of this sequence disparity in the setting of preexisting immunity are unknown and may have limited the immune modulation possible through this approach. For example, it is possible that the immunogenic determinants of hFVIII may differ significantly from ET3, and therefore liver-synthesized ET3 is not capable of inhibiting or eradicating hFVIII immunogen-specific memory B cells or plasma cells. Therefore, to test this possibility, the experiment was repeated using animals preimmunized with recombinant ET3. However again, no change in anti-ET3 ELISA or Bethesda titers was observed in animals preimmunized with ET3 (Figure 4a,b; Supplementary Figure S3).
Figure 4.

Effect of AAV-ET3 gene therapy on preexisting anti-ET3 immunity. (a) Individual anti-ET3 ELISA titers for (closed shapes) the naive hemophilia A mice preimmunized with ET3 and treated with 4 × 1012 vp/kg of AAV8-HLP-ET3 vector and (open shapes) the preimmunized controls. (b) Anti-ET3 Bethesda titers in (closed shapes) the naive hemophilia A mice preimmunized with ET3 and treated with 4 × 1012 vp/kg of AAV8-HLP-ET3 vector and (open shapes) the preimmunized controls. AAV, adeno-associated virus; BU, Bethesda units; HLP, hybrid liver-specific promoter.
Given that AAV8-ET3 administration did not modulate preexisting anti-hFVIII or ET3 immunity at the total IgG or inhibitor levels and no circulating ET3 antigen or activity was observed at any time point, three alternative explanations appeared possible. The first was that the preexisting immunity to FVIII somehow prevented AAV-FVIII transduction of hepatocytes. The second was that the adaptive immune system eliminated the transduced cells through a cytotoxic immune response. Similar observations to these have been made in clinical trials of AAV-FIX for hemophilia B where it has been shown that plasma from individuals containing anti-AAV antibodies can impair transduction of mouse liver through AAV neutralization and that CD8+ T-cell-mediated clearance of transduced hepatocytes frequently occurs 8–12 weeks post vector administration when AAV-FIX doses equal to or exceeding 2 × 1012 vp/kg are tested.33,34 Since laboratory mice generally do not possess anti-AAV neutralizing antibodies and CD8+ T-cell responses against transduced hepatocytes also are not observed in murine models, we hypothesized a third scenario wherein hepatocytes were effectively transduced by AAV-FVIII and subsequently biosynthesized ET3 molecules that were rapidly neutralized upon cellular secretion by the saturating levels of circulating anti-FVIII inhibitory antibodies. In order to test this hypothesis, AAV-treated animals harboring preexisting and ongoing humoral immunity to ET3 received lethal doses of radiation (11 Gy TBI) to pharmacologically eliminate antigen-specific effector T cells and B cells, as well as possibly some antibody-secreting plasma cells which are known to be more radiation resistant.35 Previously, we demonstrated that HSC-directed LV gene therapy in the setting of 11 Gy TBI eradicated preexisting anti-FVIII humoral immunity, provided sustained circulating FVIII activity, and established robust immune nonresponsiveness to both the immunogen, hFVIII, and the transgene product, porcine FVIII or ET3.24,26 In the current study, we sought to detect genetically modified hepatocytes and circulating transgene product (ET3) in the context of humoral immunity. A similar approach was pursued, but in this study incorporated transplantation of nongenetically modified donor HSCs to help “reset” or minimally dampen the preexisting anti-FVIII immune status. Successful transplantation and engraftment of donor HSCs was confirmed by flow cytometry and determined to be ~90% donor cell engrafted (data not shown). Animals were monitored for 10 weeks posttransplantation during which time a gradual decrease in anti-FVIII antibody titers was observed by ELISA (Figure 5a,b). However, despite the decrease in anti-FVIII IgG titer, plasma FVIII activity remained below the level of detection by chromogenic substrate assay in all animals. As a more direct approach to identify genetically modified hepatocytes expressing ET3, the experimental animals were euthanized and primary liver cells were assayed for ET3 transgene DNA copy number as well as ET3 RNA transcript levels (Figure 6). The positive detection and quantitation of ET3 transgene DNA and mRNA transcripts in liver tissue supports the conclusion that primary liver hepatocytes were transduced in vivo by administration of the AAV vector and that the transgene cassette delivered remained functional throughout the experiment despite the presence of ongoing FVIII immunity.
Figure 5.

Effect of radiation-based immune suppression following AAV-FVIII gene therapy. (a) Anti-ET3 IgG titers were measured by ELISA in naive hemophilia A mice preimmunized with ET3 and then treated with 4 × 1012 vp/kg AAV8-HLP-ET3 vector posttransplant. Mice were conditioned with 11 Gy total body irradiation and then transplanted with 5 million whole bone marrow cells (n = 4). (b) Individual anti-ET3 IgG titers measured by ELISA. AAV, adeno-associated virus; HLP, hybrid liver-specific promoter.
Figure 6.
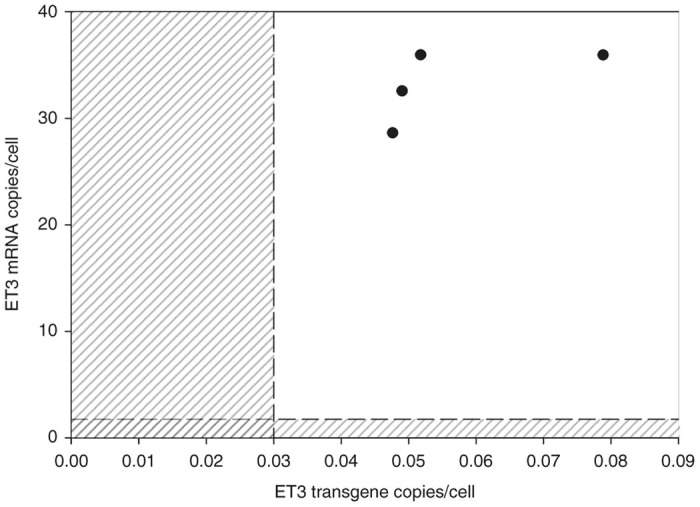
AAV-FVIII gene transfer in the presence of FVIII immunity. Copy number and mRNA levels were determined in hepatocytes isolated from the perfused livers of naive hemophilia A mice preimmunized with ET3 and treated with 4 × 1012 vp/kg AAV8-HLP-ET3 vector (n = 4). Dashed lines and shaded regions indicate the limit of detection for determining both transgene and mRNA copy number. AAV, adeno-associated virus; HLP, hybrid liver-specific promoter.
Efficacy of AAV-FVIII in the setting of low-titer anti-FVIII immunity
Liver-directed AAV-FVIII also has been shown to overcome preexisting anti-FVIII antibodies in a canine model of hemophilia A harboring low anti-FVIII inhibitory titers (≥3 Bethesda unit (BU)).36 As the preexisting inhibitory titers to ET3 in our earlier experiment ranged from 46 to 270 BU and would be considered in the high titer range, it is possible that an immune threshold exists beyond which the ability of AAV8-HLP-ET3 to modulate the anti-FVIII immune response is limited. In order to investigate this possibility, hemophilia A mice (n = 10) were immunized by weekly injection of recombinant ET3 with concomitant monitoring of the anti-FVIII humoral immune response. After three immunizations, all mice had measurable ELISA titers and Bethesda titers ranging from 0 to 25 BU. Subsequently, the animals were administered 4 × 1012 vp/kg of AAV8-HLP-ET3 and monitored biweekly for changes in ELISA titer (Figure 7a,b). Again, however, no significant differences were observed between the low-titer cohorts compared to the two previous high-titer cohorts suggesting that a preexisting immune threshold for rAAV-FVIII does not exist in hemophilia A mice.
Figure 7.

AAV-FVIII gene therapy in the presence of low-titer FVIII inhibitors. (a) Anti-ET3 IgG titers are represented as the percent of the initial titer determined by ELISA in mice with low initial anti-ET3 Bethesda titers (0–25 BU). Naive hemophilia A mice were preimmunized with ET3 and treated with 4 × 1012 vp/kg of AAV8-HLP-ET3 vector (n = 10). (b) Individual anti-ET3 IgG titers measured by ELISA in mice with low initial anti-ET3 Bethesda titers. Mice were preimmunized with ET3 and treated with 4 × 1012 vp/kg of AAV8-HLP-ET3 vector. AAV, adeno-associated virus; HLP, hybrid liver-specific promoter.
Discussion
Hemophilia A remains a promising candidate disease for clinical gene therapy. The therapeutic threshold of achieving 10–50 pmol/l circulating levels of transgene product (i.e., FVIII) is generally predicted to be achievable with current gene transfer technology. Although FVIII replacement therapy certainly is effective at maintaining hemostasis in most cases, the products are short acting, challenging to administer prophylactically and prohibitively expensive for the majority of patients. Gene therapies have the potential to overcome these challenges and transform the care of persons with hemophilia A. Of the available clinical gene transfer technologies, LV and AAV vectors appear to be at the forefront.21,37–39 Possibly the most significant unresolved issue for these approaches, other than demonstration of therapeutic success, is the effect FVIII gene transfer-based therapy will have on anti-FVIII immunity as anti-FVIII immune responses represent the greatest challenge to state of the art clinical care. The patient population targeted in all gene therapy of hemophilia A clinical trials to date has been those previously treated with FVIII replacement products without evidence of existing or prior anti-FVIII immunity. This patient exclusion criteria may preclude the acquisition of data regarding the ability of FVIII gene transfer to modulate tolerance and/or immune activation as well as the prediction of outcomes for nontolerized patients. It has been shown in several model systems, as well as clinically for other diseases, that gene transfer can either elicit new immune responses or alternatively induce a tolerogenic state depending on the platform used and disease setting. Current clinical HSC-directed gene therapy involves the transplantation of LV-transduced HSCs genetically modified to express a missing or defective protein, which may present as a neoantigen depending on the underlying causal mutation. As these cells repopulate the immune system, the resulting chimerism facilitates transplant tolerance, an immunological state in which the host immune cells are not activated by the neoantigen but maintain the ability to launch a pathogen-specific immune response.40–42 This was demonstrated in E16 f8−/− mice using HSC-directed retroviral vector gene therapy incorporating a human FVIII gene variant. Mice treated with either lethal or sublethal TBI were demonstrated to be immune-competent against tetanus toxoid mixed in adjuvant while demonstrating tolerance to hFVIII protein infusions measured by both Bethesda titers and total anti-FVIII IgG.43,44 Additionally, these findings correlated with the studies published by our group.26 Contrary to HSCT gene therapy, AAV vectors are more efficient at in vivo targeting of nonhematopoietic tissues such as liver parenchyma and therefore may possess the capability to take advantage of the liver’s role in immune homeostasis to facilitate tolerance. This hypothesis has been substantiated in preclinical hemophilia B studies in which AAV vectors incorporating a FIX transgene have been shown to induce transgene-specific regulatory T cells and inhibit reactive T-cell responses.45 Although clinical deficiencies in FVIII and FIX are indistinguishable, the proteins themselves are not homologous and the respective immune responses to them are quite dissimilar with FVIII demonstrating more potent immunogenicity at lower concentrations. These confounding differences suggest that gene transfer-facilitated tolerance may differ in terms of durability when incorporating a FVIII transgene in the context of hemophilia A gene therapy.
We previously showed and demonstrate again in the current study that both HSC-directed LV gene therapy and liver-directed AAV gene therapy incorporating a high-expression FVIII transgene can elevate plasma FVIII activity levels into the normal range.22,23 However, in both studies, anti-FVIII inhibitor formation occurred under certain experimental conditions. For HSCT LV gene therapy, we discovered that pretransplantation radiation or other pharmacological-based T-cell suppression is critical to the avoidance of an immune response to FVIII posttransplantation.25 Furthermore, we demonstrated that these same animals are immunologically nonresponsive to i.v. challenge with recombinant FVIII. In these studies, we utilized earlier generation γ-retroviral vectors encoding a high-expression porcine FVIII transgene. Therefore, to evaluate tolerance in the context of a state of the art clinical HSCT LV gene therapy platform, naive hemophilia A mice were conditioned with lethal doses of radiation to better control levels of engraftment and transplanted with sca-1+-enriched bone marrow cells transduced using an LV vector expressing ET3 via a universal (SIN-LV-EF1α-ET3) or myeloid-specific (SIN-LV-CD68-ET3) promoter. It is important to note that Moayeri et al. have shown E16 F8−/− mice treated with either lethal or sublethal doses of TBI conditioning prior to HSCT maintained immune competence while failing to develop an anti-FVIII immune responses after protein infusions of hFVIII. In the current study, all transplanted animals engrafted with >80% total donor cells and less than 10% genetically modified cells in the peripheral blood mononuclear cell population. We believe this level of engraftment to be more clinically translatable as the use of safer LV viral vectors with diminished transduction capabilities compared to gamma retroviruses and the incorporation of nonmyeloablative conditioning regimens likely will result in microchimeric levels of genetically modified cells. Interestingly, baseline FVIII activity prior to immunological challenges was only detectable in SIN-LV-CD68-ET3-transduced sca-1+ cell-treated animals and apparently was not a determinant of transplantation induced FVIII nonresponsiveness. It is possible that anti-ET3 antibodies are present in the SIN-LV-EF1α-ET3-transduced sca-1+ cell-treated mice and are binding to circulating ET3 but remain at a level below the limit of detection for our assay. After four weekly i.v. injections of recombinant FVIII, transplanted animals were assayed for the development of anti-FVIII immune responses. SIN-LV-EF1α-ET3-transduced sca-1+ cell-treated mice had no measurable ELISA titer at any time for the duration of the experiment, despite undetectable levels of circulating ET3. We hypothesize that levels of circulating FVIII required to restore hemostasis are not necessary to facilitate a nonresponsive immune response to FVIII. It is possible that the levels of FVIII antigen being expressed posttransplant, although not detectable, may be sufficient to carry out normal antigen presentation. Another possibility is that FVIII is not being effectively secreted but retained intracellularly in various immune cells. As these cells undergo normal physiological turnover, the immune system is exposed to FVIII in the absence of costimulatory signaling and therefore the immune response to FVIII becomes nonresponsive in nature rather than leaning towards CD4+ T-cell activation. Although transient, near baseline titers were observed in two of the SIN-LV-CD68-ET3-transduced sca-1+ cell-treated animals, the immune responses observed in the HSC-directed LV gene therapy cohorts spontaneously resolved. These results are similar to clinical outcomes observed in hemophilia A patients considered tolerized to prophylactic FVIII treatments, as repeated protein infusions do not result in the development of anti-FVIII antibody titers. Cell-mediated clearance of genetically modified cells is unlikely as transgene copies were detected in several tissues at 7 weeks with higher transgene copy numbers observed in the SIN-LV-CD68-ET3 cohort which is consistent with the higher multiplicity of infection used for transduction of the sca-1+ hematopoietic HSC stem and progenitor cells. These data suggest that unimolecular chimerism in the myeloid compartment is sufficient to facilitate FVIII-specific transplantation tolerance following HSC-directed LV-FVIII gene therapy. Specifically, CD68 expression has been confirmed in both thymic and liver tissues, both of which are known for their roles in T-cell education and Treg induction, respectively, suggesting a potential mechanism for CD68-mediated transplantation tolerance.46 However, further experimentation will be required to tease apart what mechanisms of transplantation tolerance are contributing to the FVIII immune nonresponsiveness.
In previous AAV-FVIII gene transfer studies, we did not evaluate specifically the durability of FVIII nonresponsiveness and/or whether reintroduction of FVIII protein could break this state, which is a potential clinical outcome. Additional studies demonstrating that AAV liver-directed expression of coagulation factor IX prevents inhibitor formation after peripheral infusions of FIX concentrate in multiple animal models of hemophilia B suggest that a similar beneficial outcome may result from AAV-FVIII gene therapy.47,48 In the FIX studies, tolerance was induced using a dose observed to evade an anticapsid CD8+ T-cell response in previous clinical trials. However, in our experiments, this dose of AAV8-HLP-ET3 (4 × 1012 vg/kg) was not shown to be tolerogenic as evident by the observation of rapid, high-titer FVIII-specific inhibitory antibody formation following serial challenges with adjuvant-free recombinant FVIII. It is plausible that higher vector doses may have shown measurable differences in immune modulation in the preimmunized cohorts as was observed in the naive setting; however, these experiments were designed prior to collection of those results. In these studies, vector dose was chosen based on previously published data and the guidelines set by the hemophilia B clinical trials using AAV-FIX. However, even at relatively high doses (2 × 1013 vp/kg), AAV8-HLP-ET3 was unable to completely inhibit a humoral immune response against FVIII during recombinant FVIII challenge, reinforcing the hypothesis that significant differences in immunogenicity exist between FVIII and FIX. These observations may be associated with the tendency of FVIII to engage in the unfolded protein response, perhaps contributing to its immunogenicity.49 Again, however, as we have shown that porcine FVIII and ET3 display diminished engagement of the unfolded protein response, this hypothesis is not supported by the current studies. Furthermore, there is a significant difference between the specific activities of FIX versus FVIII. A single unit of FIX has a plasma concentration of ~5,000 ng/ml while a single unit of FVIII only has a plasma concentration between 100 and 200 ng/ml. At steady state, liver-directed expression of FIX results in antigen molar concentrations order of magnitude greater than FVIII and could therefore modulate the response of antigen-presenting cells and immune effector cells differently. These effects may be amplified with AAV-FVIII compared to AAV-FIX as liver-directed tolerance has been shown to be dependent on expression levels of antigen.9,50 Our results parallel these conclusions in that only the very high dose of AAV8-HLP-ET3 (2 × 1013 vp/kg) was able to attenuate inhibitor formation during rigorous, but clinically modeled, immunological challenges with recombinant FVIII. Herzog et al. have shown that adoptive transfer of CD4+ CD25+ regulatory T cells from AAV-FIX-treated mice can suppress anti-FIX antibody formation in naive and subsequently immunologically challenged mice.9,45 However, it is important to note that for all of these studies, analysis of Treg responses, adoptive transfers experiments, and analysis of Foxp3 induction were performed in immune-competent F9+/+ mice, in which self-tolerance exists against murine FIX. Existing data support the concept that natural Tregs (nTregs) originating in the thymus are responsible for maintaining self-tolerance and are selected for through the interaction of peptides bound to major histocompatibility complex and T-cell receptors expressed on the surface of nTregs.51 Given that CD4+ T-cell epitopes have been demonstrated to be conserved between species, it is possible that nTreg-specific epitopes could be conserved between orthologs.52 Therefore, nTregs responsible for maintaining self-tolerance to murine FIX may also contribute to CD4+ CD25+ Treg responses observed in AAV-FIX studies using F9+/+ immune-competent mice. Subsequently, it is plausible that the AAV-Treg induction paradigm may not be applicable to mouse models of hemophilia in which self-tolerance to the murine ortholog may not be present. Lastly, long-term AAV studies incorporating a canine FVIII transgene have been shown to be tolerogenic in hemophilia A dogs suggesting that AAV-FVIII tolerance may be dependent on the use of a species-specific FVIII transgene.
In contrast to reducing protein replacement product immunogenicity, elimination of preexisting immune responses is a distinct therapeutic objective. Since, both liver-directed AAV and LV vector-based gene therapy have been shown to eliminate circulating inhibitory antibodies and transgene-specific memory B cells in preimmunized hemophilia B mice,10,11 a logical extension is that liver-directed FVIII gene transfer may provide similar therapeutic benefit to hemophilia A patients with inhibitors. Furthermore, this represents a critical un-met clinical need as long-term treatment options are limited or not available to the majority of the hemophilia A inhibitor patient population. We have previously shown that transplantation of HSCs genetically modified by gamma retroviruses into lethally and sublethally irradiated mice had the ability to eradicate preexisting anti-FVIII inhibitors and establish therapeutic levels of circulating FVIII. However, these results were not clinically translatable as high doses of radiation were required for conditioning. Therefore, in addition to studying liver-directed AAV gene therapy in the FVIII naive state, we also analyzed the therapeutic potential of AAV8-HLP-ET3, due its efficiency in gene transfer and apparent safety observed in ongoing clinical trials, in preimmunized hemophilia A mice harboring both low and high FVIII inhibitory titers. We observed no significant changes in either ELISA titer or Bethesda titers over the course of 16 weeks suggesting no modulation of anti-FVIII antibody-secreting cells or FVIII-specific T-cell responses. Both copy number and mRNA analysis confirmed transduction of hepatocytes and expression of the transgene, which likely was being instantly neutralized by the circulating inhibitors once secreted from the cell. Given that higher doses of AAV8-HLP-ET3 (2 × 1013 vp/kg) demonstrated an attenuated immune response after immunological challenge in the naive state, it is possible that higher vector doses may be effective at eradicating preexisting anti-FVIII inhibitory antibodies as observed in the FIX studies. However, recent AAV clinical trials have shown this dose to be clinically untranslatable and therefore ongoing work in our lab has been focusing on developing more potent FVIII-expressing AAV vectors that deliver higher circulating levels of antigen at lower doses of vector.
Overall, the results of the current study again highlight the differences in both the immune response to FVIII and FIX and the immune consequences of liver-directed as compared to HSC-directed gene transfer. Unlike what has been shown for liver-directed FIX expression in hemophilia B mice, liver-directed FVIII gene therapy in both the naïve and preimmunized setting did not make hemophilia A mice nonresponsive to exogenous infused recombinant FVIII. However, similar to our previous results, in the current study, HSC-directed LV-ET3 gene therapy again was effective at promoting immune nonresponsiveness to serial challenges with exogenous recombinant FVIII. Therefore, despite both ET3 gene transfer approaches delivering sustained, curative levels of plasma FVIII activity in hemophilia A mice without the initial provocation of anti-FVIII inhibitors, only the HSC-LV approach, at least under these experimental settings, appears to prevent the development of anti-FVIII IgG upon immunological challenge with peripheral infusions of recombinant FVIII. Although HSC-LV gene therapy does require preconditioning for HSC engraftment, this component also has clear suppressive effects on the immune system that likely contribute to its success. However, even with stringent pharmacological immune modulation, 11 Gy TBI followed by bone marrow transplant, AAV8-HLP-ET3 gene transfer was unable to efficiently eradicate an anti-FVIII humoral immune response. Another caveat of the current nonclinical studies is that the line of hemophilia A mice used demonstrate uniform inhibitor responses following six or less i.v. infusions of exogenous FVIII. This is in stark contrast to the human clinical situation where inhibitor frequency is less than 0.3 in severe hemophilia A and significantly less in moderate and mild cases. As all clinical hemophilia gene therapy studies to date have been conducted in the previously treated population without inhibitors, the current studies cannot model this situation. These studies were not designed to make direct comparisons in terms of the mechanisms of immune nonresponsiveness being achieved by each method of gene transfer or the kinetics of induction, however they do show that different gene therapy platforms may affect differences in clinical immune outcomes and that immune barrier in hemophilia A may be greater than hemophilia B due to differences in coagulation factor immunobiology.
Materials and Methods
Animal experiments
All animal studies were performed under the guidelines set by the Emory University Institutional Animal Care and Use Committee. Exon 16-disrupted mice back-crossed onto a C57BL/6 background were used as a model of hemophilia A.53 In the HSCT gene therapy experiments, 6–8-week-old transgenic mice expressing the enhanced green fluorescent protein from the β-actin promoter on a C57Bl/6 background were used as congenic donors of HSCs.54 TBI was administered using a vented chamber within Gammacell 40 Extractor. Immunizations and immunological challenges using purified FVIII were performed by retro-orbital injection. Lastly, AAV administration was delivered i.v. to 8–10-week-old mice by tail-vein injection.
Whole bone marrow transplants
Transplant recipients received 11 Gy TBI split over two doses 4 hours apart. Whole bone marrow was isolated from 6–8-week-old congenic green fluorescent protein-positive transgenic mice. Cells were flushed from the femurs and tibias and filtered to eliminate debris. After 10 minutes of red cell lysis, whole bone marrow cells were resuspended in sterile phosphate-buffered saline, counted, and 5 million cells per mouse were immediately infused, via retro-orbital injection, into transplant recipients.
Sca-1+-enriched bone marrow transplants
Whole bone marrow was enriched for Sca-1+ antigen as previously described using positive immunomagnetic bead selection.27 Cells were then stimulated for 24 hours in media containing murine stem cell factor (100 ng/ml), murine interleukin-3 (20 ng/ml), human interleukin-11 (100 ng/ml), and human Flt-3 ligand (100 ng/ml). After stimulation, cells were transduced twice in half volume (multiplicity of infection between 16 and 70) virus at a density of 2 million Sca-1+ cells/ml over the course of 24 hours. Cells were then resuspended in phosphate-buffered saline and 1 million transduced cells were injected retro-orbitally into lethally irradiated (11 Gy TBI) naive hemophilia A mice.
LV-FVIII production
Viral accessory plasmids along with the bioengineered high-expression FVIII transgene (designated ET3) encoding expression plasmid were transiently transfected in 293T-17 cells using a calcium phosphate transfection method to generate LV vectors pseudotyped with the VSVG envelope similar to the method described previously except for the utilization of the calcium phosphate transfection reagent (Sigma Aldrich, St. Louis, MO).55 Conditioned media was collected for 3 days beginning at 48 hours posttransfection and passed through a 0.45-µm filter. Virus was concentrated by overnight centrifugation at 10,000 × g overnight, followed by filtration using a 0.22-µm filter. Viral concentrate titers were determined using quantitative real-time PCR analysis.
AAV-FVIII production
DNA fragments encoding either the 5′ AAV inverted terminal repeat proximal to the hybrid liver-specific promoter (HLP) or a synthetic rabbit beta-globin polyadenylation signal and 3′ inverted terminal repeat were synthesized de novo (Integrated DNA Technologies and GenScript) and introduced to the flanking ends of the ET3 transgene by restriction enzyme digestion/ligation-based molecular cloning techniques to generate AAV8-HLP-ET3 (5,111 base pairs).30 The complete sequence is available as Supplementary Figure S1. Recombinant AAV8/2 vector particles carrying the HLP-ET3 cassette were produced as previously described.56 Briefly, 293T cells were triple transfected by the calcium phosphate method with the AAV8-HLP-ET3 cassette, adenovirus helper plasmid AdHP, and AAV8 packaging plasmid AAV8PK.57,58 Seventy-two hours after transfection, the cells were harvested, lysed, and clarified by centrifugation. Vector particles were precipitated using 8% polyethylene glycol, resuspended, and purified using two round of cesium chloride gradient ultracentrifugation. Vector particles were dialyzed into phosphate-buffered saline supplemented with 0.001% pluronic F68 and quantified by quantitative SDS–PAGE densitometry against a standard reference vector.
FVIII and immune response analyses
Anti-FVIII antibody titers were determined using an ELISA. Immobilized hFVIII (Recombinate, Baxalta, Bannockburn, IL) or ET3 was bound to microtiter plates and then incubated with mouse plasma. Anti-FVIII antibodies were detected using a pan goat-anti-mouse IgG secondary antibody conjugated to alkaline phosphatase and absorbance was read at 405 nm. The absorbance values of test plasmas were plotted against the logarithm of the plasma dilution, and the titers were determined to be the reciprocal of the dilution in which the optical density value was three times that of the background (naïve E16 F8−/− mouse plasma), or an optical density reading of 0.3.59 Additionally, samples at any given time point were analyzed on the same sets of plates using the same buffers and reagents in order to make valid comparisons between animals at a given time point. Inhibitory antibody titers were determined using a Bethesda assay as previously described.60,61 Titers were determined as the average dilution of test plasmas in which 50% of the FVIII activity was inhibited. Lastly, FVIII activity was determined using a commercially available COATEST SP FVIII assay (Coatest SP, Diapharma, West Chester, OH) according the manufacturer’s instruction. Activity was quantified using a standard curve was generated using pooled citrated human plasma (Georgia King, Overland Park, KS).
Molecular studies
DNA and RNA were isolated from livers perfused for 15 minutes with saline using the DNeasy Blood and Tissue Kit (Qiagen) and the RNeasy mini kit (Qiagen), respectively, according to the manufacturer’s instructions. All qPCR and data analysis was performed on a Step-One real-time PCR platform using the device’s software. Copy number was determined using a standard curve generated from dilutions of AAV8-HLP-ET3 plasmid. Standards or 100 ng of sample DNA was added to 1× SYBR Green PCR Master Mix (Life Technologies) containing 300 nm concentrations of forward and reverse primers. Copy number was normalized to the number of mammalian diploid genomes present in 100 ng of DNA. Similarly, mRNA transcript levels were determined on AAV-treated liver tissues as described previously using 100 ng of sample RNA.28
Acknowledgments
We would like to thank Shangzhen Zhou (Center for Cellular and Molecular Therapeutics, Children’s Hospital of Philadelphia and currently, Center for Advanced Retinal & Ocular Therapeutics, AAV Vector Core) for technical assistance with AAV vector production. This work was supported by funding from the National Institutes of Health/National Heart, Lung and Blood Institute (C.B.D. and H.T.S.) and Hemophilia of Georgia (C.B.D. and H.T.S.).
Footnotes
C.B.D. and H.T.S. are cofounders of Expression Therapeutics and own equity in the company. Expression Therapeutics owns the intellectual property associated with ET3. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies.
References
- Lollar, P (2004). Pathogenic antibodies to coagulation factors. Part one: factor VIII and factor IX. J Thromb Haemost 2: 1082–1095. [DOI] [PubMed] [Google Scholar]
- Meeks, SL, Chapman, RL, Kempton, C and Dunn, AL (2013). Late immune tolerance induction in haemophilia A patients. Haemophilia 19: 445–448. [DOI] [PubMed] [Google Scholar]
- Hausl, C, Ahmad, RU, Sasgary, M, Doering, CB, Lollar, P, Richter, G et al. (2005). High-dose factor VIII inhibits factor VIII-specific memory B cells in hemophilia A with factor VIII inhibitors. Blood 106: 3415–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai, Y, Shima, M, Tanaka, I, Fukuda, K, Yoshida, K and Yoshioka, A (2004). Association of anti-idiotypic antibodies with immune tolerance induction for the treatment of hemophilia A with inhibitors. Haematologica 89: 696–703. [PubMed] [Google Scholar]
- Mueller, SN and Ahmed, R (2009). High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA 106: 8623–8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, B and Lillicrap, D (2009). The molecular mechanisms of immunomodulation and tolerance induction to factor VIII. J Thromb Haemost 7: 1446–1456. [DOI] [PubMed] [Google Scholar]
- Tomita, Y, Khan, A and Sykes, M (1994). Role of intrathymic clonal deletion and peripheral anergy in transplantation tolerance induced by bone marrow transplantation in mice conditioned with a nonmyeloablative regimen. J Immunol 153: 1087–1098. [PubMed] [Google Scholar]
- Racine, J, Wang, M, Zhang, C, Lin, CL, Liu, H, Todorov, I et al. (2011). Induction of mixed chimerism with MHC-mismatched but not matched bone marrow transplants results in thymic deletion of host-type autoreactive T-cells in NOD mice. Diabetes 60: 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, O, Dobrzynski, E, Wang, L, Nayak, S, Mingle, B, Terhorst, C et al. (2007). Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood 110: 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markusic, DM, Hoffman, BE, Perrin, GQ, Nayak, S, Wang, X, LoDuca, PA et al. (2013). Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol Med 5: 1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annoni, A, Cantore, A, Della Valle, P, Goudy, K, Akbarpour, M, Russo, F et al. (2013). Liver gene therapy by lentiviral vectors reverses anti-factor IX pre-existing immunity in haemophilic mice. EMBO Mol Med 5: 1684–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Biasco, L, Scaramuzza, S, Ferrua, F, Cicalese, MP, Baricordi, C et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341: 1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi, A, Montini, E, Lorioli, L, Cesani, M, Fumagalli, F, Plati, T et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158. [DOI] [PubMed] [Google Scholar]
- Monahan, PE, Sun, J, Gui, T, Hu, G, Hannah, WB, Wichlan, DG et al. (2015). Employing a gain-of-function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: preclinical evaluation supporting an ongoing adeno-associated virus clinical trial. Hum Gene Ther 26: 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantly, ML, Chulay, JD, Wang, L, Mueller, C, Humphries, M, Spencer, LT et al. (2009). Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci USA 106: 16363–16368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Rosales, C, McIntosh, J, Rastegarlari, G, Nathwani, D, Raj, D et al. (2011). Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther 19: 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embury, JE, Charron, CE, Martynyuk, A, Zori, AG, Liu, B, Ali, SF et al. (2007). PKU is a reversible neurodegenerative process within the nigrostriatum that begins as early as 4 weeks of age in Pah(enu2) mice. Brain Res 1127: 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell, HE, McCurdy, VJ, Eaton, SC, Wilson, DU, Johnson, AK, Randle, AN et al. (2015). AAV-mediated gene delivery in a feline model of Sandhoff disease corrects lysosomal storage in the central nervous system. ASN Neuro 7: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, J, Ashtari, M, Wellman, J, Marshall, KA, Cyckowski, LL, Chung, DC et al. (2012). AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med 4: 120ra15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High, KH, Nathwani, A, Spencer, T and Lillicrap, D (2014). Current status of haemophilia gene therapy. Haemophilia 20 (suppl. 4): 43–49. [DOI] [PubMed] [Google Scholar]
- Brown, HC, Wright, JF, Zhou, S, Lytle, AM, Shields, JE, Spencer, HT et al. (2014). Bioengineered coagulation factor VIII enables long-term correction of murine hemophilia A following liver-directed adeno-associated viral vector delivery. Mol Ther Methods Clin Dev 1: 14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering, CB, Denning, G, Dooriss, K, Gangadharan, B, Johnston, JM, Kerstann, KW et al. (2009). Directed engineering of a high-expression chimeric transgene as a strategy for gene therapy of hemophilia A. Mol Ther 17: 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering, CB, Gangadharan, B, Dukart, HZ and Spencer, HT (2007). Hematopoietic stem cells encoding porcine factor VIII induce pro-coagulant activity in hemophilia A mice with pre-existing factor VIII immunity. Mol Ther 15: 1093–1099. [DOI] [PubMed] [Google Scholar]
- Ide, LM, Gangadharan, B, Chiang, KY, Doering, CB and Spencer, HT (2007). Hematopoietic stem-cell gene therapy of hemophilia A incorporating a porcine factor VIII transgene and nonmyeloablative conditioning regimens. Blood 110: 2855–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide, LM, Iwakoshi, NN, Gangadharan, B, Jobe, S, Moot, R, McCarty, D et al. (2010). Functional aspects of factor VIII expression after transplantation of genetically-modified hematopoietic stem cells for hemophilia A. J Gene Med 12: 333–344. [DOI] [PubMed] [Google Scholar]
- Gangadharan, B, Parker, ET, Ide, LM, Spencer, HT and Doering, CB (2006). High-level expression of porcine factor VIII from genetically modified bone marrow-derived stem cells. Blood 107: 3859–3864. [DOI] [PubMed] [Google Scholar]
- Doering, CB, Healey, JF, Parker, ET, Barrow, RT and Lollar, P (2002). High level expression of recombinant porcine coagulation factor VIII. J Biol Chem 277: 38345–38349. [DOI] [PubMed] [Google Scholar]
- Doering, CB, Healey, JF, Parker, ET, Barrow, RT and Lollar, P (2004). Identification of porcine coagulation factor VIII domains responsible for high level expression via enhanced secretion. J Biol Chem 279: 6546–6552. [DOI] [PubMed] [Google Scholar]
- McIntosh, J, Lenting, PJ, Rosales, C, Lee, D, Rabbanian, S, Raj, D et al. (2013). Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 121: 3335–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino, DE, Lange, AM, Altynova, ES, Sarkar, R, Zhou, S, Merricks, EP et al. (2011). Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther 19: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiwata, A, Mimuro, J, Mizukami, H, Kashiwakura, Y, Takano, K, Ohmori, T et al. (2009). Liver-restricted expression of the canine factor VIII gene facilitates prevention of inhibitor formation in factor VIII-deficient mice. J Gene Med 11: 1020–1029. [DOI] [PubMed] [Google Scholar]
- Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno, CS, Pierce, GF, Arruda, VR, Glader, B, Ragni, M, Rasko, JJ et al. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12: 342–347. [DOI] [PubMed] [Google Scholar]
- Slifka, MK, Antia, R, Whitmire, JK and Ahmed, R (1998). Humoral immunity due to long-lived plasma cells. Immunity 8: 363–372. [DOI] [PubMed] [Google Scholar]
- Finn, JD, Ozelo, MC, Sabatino, DE, Franck, HW, Merricks, EP, Crudele, JM et al. (2010). Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood 116: 5842–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL and Herzog, RW (2015). Gene therapy for hemophilia. Front Biosci (Landmark Ed) 20: 556–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori, T, Mizukami, H, Ozawa, K, Sakata, Y and Nishimura, S (2015). New approaches to gene and cell therapy for hemophilia. J Thromb Haemost 13 (suppl. 1): S133–S142. [DOI] [PubMed] [Google Scholar]
- Doering, CB and Spencer, HT (2014). Replacing bad (F)actors: hemophilia. Hematology Am Soc Hematol Educ Program 2014: 461–467. [DOI] [PubMed] [Google Scholar]
- Roncarolo, MG, Gregori, S, Lucarelli, B, Ciceri, F and Bacchetta, R (2011). Clinical tolerance in allogeneic hematopoietic stem cell transplantation. Immunol Rev 241: 145–163. [DOI] [PubMed] [Google Scholar]
- Ruiz, P, Maldonado, P, Hidalgo, Y, Gleisner, A, Sauma, D, Silva, C et al. (2013). Transplant tolerance: new insights and strategies for long-term allograft acceptance. Clin Dev Immunol 2013: 210506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs, DH (2011). Transplant tolerance: bench to bedside–26th annual Samuel Jason Mixter Lecture. Arch Surg 146: 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayeri, M, Hawley, TS and Hawley, RG (2005). Correction of murine hemophilia a by hematopoietic stem cell gene therapy. Mol Ther 12: 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayeri, M, Ramezani, A, Morgan, RA, Hawley, TS and Hawley, RG (2004). Sustained phenotypic correction of hemophilia a mice following oncoretroviral-mediated expression of a bioengineered human factor VIII gene in long-term hematopoietic repopulating cells. Mol Ther 10: 892–902. [DOI] [PubMed] [Google Scholar]
- Mingozzi, F, Liu, YL, Dobrzynski, E, Kaufhold, A, Liu, JH, Wang, Y et al. (2003). Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest 111: 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal, AJ, McNeill, E, Kapellos, TS, Regan-Komito, D, Norman, S, Burd, S et al. (2014). Human CD68 promoter GFP transgenic mice allow analysis of monocyte to macrophage differentiation in vivo. Blood 124: e33–e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer, GP, Herzog, RW, Mount, J, Arruda, VR, Tillson, DM, Hathcock, J et al. (2009). Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood 113: 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crudele, JM, Finn, JD, Siner, JI, Martin, NB, Niemeyer, GP, Zhou, S et al. (2015). AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood 125: 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, HC, Gangadharan, B and Doering, CB (2011). Enhanced biosynthesis of coagulation factor VIII through diminished engagement of the unfolded protein response. J Biol Chem 286: 24451–24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack, BK, Merchant, S, Markusic, DM, Nathwani, AC, Davidoff, AM, Byrne, BJ et al. (2012). Transient B cell depletion or improved transgene expression by codon optimization promote tolerance to factor VIII in gene therapy. PLoS One 7: e37671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, MS, Boesteanu, A, Reed, AJ, Petrone, AL, Holenbeck, AE, Lerman, MA et al. (2001). Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol 2: 301–306. [DOI] [PubMed] [Google Scholar]
- Shkap, V, Molad, T, Brayton, KA, Brown, WC and Palmer, GH (2002). Expression of major surface protein 2 variants with conserved T-cell epitopes in Anaplasma centrale vaccinates. Infect Immun 70: 642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, L, Lawler, AM, Antonarakis, SE, High, KA, Gearhart, JD and Kazazian, HH Jr. (1995). Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet 10: 119–121. [DOI] [PubMed] [Google Scholar]
- Nakanishi, T, Kuroiwa, A, Yamada, S, Isotani, A, Yamashita, A, Tairaka, A et al. (2002). FISH analysis of 142 EGFP transgene integration sites into the mouse genome. Genomics 80: 564–574. [DOI] [PubMed] [Google Scholar]
- Johnston, JM, Denning, G, Moot, R, Whitehead, D, Shields, J, Le Doux, JM et al. (2014). High-throughput screening identifies compounds that enhance lentiviral transduction. Gene Ther 21: 1008–1020. [DOI] [PubMed] [Google Scholar]
- Ayuso, E, Mingozzi, F, Montane, J, Leon, X, Anguela, XM, Haurigot, V et al. (2010). High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther 17: 503–510. [DOI] [PubMed] [Google Scholar]
- Matsushita, T, Elliger, S, Elliger, C, Podsakoff, G, Villarreal, L, Kurtzman, GJ et al. (1998). Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther 5: 938–945. [DOI] [PubMed] [Google Scholar]
- Wright, JF, Le, T, Prado, J, Bahr-Davidson, J, Smith, PH, Zhen, Z et al. (2005). Identification of factors that contribute to recombinant AAV2 particle aggregation and methods to prevent its occurrence during vector purification and formulation. Mol Ther 12: 171–178. [DOI] [PubMed] [Google Scholar]
- Parker, ET, Healey, JF, Barrow, RT, Craddock, HN and Lollar, P (2004). Reduction of the inhibitory antibody response to human factor VIII in hemophilia A mice by mutagenesis of the A2 domain B-cell epitope. Blood 104: 704–710. [DOI] [PubMed] [Google Scholar]
- Meeks, SL, Healey, JF, Parker, ET, Barrow, RT and Lollar, P (2009). Non-classical anti-factor VIII C2 domain antibodies are pathogenic in a murine in vivo bleeding model. J Thromb Haemost 7: 658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper, CK, Aledort, L, Aronson, D, Counts, R, Edson, JR, van Eys, J et al. (1975). Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 34: 612. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.