

Abstract
Epithelia are fundamental tissues that line cavities, glands, and outer body surfaces. We use three-dimensional (3D) embedded culture of primary murine mammary epithelial ducts, called “organoids,” to recapitulate in days in culture epithelial programs that occur over weeks deep within the body. Modulating the composition of the extracellular matrix (ECM) allows us to model cell- and tissue-level behaviors observed in normal development, such as branching morphogenesis, and in cancer, such as invasion and dissemination. Here, we describe a collection of protocols for 3D culture of mammary organoids in different ECMs and for immunofluorescence staining of 3D culture samples and mammary gland tissue sections. We illustrate expected phenotypic outcomes of each assay and provide troubleshooting tips for commonly encountered technical problems.
Keywords: 3D culture, Organotypic culture, Organoids, Extracellular matrix, Matrigel, Collagen I, Branching morphogenesis, Mammary epithelium, Immunofluorescence
1 Introduction
Mammary gland development occurs postnatally from a simple epithelial rudiment [1, 2]. During puberty, this rudiment undergoes stratification and initiates branching morphogenesis to form a network of epithelial tubes. The functional unit of elongation is a proliferative, multilayered front called the terminal end bud (TEB) [3, 4]. Behind the TEB, repolarization to a mature duct reestablishes a simple, bi-layered architecture, characterized by an inner layer of luminal epithelial cells and an outer layer of myoepithelial cells. These fundamental programs involve concurrent changes in cell proliferation, migration, polarity, and tissue architecture and are modulated by signaling cues from stromal cells and the extracellular matrix (ECM) [5].
Because mammary epithelium develops within a fat pad with limited optical accessibility, various groups have used 3D culture to facilitate direct observation and manipulation of epithelial cell behaviors [6–14]. Cultures of primary mammary tissues were first developed half a century ago [15, 16], but the past decade has seen significant improvements in and increasing utilization of 3D culture models [17–20]. While many conventional methods rely on immortalized cell lines or primary single cells, we use freshly isolated, murine mammary epithelial ducts, which we term “organoids.” The organoid assay arose from a series of papers published in the Bissell and Werb Labs [17–19, 21]. There are technical differences among the papers, but all involve mechanical disruption, enzymatic digestion, and differential centrifugation to separate mammary epithelial organoids from surrounding adipocytes and stromal cells. Purified organoids can be embedded in various ECMs to model distinct epithelial programs.
Our recent studies have revealed many of the cellular mechanisms driving epithelial morphogenesis and demonstrated that the ECM microenvironment regulates the migration and dissemination of mammary epithelial cells [17, 22–24]. Normal development in vivo occurs within a basement membrane, and we use Matrigel, a basement membrane-rich ECM, as an experimentally convenient model for the normal ductal microenvironment. Culture of organoids in basal medium, without supplemental growth factors, induces formation of simple, bi-layered cysts, while culture with growth factor induces a stereotyped program of branching morphogenesis. In contrast, cancer progression involves breaks in the basement membrane, and the microenvironment around a tumor is enriched in collagen I [25–30]. We demonstrated that collagen I-rich microenvironments induce a conserved program of invasion and dissemination in normal and malignant mammary epithelium [23]. Conversely, defined mixtures of Matrigel and collagen I can reproduce a more physiological organization of the elongating TEB [24].
Building on our previously published methods [17, 22, 23, 31, 33–35], this protocol seeks to provide a comprehensive guide to utilizing the mammary organoid assay (Fig. 1a). We introduce several variations on the assay in different ECMs and growth conditions that model different aspects of epithelial development and disease. We also provide optimized protocols for immunofluorescence staining of organoids in 3D embedded culture and of tissue sections of whole mammary glands.
Fig. 1.
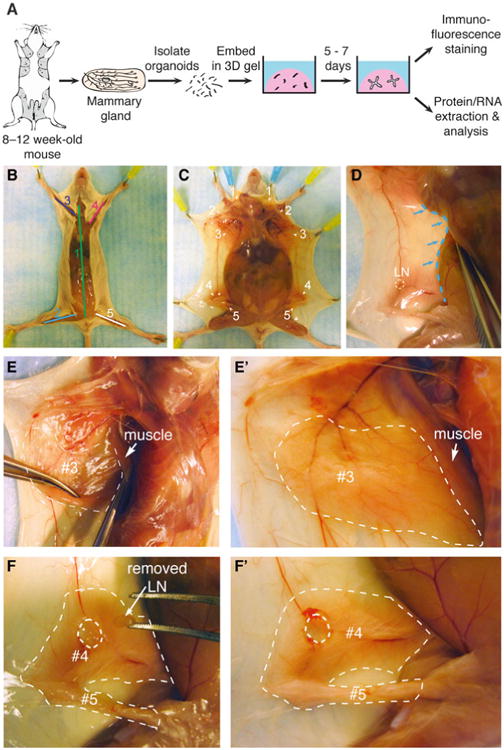
Collection of mouse mammary glands for organoid isolation and 3D culture. (a) Schematic description of isolation and 3D culture of mouse mammary organoids. (b) Scheme for surgically accessing the mammary glands. Numbers indicate the order of cuts. (c) Locations of the ten mammary glands. (d) Expose glands #3, #4, and #5 by pushing back the abdomen (blue dotted line) with the back of the Graefe forceps. (e–e′) A thin layer of muscle partially covers gland #3 (e) and should be pushed back before dissection (e′). Dotted line in (e′) indicates the region of gland #3 to be collected. (f) Use the Graefe forceps to pluck out the lymph node in gland #4. Dotted line in (f′) indicates the approximate region of glands #4 and #5 to be collected (Color figure online)
2 Materials
2.1 Mice
We have successfully isolated and cultured organoids from mice ranging in age from E18.5 through 1.5 years. There are variations in response to growth factors with age and strain. All protocols below are optimized for FVB mice between 8 and 12 weeks of age. Once euthanized, mammary tissue is optimally isolated and cultured immediately, but successful organoid cultures have been established from mice sacrificed and then kept at 4 °C overnight.
2.2 Reagents
-
Collagenase solution in DMEM/F12: 2 mg/mL collagenase,2 mg/mL trypsin, 5 % v/v fetal bovine serum (FBS), 5 μg/mL insulin, and 50 μg/mL gentamicin.
Collagenase from Clostridium histolyticum (Sigma C2139): Dissolve 1 g in 10 mL DMEM/F12, and make 200 μL aliquots. Store at −20 °C.
Trypsin: Dissolve 1 g in 10 mL DMEM/F12, and make 200 μL aliquots. Store at −20 °C.
Dulbecco's phosphate-buffered saline (DPBS, with Ca2+, Mg2+).
Phosphate-buffered saline (PBS, without Ca2+, Mg2+).
BSA solution: 2.5 % bovine serum albumin (BSA) in DPBS.
2,000 U DNase (Sigma D4263): Dissolve in 1 mL of PBS, and make 40 μL aliquots. Store at −20 °C.
Organoid medium in DMEM/F12: 1 % penicillin/streptomycin and 1 % insulin-transferrin-selenium-X (ITS) (GIBCO 51500).
FGF2, 25 μg (Sigma F0291): Dissolve in 250 μL of PBS, and make 20 μL aliquots. Store at −20 °C.
Growth Factor Reduced Matrigel (Corning 354230).
Rat tail collagen I (Corning 354236).
DMEM 10×, low glucose.
1.0 N NaOH.
4 % paraformaldehyde (PFA) in DPBS.
OCT compound.
0.5 % Triton X-100 in DPBS.
10 % FBS in DPBS.
Mounting Medium (Sigma F4680-25ML).
Primary antibodies.
Secondary antibodies conjugated to fluorescent probes.
2.3 Instructions for Preparing Solutions
Prepare solutions as follows:
Collagenase solution (10 mL per mouse): Combine 9 mL DMEM/F12, 500 μL FBS, 5 μL insulin (10 mg/mL stock), 10 μL gentamicin (50 mg/mL stock), 200 μL collagenase (100 mg/mL stock), and 200 μL trypsin (100 mg/mL stock) in a 15 mL tube. Filter sterilize through a 0.2 μm filter into a new tube (Do not filter collagenase and trypsin stocks, the filter will get clogged). This solution should be made fresh for each experiment.
BSA solution: Combine 46 mL DPBS and 4.1 mL BSA (30 % stock solution). Filter sterilize, and store at 4 °C. This solution can be reused for several experiments if kept sterile but should be monitored for contamination.
Organoid medium: Remove 10 mL of DMEM/F12 from a 500 mL bottle of medium. Add 5 mL penicillin/streptomycin (10,000 units penicillin and 10 mg streptomycin/mL stock) and 5 mL ITS. For the branching morphogenesis assays (Subheadings 3.10.2 and 3.10.3) and the invasion assay (Subheading 3.10.4), supplement organoid medium with growth factor at the desired concentration. Diverse growth factors induce branching in the 1–10 nM range, including EGF ligands (EGF, TGF-α, amphiregulin, heregulin, neuregulin), FGF ligands (FGF2, FGF7), and HGF. We typically use 2.5 nM FGF2 for 8–12-week-old FVB mice. It is necessary to optimize the growth factor concentration for the specific age and strain of mouse.
2.4 Tools and Instruments
One Spencer Ligature scissors, delicate pattern (Fine Science Tools (FST) 14028-10): For mouse exterior.
One standard forceps, narrow pattern (FST 11003–12): For mouse exterior.
One Iris scissors, straight pattern (FST 14060–09): For mouse interior (sterile).
One Graefe forceps (FST 11051–10): For mouse interior (sterile).
Sterile scalpel, #10 blade.
Polystyrene Petri dish.
Benchtop incubator orbital shaker (Thermo Scientific MaxQ 4450).
Incublock™ microtube incubator with two blocks set to 37 °C (Denville Scientific Inc. I0540).
Ice bucket.
Centrifuge tubes, 15 and 50 mL.
Chambered coverglass, 2-well and 4-well (Nunc, Lab-Tek, Thermo Scientific).
SensoPlate, black, 24 W multiwell plate, glass bottom, sterile, w/lid (Greiner Bio-One 662892).
24-Well glass plate cover (MatTek Corp P24GTOP-1.5-F).
Disposable base molds: 15 × 15 × 5 mm, 24 × 24 × 5 mm, and 30 × 24 × 5 mm (Fisher Scientific).
Superfrost® Plus Gold precleaned microscope slides (Fisher Scientific 15-188-48).
Cover glass, 50 × 22 mm.
Orbital Shaker (Reliable Scientific, Inc.).
StainTray with black lid (Simport M920-2).
Cryostat.
3 Methods
3.1 Collecting Mouse Mammary Glands
Mice have five pairs of mammary glands located beneath the skin and outside the peritoneum. This section describes how to collect glands #3, #4, and #5 for organoid isolation and how to limit contamination by other tissues.
Generally, use female mice between 8 and 12 weeks old.
Sterilize the dissecting area with 70 % ethanol.
Sterilize the dissecting tools by heat in a glass bead sterilizer.
Euthanize the mouse in a CO2-saturated chamber for 3–5 min followed by cervical dislocation.
Pin the mouse face up to a protected Styrofoam board.
Wet the mouse thoroughly with 70 % EtOH. Use the back of the standard forceps to smooth down the fur. Wipe away any feces with a 70 % EtOH-damp Kimwipe.
Use the standard forceps to grasp the skin above the groin.
Use the Spencer Ligature scissors to cut along the ventral midline from the groin to the chin (Fig. 1b). Be careful to cut only the skin and not the peritoneum underneath.
Make four incisions from the midline cut towards the four legs (Fig. 1b).
Use the standard forceps to pull back the skin one side at a time to expose the mammary glands (Fig. 1c). Use the dorsal side of the Graefe forceps to help separate the skin from the peritoneum (Fig. 1d).
Push back a thin yellow layer of muscle located on top of gland #3 to expose the mammary gland (Fig. 1e–e′).
Use the Graefe forceps to remove the inguinal lymph node located at the intersection of three blood vessels in gland #4 (Fig. 1f–f′).
Use the Graefe forceps and Iris scissors to grasp and pull out mammary glands #3, #4, and #5 from both right and left sides. Pool glands in a sterile Petri dish (Fig. 2a) (see Note 1).
Fig. 2.
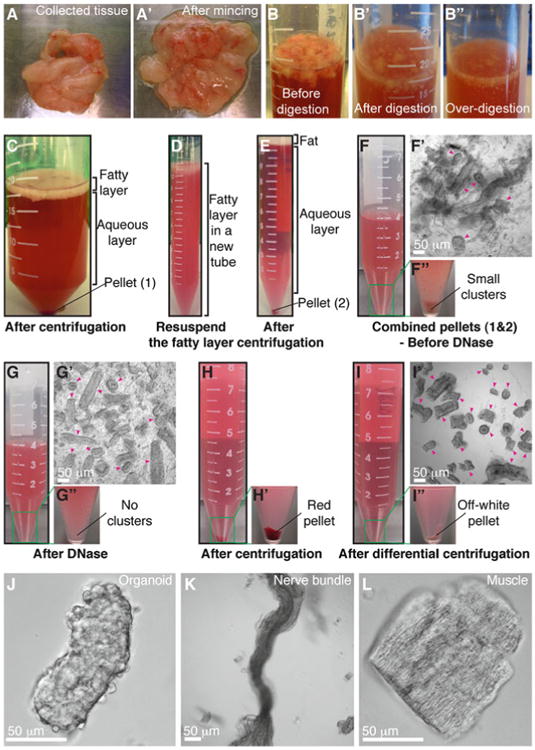
Mammary organoid isolation. (a–a′) Collected mammary glands are pooled in a Petri dish (a) and minced until the tissue relaxes, typically 25–50 cuts (a′). (b–b″) Incubation in collagenase solution breaks up the fat pad (b) into smaller pieces that are relatively dispersed (b′). Too long of a digestion (b″) will cause organoids to be too small and not grow well. (c) Following incubation in collagenase solution, centrifugation separates the suspension into three layers, with a top opaque layer of fat and a pellet (#1) of epithelium and stroma. (d) The fatty layer is transferred to a new tube and resuspended in 10 mL DMEM/F12. (e) Centrifugation of the dispersed fatty layer recovers additional epithelium in the pellet (#2). (f–f″) The combined pellets from (c) and (e) are resuspended in 4 mL DMEM/F12 with DNase (f). Before DNase treatment, organoids (pink arrowheads) are loosely attached to each other and to stromal cells (f′), forming visible clusters in the tube (f″). (g–g″) DNase treatment causes organoids (pink arrowheads) to detach from one another (g′) and the clusters to disappear (g″). (h–h′) Centrifugation of the suspension in (g) results in a compact red pellet (h′). (i–i″) Differential centrifugation removes single cells from the suspension (i′) and results in an off-white pellet of purified epithelial organoids (i″). Organoids (pink arrowheads) may appear rounded and small or more elongated and even branched (i′). Larger organoids typically survive and branch more efficiently in our assays. (j) Close-up view of an organoid. (k–l) Non-epithelial tissues can be observed in the final suspension, including nerve bundles (k) and muscle (l) (Color figure online)
3.2 Isolating Mammary Epithelial Organoids
Mammary epithelium is embedded inside a fat pad containing adipose tissue and collagen-rich stroma. This section describes how to purify fragments of mammary epithelium (“organoids”) using enzymatic and mechanical digestion. All centrifugation speeds refer to a Sorvall Legend X1R benchtop swinging bucket centrifuge (1,500 rpm, 1,250 rcf). We have achieved similar results with 1,500 rpm spins in similar benchtop centrifuges from other manufacturers.
In a sterile hood, mince mammary glands with a scalpel, ∼25–50 times per mouse, until the tissue relaxes (Fig. 2a, a′). Use a separate scalpel for each mouse type.
Use the scalpel to transfer the minced glands to collagenase solution in a 15 or a 50 mL tube (Fig. 2b). We use 10 mL of collagenase solution per mouse.
Shake the suspension at 110–150 rpm for 30–40 min (see Note 2) at 37 °C until the tissue breaks up into smaller pieces and is relatively dispersed (Fig. 2b′) but not overdigested (Fig. 2b″). We typically use a Thermo Scientific MaxQ 4450 for this purpose.
Spin the tube in a centrifuge at 1,500 rpm for 10 min at room temperature. The tube will have three layers: a fatty layer on top, an aqueous layer in the middle, and a red pellet of epithelium on the bottom (Fig. 2c).
Precoat (Fig. 3a) (see Note 3) a 15 mL tube with BSA solution. Use one tube per mouse type. For all subsequent steps, precoat all pipette tips and tubes with BSA solution prior to contact with mammary tissue (Fig. 3).
To recover additional epithelial tissue, use a BSA-coated pipette (Fig. 3b) to transfer the opaque fatty layer into the BSA-coated 15 mL tube. Add DMEM/F12 up to 10 mL. Pipette up and down vigorously to disperse the fatty layer (Fig. 2d). Spin the tube at 1,500 rpm for 10 min at room temperature. Aspirate the fat and supernatant, and save the pellet (Fig. 2e).
Aspirate (see Note 4) the aqueous layer in the tube with the original pellet.
Add 10 mL DMEM/F12 to the tube with the original pellet, and transfer to the 15 mL “fatty layer” tube (step 6). Pipette up and down vigorously to resuspend and combine the two pellets.
Spin the tube at 1,500 rpm for 10 min at room temperature.
Aspirate the supernatant, and add up to 4 mL DMEM/F12 to the combined pellet (Fig. 2f). At this stage during isolation, the suspension contains small clusters of organoids and stromal cells attached to one another (Fig. 2f–f″).
Add 40 μL DNase (2 U/μL) into the 4 mL organoid suspension, and gently invert by hand for 2–5 min at room temperature to break up the clusters and detach organoids from single cells (Fig. 2g′–g″).
Add 6 mL of DMEM/F12, and pipette up and down thoroughly.
-
Spin the tube at 1,500 rpm for 10 min at room temperature. The pellet should now appear red and more compact (Fig. 2h–h′).
Next perform differential centrifugation to wash out enzymes and separate single stromal cells from the epithelial organoids. The protocol suggests aspiration of the supernatant assuming that the stromal cells will be discarded. If recovery of mammary stromal populations is desired, then transfer the supernatant after each spin to a 50 mL tube.
Aspirate the supernatant to the 0.5 mL mark.
Resuspend the pellet in 10 mL DMEM/F12, and mix thoroughly.
Pulse to 1,500 rpm, and stop the centrifuge 3–4 s after it reaches speed.
Repeat steps 14–16 three more times (see Note 5).
The final pellet should be off-white and consist mostly of organoids, without single cells (Fig. 2i–i″, j). However, the organoid suspension may be contaminated with other tissue types, most commonly fiber bundles (Fig. 2k) and muscle (Fig. 2l).
Fig. 3.
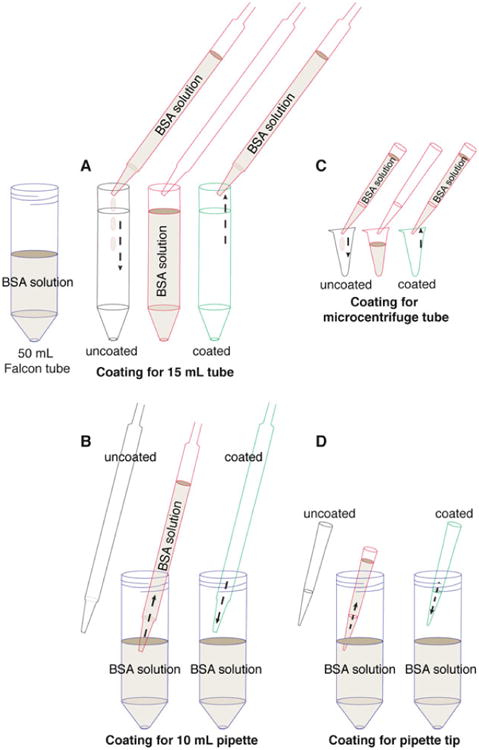
Precoating tubes and pipette tips with BSA. Fresh tissue can adhere to uncoated plastic surfaces, and this protocol involves many pipetting steps. Accordingly, it is essential to precoat the plastic surfaces with BSA solution to maximize final organoid yield. (a) Precoat a 15 mL tube by filling the tube with BSA solution, inverting the tube to precoat the cap, and removing the BSA solution. (b) Precoat a 10 mL pipette tip by taking up BSA solution to fill the entire pipette and ejecting back out. (c, d) Use the same approach to precoat a microcentrifuge tube (c) and a small pipette tip (d) with BSA solution
3.3 Organoid Density Determination
This section describes how to determine the density of the organoid suspension and the overall yield.
Resuspend the pellet in 10 mL DMEM/F12 to form a homogeneous mixture (see Note 6).
Mix thoroughly (e.g., by rocking the tube by hand), and transfer 50 μL of the suspension to a 30 mm Petri dish. Count the number of organoids in this sample volume under a microscope (Fig. 2i′, j).
-
Calculate the total number of organoids collected according to the following formula. For example, if 20 organoids were counted in a 50 μL sample removed from a 10 mL total volume (9,950 μL remaining), then the total number of organoids would be (20/50) × 9,950 = 3,980 organoids:
Calculate the organoid density (see Note 7), and readjust to 1,000 organoids/mL to simplify allocation to ECM gels.
Calculate the number of organoids and the respective volume of suspension required for each experiment.
Aliquot the required volumes of organoid suspension into BSA-coated 1.5 mL microcentrifuge tubes (Fig. 3c), and spin the tubes at 1,500 rpm for 5 min at room temperature.
Carefully remove the supernatant so as not to disturb the pellet (see Note 8).
Calculate the volume of ECM solution required to reach a final density of two organoids/μL (see Note 9).
3.4 Plating Mammary Organoids in Matrigel
Mammary epithelium develops in vivo within a basement membrane. 3D culture in Matrigel, a basement membrane-rich gel, recapitulates important features of epithelial development [17, 19, 22, 23]. This section describes how to embed organoids in 3D Matrigel.
Thaw Matrigel at 4 °C for 3–4 h prior to plating. If the Matrigel is put at 4 °C to thaw at the start of the prep, it will be ready to use by the end of the prep. During plating, always keep Matrigel on ice.
Use a plate with a cover glass bottom for time-lapse imaging.
Preincubate the plate at 37 °C for 5 min.
Set up the tissue culture hood in preparation for plating (Fig. 4a).
Set the heating block to 37 °C, and place the plate in direct contact with the block (Fig. 4b-c′) (see Note 10).
Add the required volume of liquid Matrigel to a microcentrifuge tube with organoids. Since Matrigel is quite viscous, first pipette up and down slowly a few times to coat the tip and ensure an accurate volume.
Keep the Matrigel-containing tube on ice or in a cold block. Resuspend the organoid pellet gently to avoid introducing air bubbles. Do not try to take up the entire volume into the pipette tip while mixing.
-
Plate the appropriate volume of Matrigel/organoid suspension into the wells according to the following table (Fig. 5a). Pipette up and down to resuspend the organoids before plating each sample, and pipette out only until the first stop.
Type of plate Volume of gel/well (μL) Volume of medium/well (μL)
24-Well plate 50–150 750–1,000
4-Well chamber 50–75 750–1,000
2-Well chamber 150–300 1,500–2,000
Keep the plate on the heating block for several minutes to allow further gelation before returning it to the incubator.
Incubate the plate at 37 °C, 5 % CO2, for 30–60 min.
Gently add pre-warmed organoid medium to the wells. For the cyst formation assay (Subheading 3.10.1), use basal medium without supplemental growth factors. For the branching morphogenesis assay (Subheading 3.10.2 and 3.10.3), supplement the medium with nanomolar concentrations of growth factor. A variety of growth factors may be used, including EGF ligands (EGF, TGF-α, amphiregulin, heregulin, neuregulin), FGF ligands (FGF2, FGF7), and HGF. We most commonly use 2.5 nM FGF2.
Add sterile water or PBS to the empty wells to prevent desiccation.
Label the wells. Return the plate to the incubator.
If the plate will be used for DIC imaging, use a glass plate cover for better image quality.
Fig. 4.

Setting up the tissue culture hood for plating. (a) Sample layout of reagents, tools, and equipment used for plating 3D culture samples. (b) Heating block setup for plating in 2-well or 4-well chambers. (c, c′) To plate in a 24-well dish, remove one of the blocks from the heating block (c) to establish direct contact between the remaining block and the plate bottom (c′)
Fig. 5.
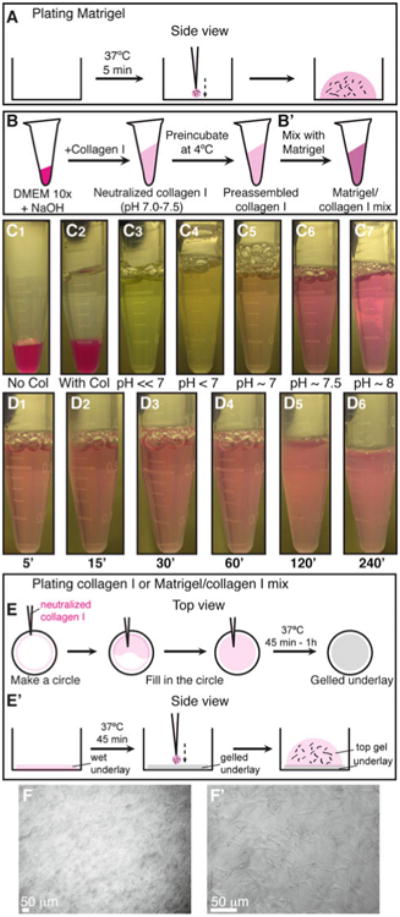
Plating organoids in 3D Matrigel and collagen I. (a) Schematic description of plating organoids in Matrigel. (b–b′) Schematic description of preparing preassembled collagen I (b), which can be used alone or mixed with Matrigel (b′). (c1–c7) Color indicators for the pH of the collagen I solution during neutralization. (d1–d6) Decreasing transparency of the collagen I solution during preincubation on ice. (e–e′) Schematic description of plating organoids in 3D collagen I or in a mixture of Matrigel and collagen I. (e) Shows a top view for making an underlay on the cover glass. (e′) Shows a side view of how to plate the organoid/collagen I suspension on top of the gelled underlay. (f–f′) Representative DIC images of collagen I fibers at low (f) and high (f′) magnification (Color figure online)
3.5 Preparing Collagen I Solution
Collagen I solubilized from rat tail is commonly used to study 3D migration of many cell types [32]. However, the properties of collagen I gels vary depending on multiple factors during preparation, such as temperature, pH, and collagen concentration. We demonstrated that the extent of collagen fiber assembly correlated strongly with invasive behavior [24]. This section describes how we prepare collagen I (Fig. 5b).
-
Rat tail collagen I is used to prepare a collagen solution according to the following formula. The steps below describe how to make a 250 μL solution. Scale up the volume as needed:
Total volume (μL) 250 500 1,000 2,000 5,000
1.0 N NaOH (μL) 8 16 32 64 160
DMEM 10 × (μL) 25 50 100 200 500
Collagen I stock (μL) 217 434 868 1,763 4,340
Perform all steps on ice. To work with a large volume of collagen solution, use a 1,000 μL extra long pipette tip to avoid the collagen solution getting stuck to the filter barrier during pipetting.
First, combine 25 μL DMEM 10 × and 8 μL NaOH, and mix well. The solution will turn a dark pink color (Fig. 5c1).
Add 217 μL collagen I (Fig. 5c2) (see Note 11). Since collagen I is quite viscous, pipette up and down slowly a few times to coat the pipette tip.
Mix the solution well until the color remains stable. When the pH changes from acidic → neutral → basic, the color changes from light green/yellow → light pink/orange → dark pink, respectively (Fig. 5c3–7). The desired color is light pink or salmon, which corresponds to a pH of 7.0–7.5 (Fig. 5c6) (see Note 12). The pH can be tested using pH strips.
Use DMEM 1 × to adjust the neutralized collagen I solution to the desired collagen concentration (see Note 13). For the invasion assay (Subheading 3.10.4), we use a collagen concentration of 3 mg/mL.
3.6 Plating Mammary Organoids in Collagen I
Fibrillar collagen I, the most abundant structural protein in mammary glands, plays an important role in normal development as well as in breast cancer. Our previous studies have demonstrated that collagen I induces a conserved response of protrusive invasion in both normal and tumor organoids [23]. This section describes how to properly prepare preassembled collagen I and embed mammary organoids in a 3D gel.
Use a plate with a glass bottom for time-lapse imaging.
Use 20-30 μL of neutralized collagen to make a thin underlay on the cover glass of the well at room temperature (Fig. 5e). The underlay helps the top collagen/organoid suspension attach better to the cover glass.
Incubate the plate with the underlays at 37 °C until ready for plating.
Preincubate the neutralized collagen I solution (used for the top gel) at 4 °C for 60–120 min for preassembly [24] (see Note 14). The collagen I solution will turn cloudy and fibrous (Fig. 5d1–6) (see Note 15), a state we term preassembled collagen I.
Set up the tissue culture hood in preparation for plating (Fig. 4a).
Set the heating block to 37 °C, and place the plate on top, in direct contact with the block (Fig. 4b–c′) (see Note 10).
Always keep the collagen I solution on ice. Add the desired amount of preassembled collagen I to the organoid pellet in a microcentrifuge tube. Since collagen I is quite viscous, first pipette up and down slowly a few times to coat the tip and ensure an accurate volume.
Keep the tube on ice or in a cold block. Resuspend the organoid pellet gently to avoid introducing air bubbles. Do not try to take up the entire volume into the pipette tip while mixing.
Plate the appropriate volume of collagen/organoid suspension (see table in Subheading 3.4) on top of the underlay (Fig. 5e′). Pipette up and down to resuspend the organoids before plating each sample, and pipette out only until the first stop.
Keep the plate on the heating block for several minutes to allow further gelation before returning it to the incubator (see Note 16).
Incubate the plate at 37 °C, 5 % CO2, for 45–60 min. After gelation, collagen I fibrils are visible under the microscope at 10× and 40× (Fig. 5f-f′).
Gently add pre-warmed organoid medium supplemented with growth factor to the wells. A variety of growth factors may be used, including EGF ligands (EGF, TGF-α, amphiregulin, heregulin, neuregulin), FGF ligands (FGF2, FGF7), and HGF. We most commonly use 2.5 nM FGF2.
Add sterile water or PBS to the empty wells to prevent desiccation.
Label the wells. Return the plate to the incubator.
If the plate will be used for DIC imaging, use a glass plate cover for better image quality.
3.7 Plating Mammary Organoids in a Mixture of Matrigel and Collagen I
A mixture of Matrigel and collagen I represents a more physiological ECM microenvironment for mammary branching morphogenesis. The presence of collagen I significantly improves epithelial ductal elongation and myoepithelial coverage [24]. We do not observe epithelial protrusions into mixed Matrigel/collagen I gels [24]. This section describes how to properly prepare a mixture of Matrigel and preassembled collagen I and how to embed mammary organoids in this mixed matrix.
Prepare collagen I solution as described in Subheading 3.5.
Repeat steps 1–4 in Subheading 3.6 to make underlays and prepare preassembled collagen I.
Combine Matrigel and preassembled collagen I at the desired ratio. Gently pipette up and down a few times to form a homogeneous solution (Fig. 5b′).
Always keep the mixed matrix solution on ice. Add the desired amount to the organoid pellet in a microcentrifuge tube.
Plate the mixed matrix/organoid suspension as described in steps 5–15 in Subheading 3.6 (Fig. 5e′).
3.8 Immuno-fluorescence Staining of 3D Culture Samples
The thickness of 3D gels and the multicellular structure of mammary organoids often result in reduced antibody accessibility for immunofluorescence (IF) staining and poor visualization during imaging. This section describes two methods for performing IF staining in 3D culture samples. First, fix gels as follows:
Remove organoid medium from the wells.
Fix samples with 4 % PFA for 10–15 min at room temperature (see Note 17) on an orbital shaker at 20 rpm.
Remove PFA, and wash samples 2–3× 10 min with DPBS.
From here, you can perform antibody staining directly in intact 3D gels or on cut sections on slides. The whole-gel staining works well with high-quality antibodies and probes such as phalloidin (stains for F-actin), smooth muscle actin (SMA), and keratin 14. For many other antibodies, staining sections on slides is preferable.
3.8.1 Staining Whole Gels
Permeabilize the gel with 0.5 % Triton X-100 for 30–60 min.
After permeabilization, immediately block samples with 10 % FBS (see Note 18) for 1–3 h at room temperature or overnight at 4 °C.
Remove the blocking solution, add primary antibody in 10 % FBS (see Note 18) at the desired ratio, and incubate for 2–3 h at room temperature or overnight at 4 °C.
Remove the primary antibody solution, and wash samples 3× 10 min with 10 % FBS in DPBS at room temperature.
Optional: Block the samples again with 10 % FBS for 30–60 min at room temperature.
Add secondary antibody in 10 % FBS (see Note 18) at the desired ratio, and incubate for 1–2 h at room temperature.
Wash 3× 10 min with DPBS at room temperature.
Store samples in DPBS at 4 °C, but remove DPBS before imaging. If the gel has detached from the cover slip, leaving the DPBS in the well causes the gel to wiggle or float and go out of focus.
3.8.2 Staining Sections on Slides
Gently detach the gel from the culture plate and transfer to a small disposable base mold (15 × 15 × 5 mm) filled with a thin layer of OCT.
Freeze the mold at −80 °C for 5–10 min.
Fill up the mold with OCT to cover the sample, and return to −80 °C for long-term storage.
During sectioning, store molds on dry ice. Set up the cryostat with OT at −20 °C and CT at −20 °C.
Remove an OCT block from its mold, and cut sections at 20–100 μm thickness.
Transfer sections to slides using a fine camel hair brush or a pair of forceps.
Keep slides at −80 °C for long-term storage.
For antibody staining, thaw slides at room temperature (see Note 19).
Wash slides 2–3 × 10 min with DPBS to remove OCT.
Permeabilize with 0.5 % Triton X-100 for 30–60 min (see Note 20).
Wash 2× 10 min with DPBS to remove Triton.
Block slides with 10 % FBS for 2 h at room temperature or overnight at 4 °C.
Remove the blocking solution, add primary antibody in 10 % FBS, and incubate for 2–3 h at room temperature or overnight at 4 °C (see Note 21).
Wash 3× 10 min with 10 % FBS or DPBS.
Add secondary antibody in 10 % FBS, and incubate for 2 h at room temperature or overnight at 4 °C.
Wash 3× 10 min with DPBS.
Mount slides with mounting medium and 50 × 22 mm cover-slips. Let the slides dry at room temperature in a dry StainTray or in a dark drawer before imaging.
3.9 Immuno-fluorescence Staining of Mammary Gland Tissue Sections
The opacity and thickness of the mammary fat pad limit the accessibility of mammary epithelium to whole-gland staining and imaging. This section describes how to perform IF staining in mammary gland tissue sections.
Collect mouse mammary glands #3 and/or #4, as described in Subheading 3.1, taking care to keep the entire gland intact (see Note 22). Spread out the gland on the bottom of a 1- or a 2-well chambered cover glass.
Fix the tissue with 4 % PFA for 4 h at room temperature or overnight at 4 °C.
Wash 3× 15 min with DPBS to remove PFA.
Transfer the gland to a medium (24 × 24 × 5 mm) or a large (30 × 24 × 5 mm) disposable base mold filled with a thin layer of OCT.
Freeze the mold at −80 °C for 5–10 min.
Fill up the mold with OCT to cover the gland, and return to −80 °C for long-term storage.
During sectioning, store molds on dry ice. Set up the cryostat with OT at −40 °C and CT at −30 °C.
Remove an OCT block from its mold, and cut sections at 50–200 μm thickness.
Transfer sections to slides using a fine camel hair brush or a pair of forceps.
Keep slides at −80 °C for long-term storage.
Repeat steps 8–17 in Subheading 3.8.2 for IF staining (see Note 23).
3.10 Assays
In vivo, mammary epithelium develops within a basement membrane surrounded by collagen-rich stromal tissue. The ability to manipulate the ECM microenvironment in 3D organotypic culture allows us to isolate the effects of individual matrix components on mammary epithelial cell behaviors. This section describes four assays that use different ECM compositions or growth conditions to model distinct epithelial programs (Fig. 6a).
Fig. 6.
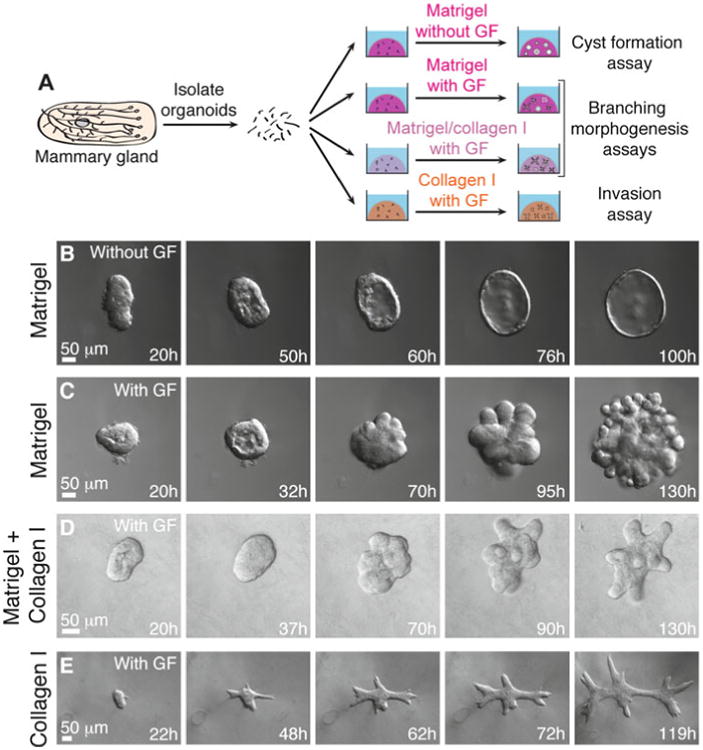
3D organotypic culture assays. (a) Schematic description of four assays that use different extracellular matrix compositions to model specific epithelial behaviors. (b–e) Representative frames of DIC time-lapse movies showing cyst formation in Matrigel in basal medium (b), branching morphogenesis in Matrigel induced by FGF2 (c), branching morphogenesis in a mixture of Matrigel and collagen I induced by FGF2 (d), and epithelial cell invasion into pure collagen I induced by FGF2 (e)
3.10.1 Cyst Formation Assay
In 3D Matrigel in basal medium, mammary organoids reorganize from a multilayered fragment to establish a simple bi-layered epithelium with an internal lumen, termed a cyst (Figs. 6b and 7a). The extent of lumen formation varies with the initial size of the organoid and with the mouse strain (Fig. 7a1–a4). The resulting morphologies include a minimal or a barely detectable lumen (Fig. 7a1), a partial lumen (Fig. 7a2), a complete lumen in a small cyst (Fig. 7a3), and a complete lumen in a large cyst (Fig. 7a4). Epithelial cells in the cyst always maintain a smooth basal surface with the ECM (Fig. 7a4′). We have observed that C57BL6 organoids form cysts with complete lumens (Fig. 6b) more efficiently than FVB organoids. Although the appearance of the lumen varies by light microscopy, immunofluorescence staining for SMA and F-actin can confirm the establishment of a simple bi-layered structure of internal luminal epithelial cells and basal myoepithelial cells (Fig. 8a). We use this assay to model the formation of mammary epithelial ducts in vivo (Fig. 8d).
Fig. 7.
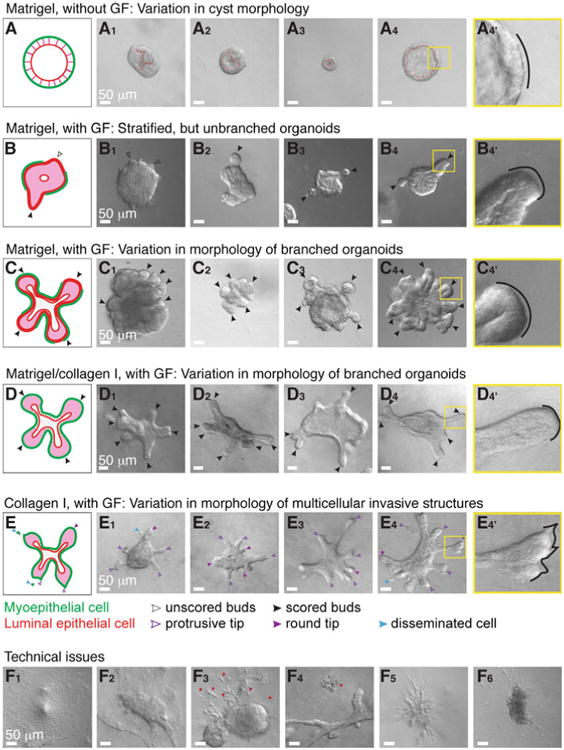
Phenotypic variability in assay outcomes. (a) Schematic description of a cyst. (a1–a4) DIC images showing variation in cyst morphology. (a4′) An inset of (a4) showing a smooth basal surface with Matrigel. (b) Schematic description of a stratified, unbranched organoid. (b1–b4) DIC images showing examples of stratified, unbranched organoids in Matrigel. (b4′) An inset of (b4) showing a smooth basal surface with Matrigel. (c) Schematic description of a branched organoid in Matrigel. (c1–c4) DIC images showing variation in branching morphology. (c4′) An inset of (c4) showing a smooth basal surface with Matrigel. (d) Schematic description of a branched organoid in a mixture of Matrigel and collagen I. (d1–d4) DIC images showing variation in branching morphology. (d4′) An inset of (d4) showing a smooth basal surface with the mixed matrix. (e) Schematic description of an organoid with protrusive tips in collagen I. (e1–e4) DIC images showing variation in protrusive invasion. (e4′) An inset of (e4) showing protrusive tips into collagen I. (f) DIC images showing commonly observed technical issues. (f1–f2) Organoids lose their 3D organization in Matrigel (f1) and collagen I gels (f2) when they make contact with the cover glass. (f3) Non-epithelial species (red arrowheads) attached to organoids may appear elongated and mesenchymal (ECM: Matrigel). (f4) A group of dead cells beside a branching organoid (ECM: collagen I). (f5) A cluster of elongated, non-epithelial cells (ECM: Matrigel). (f6) A nerve bundle disseminating single cells into the surrounding matrix (ECM: Matrigel) (Color figure online)
Fig. 8.
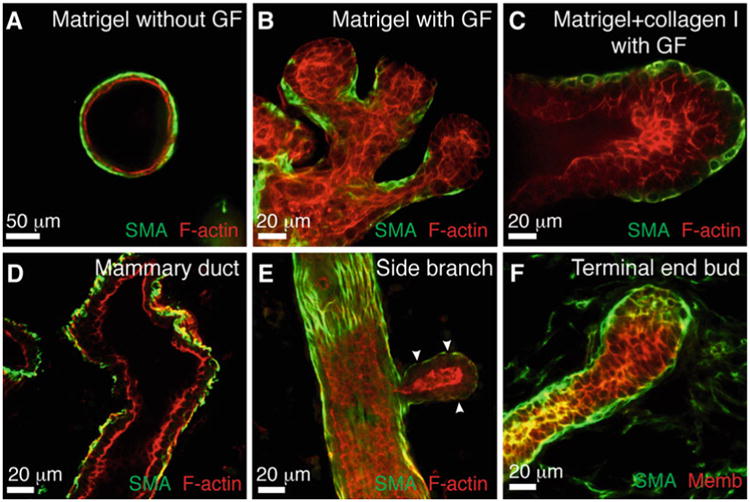
Correlation between epithelial morphologies in 3D organotypic assays and in vivo. (a–c) Representative confocal images of a cyst in Matrigel (a), branched buds in Matrigel (b), and a stratified, elongating bud in a mixture of Matrigel and collagen I (c). (d–f) Representative confocal images from mammary gland tissue sections of a bilayered duct (d), a side branch (e), and a terminal end bud (f)
3.10.2 Branching Morphogenesis Assay in Matrigel
In 3D Matrigel, nanomolar concentrations of growth factor induce mammary organoids to undergo branching morphogenesis (Fig. 6c). The branching program includes sequential steps of lumen clearing, stratification, bud initiation, and bud elongation (Fig. 6c) [17]. We observe variation in morphology depending on the extent of progression through this program. Organoids that only complete stratification or bud initiation (Fig. 7b1), or that form fewer than three buds (Fig. 7b2–b4), are not scored as “branched” (Fig. 7b). Only organoids with three or more elongated buds are scored as “branched” (Fig. 7c). However, the morphology of branched organoids varies based on the initial size of the organoid, the mouse strain, and the types of growth factors added. Here, we present four examples of branching in Matrigel (Fig. 7c1–c4). The first two show organoids with multiple multilayered, elongating buds without any regions of repolarization (Fig. 7c1–c2). In contrast, the second two show organoids that have reestablished simple epithelial architecture in the central lumen (Fig. 7c3) or within buds (Fig. 7c4). During bud elongation in Matrigel, the leading front of the bud is always non-protrusive (Fig. 7b4′, c4′). Notably, these buds lack or are incompletely covered by myoepithelial cells (SMA+, Fig. 8b). These gaps in myoepithelial coverage can be observed in vivo, particularly in side branches of the mammary ductal tree (Fig. 8e).
3.10.3 Branching Morphogenesis Assay in a Mix of Matrigel and Collagen I
In a mixture of Matrigel and collagen I, mammary organoids undergo a similar program of branching morphogenesis to that in Matrigel alone, with several notable differences [24] (Fig. 6d). Branched organoids in the mixed matrix generally have fewer buds, but the buds elongate much further into the ECM. We have demonstrated that ratios of 5:5 and 3:7 Matrigel to collagen I induce the highest average bud lengths [24]. Here, we present four examples of branching in the mixed matrices. Organoids may contain both short and long buds, without repolarization (Fig. 7d1) or with partial repolarization (Fig. 7d2, d4). We also observe bifurcation at the ends of elongated buds (Fig. 7d3). As in Matrigel alone, the leading front of the bud is always non-protrusive (Fig. 7d4′). Importantly, a mix of 3:7 Matrigel to collagen I yields a high percentage of epithelial buds that maintain complete myoepithelial coverage throughout bud initiation and elongation (Fig. 8c). This phenomenon more closely models elongation of the terminal end bud in vivo (Fig. 8f).
3.10.4 Invasion Assay
Collagen I induces a conserved protrusive response in mammary epithelium (48 h, Fig. 6e). Organoids invade collectively into collagen I, with branches varying in shape and length, from short and thin (Fig. 7e1–e2) to elongated and wide (Fig. 7e3–e4). Initially, we observe extensive subcellular protrusions. However, these protrusions cease, and the epithelium reestablishes a smooth basal surface upon formation of a basement membrane between the epithelium and ECM (Fig. 6e) [23]. The extent of invasion and epithelial reorganization varies, even within the same organoid. At day 5 in culture, we observe both protrusive (violet arrowheads, Fig. 7e4′) and non-protrusive, round tips (violet filled arrowheads, Fig. 7e1–e4) at the ends of multicellular invasive structures. We also observe single cell dissemination into collagen I (blue arrowheads, Fig. 7e1,e4).
3.11 Technical Issues
Here we present several technical problems that we commonly encounter during 3D culture. First, epithelial organoids located very close to the cover glass tend to lose their 3D structure and spread out in 2D as sheets of cells (Fig. 7f1–f2). Second, organoids may be surrounded by protrusive or stringy cells, which likely results from stromal or other non-epithelial cells attaching to organoids during their isolation (Fig. 7f3). Non-epithelial contaminating species appear distinct and behave differently from organoids.
We have observed groups of dead cells (Fig. 7f4), clusters of protrusive stromal cells (Fig. 7f5), and nerve bundles (Fig. 7f6), which tend to locally disseminate cells into the matrix.
4 Notes
Mammary gland #1 is very small. Mammary gland #2 is located in the neck and is hard to distinguish from other tissues. Generally, do not collect these glands so as to avoid contamination by other tissues (e.g., muscle or other epithelial glands) (Fig. 2l).
Incubation in collagenase solution can require up to 60 min to adequately break up the fat pad. Check the status of the suspension after 30 min of shaking. We have observed that inappropriate incubation times increase the amount of contaminating tissues in the final organoid suspension (Fig. 2k). If shaking is done in an incubator that is also used for bacterial cultures, wipe the outside of the tube with 70 % ethanol before bringing it into the tissue culture hood.
Always precoat new pipette tips and tubes with BSA solution to prevent organoids from sticking to the plastic. This precoating (Fig. 3) is critical to achieving a high final yield of organoids, especially with mice at younger ages or mice on a C57BL6 background.
Never aspirate the supernatant completely to avoid sucking up the pellet.
Carefully examine the pellet after each quick spin before aspirating the supernatant. If the organoids are not well pelleted, mix the suspension thoroughly again, and increase the centrifugation time.
The appropriate volume of DMEM/F12 to use for counting varies depending on the estimated yield. If the yield is low, add less medium. If the yield is high, dilute the suspension further 2–10×.
The yield varies significantly with mouse strain and age. We generally obtain 2,000–4,000 organoids per FVB mouse and 500–2,000 organoids per C57BL6 mouse.
Always check the pellet after every centrifugation. Be careful not to disturb the pellet when removing the supernatant. Use small pipette tips if necessary.
The optimal density of organoids in the gel differs for different ECMs and mouse strains. For example, C57BL6 organoids tend to be more contractile than FVB organoids when embedded in collagen I, resulting in contraction of the gel and detachment from the glass bottom if plated too densely.
In a 24-well plate, the glass bottom is slightly recessed from the edge of the plastic wall. When both blocks are present in the heating block (Fig. 4b), there is a small gap between the plate and the heating block surface, resulting in a temperature at the glass bottom less than 37 °C. To establish direct contact between the glass bottom and the heating block, remove one of the blocks, and set up the plate as in Fig. 4c–c′.
When preparing the collagen I solution, always wait for the solution to come down to the tip, and pipette it out completely. This is particularly important during collagen I neutralization to ensure an accurate volume and concentration of collagen.
-
Since the concentration and pH of rat tail collagen I vary among batches, adjust the pH using small volumes of collagen stock (up to 30 μL) or small amounts of 1.0 N NaOH (<0.5 μL).
If the adjustment requires addition of a large amount of collagen I stock, you will need to add more DMEM 10× to maintain ionic balance. However, this complicates calculation of the final concentration of neutralized collagen solution.
If you find that your collagen I stock is more basic, prepare the collagen solution with 7.0–7.5 μL of 1.0 N NaOH per 250 μL, and then adjust the final pH with small volumes of 1.0 N NaOH. This will avoid the need to add large volumes of collagen I stock to achieve the appropriate pH.
If you are concerned about the accuracy of the final collagen concentration, try to use the same pipette tip for mixing throughout neutralization and pH adjustment to limit the loss of collagen solution inside the pipette tip.
The preincubation time will determine the density of preassembled collagen fibrils. Due to batch variability in collagen stocks, the time required to obtain a gel with visible collagen fibrils varies considerably from 45 to 120 min. To examine the extent of fibril formation during preincubation, plate 30 μL of collagen solution onto a small Petri dish, let it gel for several minutes, and examine under the microscope.
If the neutralized collagen I solution is preincubated for more than 3–4 h on ice, it will become very cloudy and fibrous (Fig. 5d6), and the resulting gel will be less transparent, impairing visibility during imaging.
Collagen I gels tend to detach from the cover glass when kept too long on the heating block. Therefore, if you have Matrigel and collagen I gels on the same plate, plate the Matrigel samples first and the collagen I samples last.
In PFA, Matrigel becomes very fragile, especially after more than 4 days in culture. To avoid disintegration of the gel, reduce the PFA concentration to 2 % with lighter shaking or incubate the gel with 4 % PFA for 8–10 min.
In our lab, we have identified two successful approaches for performing antibody staining that use slightly different solutions and incubation times. The first one, described in Subheadings 3.8 and 3.9, uses 10 % FBS in DPBS as both the blocking buffer and the dilution buffer for antibodies. The other method uses 10 % FBS and 1 % BSA in DPBS as a blocking buffer and 1 % FBS and 1 % BSA in DPBS as the dilution buffer for antibodies.
From this step on, slides are kept in a StainTray with a black lid filled with a shallow layer of water to prevent desiccation and photo-bleaching of fluorescent probes.
If you plan to stain for extracellular proteins, such as basement membrane components, permeabilize the samples before embedding into OCT and sectioning. Direct permeabilization on slides can extract too many of these proteins.
To conserve primary antibodies, especially ones that require a high concentration, use a PAP pen to draw a hydrophobic border around the section, and add primary antibody solution within this area.
For mice less than 4 weeks of age, we typically use only gland #4 and remove the fat pad distal to the lymph nodes. Since the glands at this age are very small, pool several glands into one OCT block for sectioning.
To improve antibody staining in mammary gland tissue sections, it is sometimes useful to significantly increase the incubation times. For example, we sometimes permeabilize with Triton X-100 for 1 h at room temperature; incubate with primary antibody for 48 h at 4 °C; and incubate with secondary antibody for 6 h at room temperature or overnight at 4 °C. In addition, for incubation with antibodies, it is preferable to draw a hydrophobic border around the tissue with a PAP pen to reduce the volume of solution required and to ensure that the tissue is always immersed in solution. Do not let samples air-dry.
Acknowledgments
A.J.E. was supported by a Research Scholar Grant, RSG-12-141-01-CSM from the American Cancer Society, E.R.S. was supported by the Isaac Morris Hay and Lucille Elizabeth Hay Graduate Fellowship Award, R.J.H. was supported by an NIH/NIGMS training grant (2T32GM007445), and K.J.C. was supported by the US Department of Defense (W81XWH-12-1-0018).
References
- 1.Sternlicht MD. Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res. 2006;8:201. doi: 10.1186/bcr1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogg NA, Harrison CJ, Tickle C. Lumen formation in the developing mouse mammary gland. J Embryol Exp Morphol. 1983;73:39–57. [PubMed] [Google Scholar]
- 3.Williams JM, Daniel CW. Mammary ductal elongation: differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev Biol. 1983;97:274–290. doi: 10.1016/0012-1606(83)90086-6. [DOI] [PubMed] [Google Scholar]
- 4.Hinck L, Silberstein GB. Key stages in mammary gland development: the mammary end bud as a motile organ. Breast Cancer Res. 2005;7:245–251. doi: 10.1186/bcr1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sternlicht MD, Kouros-Mehr H, Lu P, Werb Z. Hormonal and local control of mammary branching morphogenesis. Differentiation. 2006;74:365–381. doi: 10.1111/j.1432-0436.2006.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mroue R, Bissell MJ. Three-dimensional cultures of mouse mammary epithelial cells. Methods Mol Biol. 2012;945:221–250. doi: 10.1007/978-1-62703-125-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vidi PA, Bissell MJ, Lelievre SA. Three-dimensional culture of human breast epithelial cells: the how and the why. Methods Mol Biol. 2012;945:193–219. doi: 10.1007/978-1-62703-125-7_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 9.Gudjonsson T, Ronnov-Jessen L, Villadsen R, Bissell MJ, Petersen OW. To create the correct microenvironment: three-dimensional heterotypic collagen assays for human breast epithelial morphogenesis and neoplasia. Methods. 2003;30:247–255. doi: 10.1016/s1046-2023(03)00031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson CM, Bissell MJ. Modeling dynamic reciprocity: engineering three-dimensional culture models of breast architecture, function, and neoplastic transformation. Semin Cancer Biol. 2005;15:342–352. doi: 10.1016/j.semcancer.2005.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson CM, Inman JL, Bissell MJ. Three-dimensional lithographically defined organotypic tissue arrays for quantitative analysis of morphogenesis and neoplastic progression. Nat Protoc. 2008;3:674–678. doi: 10.1038/nprot.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wozniak MA, Keely PJ. Use of three-dimensional collagen gels to study mechanotransduction in T47D breast epithelial cells. Biol Proced Online. 2005;7:144–161. doi: 10.1251/bpo112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Provenzano PP, Eliceiri KW, Inman DR, Keely PJ. Engineering three-dimensional collagen matrices to provide contact guidance during 3D cell migration. Curr Protoc Cell Biol. 2010;Chapter 10 doi: 10.1002/0471143030.cb1017s47. Unit 10 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 15.Ichinose RR, Nandi S. Lobuloalveolar differentiation in mouse mammary tissues in vitro. Science. 1964;145:496–497. doi: 10.1126/science.145.3631.496. [DOI] [PubMed] [Google Scholar]
- 16.Ichinose RR, Nandi S. Influence of hormones on lobulo-alveolar differentiation of mouse mammary glands in vitro. J Endocrinol. 1966;35:331–340. doi: 10.1677/joe.0.0350331. [DOI] [PubMed] [Google Scholar]
- 17.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simian M, Hirai Y, Navre M, Werb Z, Lochter A, Bissell MJ. The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development. 2001;128:3117–3131. doi: 10.1242/dev.128.16.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fata JE, Mori H, Ewald AJ, Zhang H, Yao E, Werb Z, Bissell MJ. The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev Biol. 2007;306:193–207. doi: 10.1016/j.ydbio.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132:3923–3933. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ewald AJ, Huebner RJ, Palsdottir H, Lee JK, Perez MJ, Jorgens DM, Tauscher AN, Cheung KJ, Werb Z, Auer M. Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J Cell Sci. 2012;125:2638–2654. doi: 10.1242/jcs.096875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, Yaswen P, Werb Z, Ewald AJ. The ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A. 2012;109:E2595–E2604. doi: 10.1073/pnas.1212834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen-Ngoc KV, Ewald AJ. Mammary ductal elongation and myoepithelial migration are regulated by the composition of the extracellular matrix. J Microsc. 2013;251(3):212–223. doi: 10.1111/jmi.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ewald AJ. Isolation of mouse mammary organoids for long-term time-lapse imaging. Cold Spring Harb Protoc. 2013 doi: 10.1101/pdb.prot072892. 2013. [DOI] [PubMed] [Google Scholar]
- 32.Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, Deryugina E, Friedl P. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol. 2009;20:931–941. doi: 10.1016/j.semcdb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013 Dec 19;155(7):1639–51. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shamir ER, Papallardo E, Jorgens DM, Coutinho K, Tsai WT, Aziz K, Auer M, Tran PT, Bader JS, Ewald AJ. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. The Journal of Cell Biology. 2014 Mar 3;204(5):839–56. doi: 10.1083/jcb.201306088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huebner RJ, Lechler T, Ewald AJ. Mammary epithelial stratification occurs through symmetry breaking vertical divisions of luminal cells. Development. 2014 Mar;141(5):1085–94. doi: 10.1242/dev.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]