

Summary
T cells have a central pathogenic role in the aetiopathogenesis of rheumatoid arthritis (RA), and are therefore a favoured target of immunotherapy aiming at physical or functional elimination. Here we report an efficacy test of FR104, a new co‐stimulation inhibitor directly targeting CD28 on T cells, in a translationally relevant model, the rhesus monkey model of collagen‐induced arthritis (CIA). As a relevant comparator we used abatacept [cytotoxic T lymphocyte antigen immunoglobulin (CTLA Ig)], an antagonist of CTLA‐4 binding to CD80/86 clinically approved for treatment of RA. Treatment with either compound was started at the day of CIA induction. Although FR104 previously demonstrated a higher control of T cell responses in vitro than abatacept, both compounds were equally potent in the suppression of CIA symptoms and biomarkers, such as the production of C‐reactive protein (CRP) and interleukin (IL)‐6 and anti‐collagen type II (CII) serum antibody (IgM/IgG). However, in contrast to abatacept, FR104 showed effective suppression of CII‐induced peripheral blood mononuclear cell (PBMC) proliferation. The current study demonstrates a strong potential of the new selective CD28 antagonist FR104 for treatment of RA.
Keywords: CD28, CIA, co‐stimulation blockade, rhesus monkey, T cell
Introduction
The central pathogenic role of T cells in rheumatoid arthritis (RA) is illustrated by the presence of T cells in synovial infiltrates 1 and the sensitivity of RA for T cell‐targeted therapies [methotrexate 2, cyclosporin A 3, 4, leflunomide 5, 6 and cytotoxic T lymphocyte‐4 immunoglobulin (CTLA 4‐Ig) 7]. Moreover, susceptibility to RA is linked strongly with certain major histocompatibility complex (MHC) class II alleles 8. A pivotal target that is involved critically in T cell activation is the interaction of CD28 on the T cells with two molecules of the B7 family, CD80 and CD86, on antigen‐presenting cells (APC). The interaction relays essential co‐stimulation to T cells 9. Activation of T cells requires the binding of the T cell receptor with an MHC–peptide complex (signal 1) and the additional interaction of CD28 with CD80/86 (signal 2). Specifically in primates, but not in mice, interaction of CD28 with inducible T cell co‐stimulator ligand (ICOS‐L) has also been described 10. These two activation signals are supplemented with a cytokine‐mediated third signal that determines the functional polarization of the newly activated T cells 11. Dysregulation of the T cell response is avoided by shut‐down signals from counter‐regulatory molecules, such as CTLA‐4 interaction with CD80/86 and programmed death‐1 (PD‐1) interaction with PD‐1 ligand 12, 13. In addition, PD‐1 ligand can interact with CD80 to regulate T cell responses 14.
Conceptually, the ideal approach for controlling autoreactive T cells would be to block T cell activation signals, without affecting shut‐down signals. For blockade of CD80/86 interaction with CD28 in the clinic, CTLA‐4 Ig (abatacept; Orencia®) has been used. However, binding of soluble CTLA‐4 Ig to CD80/86 not only prevented the relay of co‐stimulation signals to T cells, but also blocked T cell co‐inhibitory signals from the interaction of CD80/86 with cell‐bound CTLA‐4 and CD80 interaction with the PD‐1 ligand. The novel approach tested in this study is the prevention of the CD28 interaction with CD80/86 using FR104, which is a pegylated (PEG) monovalent anti‐CD28 antibody. FR104 has been developed to prevent CD28 interaction with CD80/86, while leaving shut‐down signals relayed through CD80/86 unaffected. Importantly, FR104 did not induce human T cell proliferation and cytokines secretion in vitro, even in the presence of anti‐CD3 antibodies or when cross‐linked with secondary antibodies 15. Abatacept, showing remarkable clinical effects in RA 7, was included as a clinically relevant comparator in the current experiment.
RA is a systemic autoimmune disease, which involves the activation of autoreactive T cells 16, 17, 18 and production of antibodies by autoreactive B cells. Antibodies binding citrullinated self‐proteins, indicated collectively as anti‐citrullinated protein antibodies (ACPA), and against IgG (rheumatoid factor: RF) are potent biomarkers for prognosis and early diagnosis of RA risk in genetically predisposed individuals 19. Approximately 40% of RA patients are ACPA‐negative 19. In this subgroup an association was found between disease severity and presence of serum antibodies binding carbamylated protein 20. Also, reactivity of T cells and antibodies against type II collagen (CII) has been implicated in the initiation and persistence of RA 21, 22, 23. In the rhesus monkey collagen‐induced arthritis (CIA) model used in the current experiment, cellular and humoral autoimmune reactions against CII‐specific antibodies have a central pathogenic role. The potential relevance for human RA is illustrated by the recent observation that serum autoantibodies from RA patients and CIA monkeys are directed to the same major epitopes 24.
Treatment with a monovalent anti‐mouse CD28 monoclonal antibody (mAb) was found to be clinically effective in mouse models of autoimmune disease and provided proof of concept that preventing the interaction between CD28 and CD80/86, thereby blocking signal 2 in the T cell activation cascade, ameliorates autoimmune‐mediated inflammatory diseases 25, 26. The CIA model in the rhesus monkey resembles the human condition more closely than murine models. In primates (man included) approximately 20% of CD4+ T cells and 50% of CD8+ T cells do not express CD28, whereas nearly all mouse T cells express CD28. The clinical implication of this discrepancy is illustrated by the observation that CTLA‐4 Ig first demonstrated efficacy to control transplant rejection in mice 27, but follow‐up studies in primates showed substantially less efficacy 28.
The current study shows that repetitive longitudinal dosing of FR104 in the rhesus monkey CIA model was well tolerated, and exerted a similar beneficial effect on CIA parameters to abatacept. Interestingly, FR104 was superior to abatacept in the control of CII‐induced ex‐vivo proliferation of autoreactive T cells.
Materials and methods
Animals
Healthy experimentally naive rhesus monkeys were purchased from the purpose‐bred colony of the Biomedical Primate Research Centre (BPRC) in Rijswijk, the Netherlands. CIA‐susceptible rhesus monkeys were preselected on the basis of absence of the dominant class I major histocompatibility complex (MHC) CIA resistance marker Mamu‐B*001 29, 30, 31. The total study group comprised 19 young adult, healthy rhesus monkeys (Macaca mulatta) of either sex. Both male and female monkeys (5·25–8·50 kg) were used. Individual data are given in Table 1. Before entry into the study, the animals were examined by the BPRC veterinarian staff. All animals were declared in good health and showed no signs of (infectious) diseases. Haematological and clinical chemistry values were all within the normal range. During the study the monkeys were housed socially, in spacious cages designed for the housing of macaques.
Table 1.
Animal identification, gender, age and starting weight at day of stratification.
| Animal ID | Gender | Age (years) | Starting weight | |
|---|---|---|---|---|
| Group I (placebo) | R00062 | F | 12·2 | 5·25 |
| R02049 | F | 10·3 | 7·70 | |
| R05068 | F | 7·3 | 5·92 | |
| R06045 | M | 6·3 | 7·26 | |
| R07111 | M | 5·2 | 5·60 | |
| Mean ± s.d. | 9·92 ± 4·00 | 6·78 ± 1·27 | ||
| Group II (FR104) | 96076 | F | 16·2 | 5·59 |
| R01030 | F | 11·4 | 7·80 | |
| R04091 | F | 8·3 | 7·01 | |
| R06046 | F | 6·3 | 5·84 | |
| R06100 | M | 6·3 | 8·17 | |
| R07050 | M | 5·3 | 5·88 | |
| R07061 | F | 5·3 | 5·51 | |
| Mean ± s.d. | 7·2 ± 2·74 | 5·72 ± 0·43 | ||
| Group III (abatacept) | 96089 | F | 16·3 | 8·50 |
| R01091 | F | 11·1 | 5·00 | |
| R05084 | F | 7·3 | 6·88 | |
| R07003 | M | 5·5 | 6·50 | |
| R07031 | F | 5·4 | 4·66 | |
| R07068 | M | 5·3 | 7·65 | |
| R07075 | F | 5·3 | 5·25 | |
| Mean ± s.d. | 8·15 ± 4·04 | 6·67 ± 1·13 |
M = male; F = female; s.d. = standard deviation.
The daily diet consisted of monkey food pellets (Hope Farms, Woerden, the Netherlands), seasonal fresh fruit and vegetables and bread. Drinking water was available ad libitum. Analgesic medication (Buprecare®, 0·3 mg/ml buprenorphine base; 20–100 µg/kg; Schering‐Plough BV, Maarssen, the Netherlands) was given based on the assessment of the animal caretakers and BPRC's veterinary staff (start and dosing increased or stopped on the basis of behavioural changes). Ulcerative skin lesions developing at the immunization sites were treated with wound spray (Acederm; Intervet, Boxmeer, the Netherlands) each time that an animal was sedated, in order to prevent infection.
In accordance with the Dutch law on animal experimentation, the study protocol and experimental procedures were reviewed and approved by the Experimental Animal Care and Use Committee of the BPRC before the experiments started.
Induction and clinical assessment of CIA
Prior to induction of CIA, chicken collagen type II (chCII; MD Biosciences, Zurich, Switzerland) was dissolved in 0·1 M acetic acid at 4°C for 72 h to a final concentration of 10 mg/ml. The clear solution was emulsified with an equal volume of complete Freund's adjuvant (CFA) (Difco, Detroit, MI, USA). CIA was induced by injection of 1·0 ml emulsion (5 mg chCII/animal) into the dorsal skin distributed over 10 spots of 100 µl.
Gross clinical signs were recorded by daily cage‐side inspection of behavioural changes (apathy, loss of appetite) or pain (avoidance of limb usage). Close inspection of individual joints was performed twice weekly on monkeys that were sedated by intramuscular injection of 0·1 ml/kg ketamine (10 mg/ml). At such occasions we also measured the body weight as an accepted surrogate disease marker for the CIA model and the body temperature. In addition, we collected a venous blood sample and inspected large joints (knee, elbow, ankle) and the small joints of toes and fingers for redness and/or swelling.
For the clinical and ethical management signs of clinical arthritis, soft tissue swelling (STS) and redness of affected joints were scored twice‐weekly using a previously published semiquantitative clinical score (CS; 32): 0 = no disease symptoms; 0·5 = fever; 1 = apathy and loss of appetite, weight loss; 2 = warm and tender joints, but without STS; 3 = moderate STS but normal flexibility of affected joints; 4 = severe STS with joint stiffness; 5 = such severe disease that euthanasia is necessary. Note that scores 0·5, 1 and 2 are subclinical CIA scores; CIA scores ≥ 3 represent clinically evident arthritis.
Simultaneously, the number of joints with visible STS was determined and the severity of swelling was graded on an arbitrary scale (0; ± = 0.5; + = 1; ++ = 2). This composite score of the number of swollen joints with severity of swelling was termed the total joint score (=TJS).
To ensure objectivity during clinical scoring, the investigators performing the physical examination and rating clinical scores were blinded to the different treatments during the in‐vivo part of the study.
Study design
Animals were stratified over the three experimental groups to ensure that all groups had a similar mean age, weight and sex distribution (Table 1). The test substances were FR104, a monovalent, pegylated humanized Fab' antibody fragment directed against human CD28 15, and abatacept, a clinically validated benchmark 7, which was included as positive control and as comparator. A prophylactic study design was used with treatment starting on the day of CIA induction (=day 0). One group of seven animals was treated with 10 mg/kg FR104 on days 0, 7, 14, 21, 28, 35 and 42 after immunization. A second group of seven animals was treated on the same days with 10 mg/kg abatacept. A third group of five animals received placebo treatment on the same days. All test substances were administered via the intravenous route. In previous studies these group sizes ensured sufficient statistical power for data analysis.
Power calculation
For power calculation of the minimal group size a historical data set of 22 control animals was used, all of which were immunized with chCII. The start of treatment was set on day 0 before the onset of disease. Treatment was expected to interfere with the start of the disease and to result in a delayed onset, being the time to reach CS ≥ 3. The average day of onset in this historical group of 22 animals was 18 ± 7 days. Power analysis was performed with two different effect sizes (ES; delay in onset of 14 days and 15 days), two different control group sizes (n = 4 and n = 5) and three different treatment group sizes (n = 5; n = 6 and n = 7) to comply with acute responders or non‐responders to CIA induction. For almost all conditions tested the power was 0·8 or higher using seven animals in the treated group and five animals treated with placebo.
A second important readout out was a reduction in the severity of clinically evident disease as measured by the TJS. The TJS is a composite score of the number of joints that are affected combined with severity of inflammation. Severity of swelling is determined on an arbitrary scale: no swelling, − = 0; minimal swelling, ± = 0·5; mild swelling, + = 1; and severe swelling, ++ = 2.
Additional parameters that were evaluated were validated biomarkers, including serum C‐reactive protein (CRP) levels, body weight loss, levels of collagen‐specific IgG/IgM and cellular responses and histopathology. Exploratory end‐points were cellular and humoral immune functions. Results of FR104‐ or abatacept‐treated animals were compared with placebo‐treated control animals.
Haematology and clinical chemistry
All haematological and clinical chemistry analyses were performed by the diagnostic laboratory at BPRC on a Sysmex Sf‐3000 (Goffin Meyvis, Etten‐Leur, the Netherlands) and a Cobas Integra‐400+ (Roche, Almere, the Netherlands), respectively.
Enzyme‐linked immunosorbent assay (ELISA)
Measurement of FR104 drug levels through ELISA. Microtitre plates (Maxisorp Nunc, Dutscher, Issy‐les‐moulineaux, France) were coated with recombinant human CD28‐Fc (R&D Systems, Abingdon, UK) at 2 µg/ml in carbonate buffer (pH 9·2) Empty binding sites were blocked with 1% bovine serum albumin (Sigma Aldrich, L'Isle D'Abeau Chesne, France) in 0·1% Tween‐phosphate‐buffered saline (PBS) for 2 h at 37°C. Serial dilutions of serum samples were incubated for 2 h at 37°C and detected by monoclonal mouse anti‐human kappa chain antibody (BioAtlantique, Nantes, France) at 0·2 µg/ml (1 h, 37°C) and by horseradish peroxidase (HRP)‐conjugated donkey anti‐mouse Ig antibody (0·4 µg/ml for 1 h at 37°C; Jackson Immunoresearch, Marseille, France). Optical density was recorded at 450 nm after tetramethyl benzidine (TMB) revelation. A standard curve of FR104 was analysed in parallel and concentrations were deduced from this standard curve.
Serum interleukin (IL)‐6 levels
Serum samples (150 µl) were collected twice weekly for the analysis of IL‐6 levels. IL‐6 levels were measured using a commercial IL‐6 ELISA kit, according to the manufacturer's protocol (Old World monkey IL‐6 ELISA; U‐CyTech, Utrecht, the Netherlands).
Rhesus monkey anti‐chCII antibody serum levels
Plates (96‐wells; Greiner Bio‐One, Stonehouse, UK; F‐form; microlon) were coated with chCII/PBS (10 µg/ml) for 2 h at 37ºC in a humidified atmosphere and subsequently blocked with 1% bovine serum albumin (BSA) for 1 h. Serum was diluted (IgG and IgM =>1 : 100 and 1 : 1000) in PBS + 1% BSA and added to the wells followed by incubation for 2 h at 37˚C in a humidified atmosphere. A serial dilution of an anti‐CII‐positive serum standard was analysed in parallel and relative concentrations (arbitrary units = AU) were deduced comparatively. Anti‐chCII antibodies were detected with a secondary antibody [alkaline phosphatase (AP)‐conjugated goat anti‐hIgM or hIgG; Biosource, Camarillo, CA, USA] for 1 h. ELISA was developed by adding 10 μl of poly‐nitrophenylphosphate diluted in Tris buffer (Sigma Chemicals, Zwijndrecht, the Netherlands). Colour development was determined by absorbance at 405 nm.
Effect of the test substances on in‐vitro cellular immune parameters
Mixed lymphocyte reaction (MLR). PBMC of two monkeys (responder) were cultured with 30 Gy irradiated stimulator peripheral blood mononuclear cells (PBMC) from two Mamu‐DR mismatched monkeys. The following conditions were tested. For controls: (1) responder PBMC, no inhibitor, no autologous stimulator cells (background proliferation); (2) responder PBMC, no inhibitor, with allogeneic stimulator PBMC (maximum allo response); (3) condition 2 with FR104 (10 µg/ml); (4) condition 2 with CTLA‐4 Ig (abatacept; 10 µg/ml); (5) condition 2 with cyclosporin A (CsA) (Sandimmune; Novartis Pharmaceuticals UK Ltd, Camberley, UK; 400 ng/ml); and (6) condition 1 with concanavalin A (ConA) (5 µg/ml; immunocompetence).
chCII‐induced proliferation
At the day of necropsy mononuclear cells (MNC) were isolated from the blood and spleen as well as the axillary and inguinal lymph nodes. The isolated cells were cultured (50 000/well) under different conditions: (1) anti‐CD3/CD28; (2) phorbol myristate acetate (PMA)/ionomycin stimulation (immunocompetence); and (3) chCII (10 µg/ml; specific stimulation), ovalbumin (10 µg/ml; aspecific stimulation) and medium alone (background proliferation)
MNC were cultured for 72 h at 37°C in a humidified atmosphere in 96‐well U‐bottomed microtitre plates. Supernatants were harvested (for cytokine production) and proliferation was quantified by the incorporation of tritiated [3H]‐thymidine (0·5 µCi/well/25 µl) during the final 18 h of the cultures. Incorporation of radiolabel was counted using a matrix 9600 β‐counter (Packard 9600; Packard Instrument Company, Meriden, CT, USA).
Flow cytometry
For selection of the suitable non‐human primate CIA model we determined the binding of FR104 to PBMC from two common marmosets and two rhesus monkeys. PBMC isolated from whole blood were stained with CD3PerCP (SP34.2; BD Biosciences, San Jose, CA, USA) and unlabelled FR104. FR104 binding was detected with a rabbit anti‐PEG antibody (Epitomics, Burlingame, CA, USA; clone PEG‐B‐47; cat. 2061‐1) that was stained subsequently with a goat anti‐rabbit AF405 (Invitrogen A‐31556; Invitrogen, Carlsbad, CA, USA; See Fig. 1).
Figure 1.
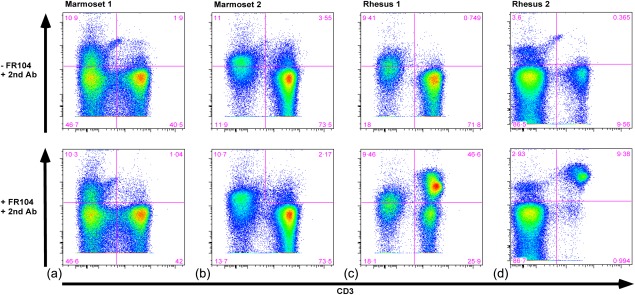
Staining of peripheral blood mononuclear cells (PBMC) from the rhesus monkey and the common marmoset with FR104. Flow cytometry was used to demonstrate selective staining of FR104. (a,b) Dot‐plots show staining of PBMC of two common marmosets (marmosets 1, 2) and (c,d) two rhesus monkeys (rhesus 1, 2). PBMC were stained without (upper row) and with (lower row) FR104 in combination with a rabbit anti‐pegylated (PEG) antibody visualized with AF405‐labelled goat anti‐rabbit antibody. Selective staining with FR104 is observed in the lower right‐hand panels displaying rhesus PBMC. For gating strategy see Supporting information, Fig. S1.
On days 0, 7, 14 and 21 and on the day of necropsy whole venous blood samples were collected into heparinized vacutainers and processed for analysis of the MNC fraction by fluorescence activated cell sorter (FACS) for evaluating the effect of the treatments on different MNC subpopulations during the course of the disease. At the day of necropsy MNC were also isolated from axillary and inguinal lymph nodes and analysed by FACS.
The following mAb staining panels were used for FACS analysis: general subset panel, CD3AF700 (SP34·2; BD Biosciences), CD4PE‐CY7 (SK3; BD Biosciences), CD8APC‐H7 (SK1; BD Biosciences), CD16PE (3G8; BD Biosciences), CD14ECD (RMO52; Beckman Coulter, Brea, CA, USA), human leucocyte antigen D‐related (HLA‐DR)PerCP (L243; BD Biosciences) and CD20V450 (L27; BD Biosciences); memory cell panel, CD3AF700, CD4PE‐CY7, CD8APC‐H7, CD197FITC (150503; ITK Diagnostics BV, Uithoorn, the Netherlands), CD95PE (DX2; BD Biosciences), CD28ECD (CD28·2; Beckman Coulter) and CD45RAbiot‐PerCP (5H9; BD Biosciences); regulatory T cells panel I, CD3AF700, CD4PE‐CY7, CD8APC‐H7, CD25PE (4E3; Miltenyi Biotec, Bergisch Gladbach, Germany) and forkhead box protein 3 (FoxP3)APC (236A/E7; eBioscience, San Diego, CA, USA); regulatory T cells panel II, CD3AF700, CD4PE‐CY7, CD8APC‐H7, CD25PE and CD152APC (BN13; BD Biosciences). Cells were fixed in 1% cytofix (BD Biosciences). Analysis of cell staining was performed on a FACS LSRII (BD Biosciences) using FlowJo software (Treestar, Ashland, OR, USA).
Joint histology
Histopathology was performed as described previously 33. The severity of histological changes was graded by a pathologist blinded to the study as published by Pettit et al. 34. This system quantifies the degree of inflammation, cartilage damage and bone damage on a scale ranging from 0 to 5.
Statistics
For statistical analyses we used Prism version 6·0e software (GraphPad Software Inc., La Jolla, CA, USA).
For area under the curve analysis, an unpaired t‐test (Mann–Whitney U‐test) was performed comparing the FR104‐ and abatacept‐treated groups to the placebo group. All tests were two‐tailed, and results with P < 0·05 were considered to be statistically significant.
Results
In‐vitro cross‐reaction of FR104 with non‐human primate T cells
The prior observation that FR104 did not bind rodent CD28 precludes efficacy testing in mouse or rat models. Two non‐human primate RA models are available in which preclinical proof of concept may be obtained; namely, the well‐established collagen‐induced arthritis (CIA) models in the common marmoset (Callithrix jacchus 35) or in the rhesus monkey (M. mulatta 32, 36). We first tested the binding of FR104 to freshly isolated PBMC from two common marmosets and two rhesus monkeys. FR104 did not bind T cells from the common marmosets (Fig. 1a,b: 2nd quadrant lower panels), but showed selective binding to T cells from both of the rhesus monkeys (Fig. 1c,d: 2nd quadrant lower panels). As reported previously 37, we noticed high variability in T cell count and in CD28 expression by T cells between individual monkeys.
Next we tested whether binding of FR104 blocked the primary response of rhesus monkey T cells in an MLR and the secondary T cell response assayed by stimulating PBMC from CII‐immunized monkeys with the immunizing antigen. Good inhibition of both the MLR (Fig. 2a) and CII‐specific proliferation (Fig. 2b) was observed for FR104. The inhibition was comparable with abatacept and CsA.
Figure 2.
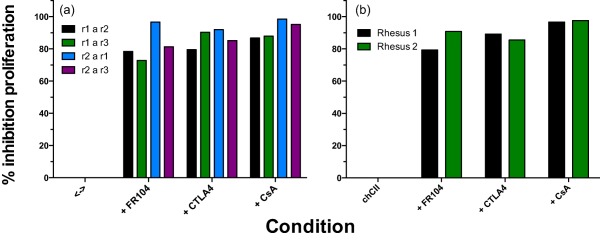
A proliferation assay was used to demonstrate functional blocking by FR104. This was compared to proliferation in the presence of cytotoxic T lymphocyte antigen 4 immunoglobulin (CTLA‐4 Ig) and cyclosporin A (CsA). The data are presented as the calculated percentage of inhibition relative to cultures with medium alone. (a) Inhibition of allogeneic responses of four major histocompatibility complex (MHC) class II mismatched peripheral blood mononuclear cells (PBMC) was calculated relative to medium alone for FR104, CTLA‐4 Ig and CsA (r1 a r2 = PBMC of rhesus monkey 1 responds to irradiated PBMC of rhesus monkey 2). (b) Inhibition of CII‐specific responses by FR104, CTLA‐4 Ig and CsA was calculated relative to medium alone for two animals that were immunized with collagen type II (CII) (rhesus 1, 2). Supporting information, Fig. S2 presents the data as stimulation indices.
These experiments show that FR104 not only binds to rhesus monkey CD28, but also blocks T cell activation and reactivation.
Serum levels of FR104 in the rhesus monkey CIA model
The clinical efficacy of FR104 and abatacept was tested in 19 CIA‐susceptible, i.e. Mamu‐B*001‐negative rhesus monkeys 30, 31. The selected animals were stratified according to age, sex and weight over three experimental groups. A placebo‐treated group, consisting of five animals, received vehicle only, seven animals were treated with FR104 and seven animals received abatacept. The first test substance dose was injected (intravenously) immediately after the CIA induction on day 0 and continued by once‐weekly dosing until post‐sensitization day (psd) 42; each monkey thus received seven doses. The FR104 dose of 10 mg/kg had been chosen based on previous pharmacokinetic, pharmacodynamics and efficacy experiments in kidney graft primate recipients 38. The animals were monitored until psd 70 days to assess a possible delayed onset or rebound of CIA after cessation of the treatment.
FR104 drug levels were determined by ELISA (see Materials and methods). FR104 levels of approximately 100 µg/ml were recorded in the first 4 days after administration (Supporting information, Fig. S3a). In order to determine the minimal level of FR104, sera were collected just before the animals were dosed. Minimal FR104 levels ≥ 30 µg/ml were detected until day 49 (Supporting information, Fig. S3b). These levels of FR104 demonstrated effective suppression of a pathogenic autoimmune response in a rhesus monkey experimental autoimmune encephalitis (EAE) model 39. Anti‐FR104 antibodies were not detected during the period of treatment (data not shown). Hence, all animals were included in the analysis.
Treatment with FR104 and abatacept mitigates the induction of anti‐chCII autoimmune reactions
The initiation and progression of CIA in rhesus monkeys is driven by the synergistic activity of anti‐CII antibodies and T cells 40. Hence, we tested the effect of the two treatments on the ex‐vivo proliferative response of MNC isolated from blood, spleen and lymph nodes at necropsy in cultures stimulated with the immunizing antigen chCII. Moreover, we analysed longitudinally collected sera for the presence of chCII‐specific IgM and IgG antibodies (see Materials and methods). It should be noted that the isotype switch from IgM to IgG requires the presence of T helper effector cells.
Antibodies (Fig. 3)
Figure 3.

Comparison of serum levels of chicken collagen type II (CII)‐specific antibodies after treatment with placebo, FR104 and abatacept. Serum samples were collected twice and subsequently analysed for the presence of immunoglobulin (Ig)M and IgG antibodies directed against chCII. Antibody levels are expressed as arbitrary units that were calculated from a standard curve obtained for chCII‐specific antibody‐positive pooled serum. Colour‐coding for each animal as presented in Fig. 4 was applied here. (a–c) chCII‐specific IgM antibody levels over time in animals treated with placebo, FR104 and abatacept. (d–f) chCII‐specific IgG antibody levels over time in animals treated with placebo, FR104 and abatacept. (g,h) Average serum levels per day for chCII‐specific IgM and IgG were calculated from the total production as determined by area under the curve analysis divided by the number of days the animals were in experiment. Significant suppression of the mean production/day of both chCII‐specific IgM and IgG compared to the placebo‐treated group was tested with the Mann–Whitney U‐test. *P < 0·05; **P < 0·01. PID = post‐induction day.
Longitudinal serum levels of chCII‐specific IgM and IgG were clearly reduced in both treatment arms (Fig. 3b,c for IgM and Fig. 3e,f for IgG). For statistical analysis, for each monkey we calculated the AUC normalized for the number of days the animals were in experiment. This analysis shows that the suppression of IgM (Fig. 3g) and IgG antibodies (Fig. 3h) by the treatments is statistically significant.
Cellular responses
At the day of necropsy MNC were isolated from venous blood (PBMC) and from axillary LN, inguinal LN and the spleen. MNC were restimulated ex vivo with chCII and additional control stimuli. There was a clear trend towards suppressed MNC proliferation in monkeys treated with FR104. Statistical significance was only reached in cultures containing MNC from axillary lymph nodes (aLN) (Fig. 4). Remarkably, treatment of the monkeys with abatacept had no detectable effect on the ex‐vivo proliferative response of mononuclear cells against chCII (Fig. 4 and Supporting information, Table S1).
Figure 4.
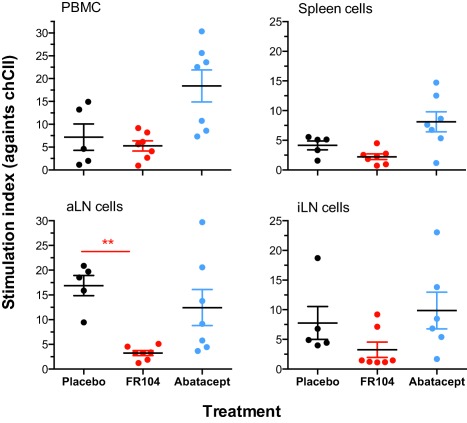
Ex‐vivo determination of chicken collagen type II (CII)‐induced proliferation of mononuclear cells (MNC) after treatment with FR104 and abatacept compared to placebo treatment. Proliferation was measured for cells isolated from peripheral blood mononuclear cells (PBMC), the spleen (spleen cells), axillary lymph node (aLN) and inguinal lymph node (iLN; inguinal cultures of one FR104‐treated animal could not be assayed). Depicted is the stimulation index [SI = proliferation of experimental sample counts per minute (CPM)/proliferation of medium control (CPM)]. An example of CII‐specific proliferation as measured in CPM is given in Supporting information, Table 1. Significance of suppression relative to the placebo‐treated group was tested with the Mann–Whitney U‐test. **P < 0·01 (in red).
We also asked whether the treatment with FR104 and abatacept induced changes in lymphocyte subsets. For that analysis we sampled PBMC on days 0, 7, 14, 21 and at the day of necropsy. In addition, we performed the same analysis on cells isolated from axillary and inguinal lymph nodes at the day of necropsy. No differences were found for major lymphoid subset populations such as B cells, T cells or natural killer (NK) cells (data not shown). In all animals, only in the subset of CD3/CD4/CD25high/FoxP3 cells did we find an effect of the treatment with FR104, namely a significant reduction in the percentage of cells with this phenotype on day 21 and at the day of necropsy for PBMC and aLN (Fig. 5a). The corresponding analysis of the CD3/CD4/CD25high/CTLA‐4 subpopulation could not confirm whether these cells were genuinely regulatory T cells, as we did not observe an effect of FR104 treatment (Fig. 5b).
Figure 5.
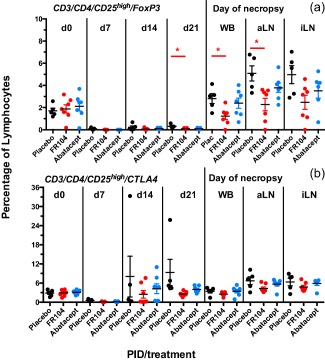
Regulatory T cells during treatment with placebo, FR104 and abatacept. Flow cytometry was used to determine the percentage of regulatory T cells as defined by the expression of forkhead box protein 3 (FoxP3) (a) or cytotoxic T lymphocyte antigen 4 (CTLA‐4) (b) on CD3/CD4/CD25high positive cells. aLN = axillary lymph nodes; iLN = inguinal lymph nodes; WB = whole blood. Significance compared to placebo‐treated animals was tested with the Mann–Whitney U‐test. *P < 0·05 (in red). For gating strategy see Supporting information, Fig. 4.
Comparable beneficial effects of FR104 and abatacept on CIA
The clinical and pathological presentation of CIA in the genetically heterogeneous rhesus monkey is complex and variable. Historical data show that the time to reach arthritis onset CIA is variable. Moreover, in monkeys that do not show outward signs of joint inflammation, subclinical arthritis can nevertheless be present in the form of CIA‐related biomarkers, body weight loss or histological synovitis 41, 42.
Gross CIA score and body weight loss. Fig. 6a–c shows the macroscopic clinical scores (CS; see Material and methods for scoring criteria) and Fig. 6d–f shows the percentages of weight loss (percentage relative to the day of CIA induction). The clinical threshold CS ≥ 3 reflects macroscopically visible joint inflammation. The combined data indicate that in the placebo group two monkeys developed severe CIA (CS ≥ 4, weight loss > 20%), two monkeys developed moderate CIA (CS ≤ 3 and/or weight loss between 10 and 20%) and one monkey developed minor CIA (CS = 0, body weight loss < 10%). Based on the same criteria, three monkeys in the group treated with FR104 developed moderate CIA and four minor CIA. In the abatacept group two monkeys developed moderate CIA and five minor CIA.
Figure 6.

Clinical parameters of collagen‐induced arthritis (CIA) in the rhesus monkey after treatment with placebo, FR104 and abatacept. The animals within each treatment group are colour‐coded. A colour represents the same animals for each graph within each treatment group. (a–c) The clinical score was measured 2×/week for each animal. (d–f) Body weight was measured 2×/week and is presented as percentage relative to day 0. (g–i) The total joints score is a composite score and was calculated from the number of joints combined with the severity of swelling (an arbitrary scale: 0 = no swelling; 0·5 = minimal swelling; 1 = mild swelling and 2 = severe swelling).
Group scores of CIA onset (a) and overall survival (b) were plotted as survival curves in Fig. 7. The figure shows a clearly delayed, albeit statistically not significant, CIA onset in the monkeys treated with FR104 or abatacept. Two monkeys of the placebo group experienced such severe CIA that they had to be killed humanely. Three placebo‐treated monkeys were killed at the end of the 70 days observation period. In the groups treated with FR104 or abatacept, none of the monkeys needed to be killed before the end of the 70 days observation period, yielding a statistically significant difference in overall survival.
Figure 7.

Disease‐free survival [time until clinical score (CS) ≥ 3] and survival after treatment with FR104 and abatacept. (a) Disease‐free survival (CS ≥ 3) after treatment with FR104 [red; log‐rank (Mantel–Cox) test; P = 0·11] and abatacept [blue; log‐rank (Mantel‐Cox) test; P = 0·12] compared to placebo‐treated controls (black). (b) Survival after treatment with FR104 (red; log‐rank (Mantel–Cox) test; P = 0·044, red asterisk) or abatacept (blue; log‐rank (Mantel–Cox) test; P = 0·044, blue asterisk) compared to placebo‐treated controls (black; two vehicle‐treated control animals were killed earlier because they reached their clinical score).
The clinical score does not take into account the number of joints that are affected and the severity of swelling in individual joints. The TJS (see Materials and methods for criteria) is used to express these clinical aspects. The clinical improvement in FR104‐ and abatacept‐treated animals is reflected by the strongly reduced total joint score (Fig. 6g–i).
Acute‐phase reaction. Onset of CIA is associated with a strongly increased serum level of IL‐6, eliciting increased production of the acute phase reactant CRP by the liver. Serum samples for the measurement of IL‐6 levels (see Materials and methods) and CRP levels were collected twice a week. CRP was measured in‐house on fresh serum using a COBAS INTEGRA 400 plus analyser (Roche Diagnostics Limited, Rotkreuz, Switzerland). IL‐6 serum levels were measured with ELISA. In the three placebo‐treated monkeys with outward detectable joint inflammation, IL‐6 (Fig. 8a) and CRP (Fig. 8d) serum levels were increased strongly. IL‐6 and CRP levels were reduced substantially in the group treated with FR104 (Fig. 8b,e), while reduction was lower in the group treated with abatacept (Fig. 8c,f).
Figure 8.

Serum C‐reactive protein (CRP) and interleukin (IL)‐6 levels after treatment with placebo, FR104 and abatacept. The animals within each treatment group are colour‐coded. A colour represents the same animals for each graph within each treatment group (see also Fig. 6). (a–c) Longitudinal analysis of serum IL‐6 levels. Serum was collected 2×/week for each animal. (d–f) Longitudinal analysis of serum CRP levels. Serum was collected 2×/week for each animal. (g) The average serum IL‐6 level/day was calculated from the total production as determined by area under the curve analysis divided by the number of days the animals were in experiment. (h) The average serum CRP level/day was calculated from the total production as determined by area under the curve analysis divided by the number of days the animals were in experiment. Significance relative to placebo‐treated group was tested with the Mann–Whitney U‐test. *P < 0·05.
For statistical evaluation we determined the mean levels per day for both parameters by dividing the AUC by the number of days the animals were in experiment. It was shown that treatment with FR104 resulted in a significant reduction in CRP levels (Fig. 8h; P = 0·017), while the reduction in CRP levels after treatment with abatacept was lower, and not significant (P = 0·26). A similar trend was observed for the mean IL‐6 levels in both the FR104‐ and abatacept‐treated animals compared to placebo‐treated animals; significance was not reached (Fig. 8g).
Histology
CIA is characterized by severe inflammatory and erosive changes in the synovial joints. In total, eight peripheral small joints/animal [four proximal interphalangeal (PIPs) and four distal interphalangeal (DIPs); one finger/extremity] were analysed by histopathology. Histopathology scores of all selected small joints were graded on an arbitrary scale adapted from Pettit et al. 34, quantifying the degree of inflammation, cartilage damage and bone damage on a scale ranging from 0 to 5. Figure 9 summarizes the effect of FR104 and abatacept on the histological aspects of CIA. Each dot represents the sum of scores for one of the eight peripheral small joints. The number of joints with increased infiltration of immune cells (inflammation = synovitis) was reduced significantly in the two treatments groups. Although the effects of treatment with FR104 and abatacept on joints erosion of cartilage and subchondral bone are substantial, they did not differ significantly from the control group.
Figure 9.
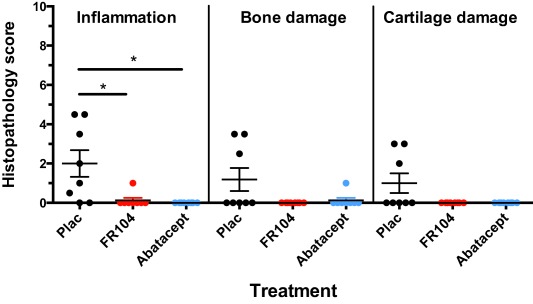
The effect of treatment with FR104 and abatacept on joint pathology as determined by a histopathology scoring system described by Petit et al. 34. The level of synovial inflammation, cartilage damage and bone damage is graded from 0 to 5. Each dot represents the sum of histopathology scores per treatment group for each of the eight joints (one proximal interphalangeal joint and one distal interphalangeal joint for both hands and feet). Significance relative to placebo‐treated group was tested with the Mann–Whitney U‐test. *P < 0·05.
Discussion
The ‘Holy Grail’ in the treatment of human autoimmune‐mediated inflammatory disease, RA included, is the restoration of immunological non‐responsiveness to self. A well‐established way to achieve this is to block the relay of co‐stimulatory signals from CD80/86 molecules on APCs to CD28 on T cells. In the current study two treatment strategies, both targeting the CD28–CD80/86 co‐stimulation T cell pathway, were explored in a relevant experimental model, the rhesus monkey model of CIA. The pegylated monovalent antibody FR104 is a new experimental treatment designed for blocking the binding sites of CD80/86 at the CD28 molecule expressed by T cells. The safety profile of monovalent FR104 was analysed extensively in vitro and in vivo and showed no signs of a superagonistic effect 15, 37. Conceptually, the value of blocking T cell co‐stimulation at the side of CD28 rather than CD80/86 is that the APC–T cell cross‐talk via interaction between CD80/86 and CTLA‐4 remains unhindered, leaving CTLA‐4 with the possibility to exert its regulatory function on effector and regulatory T cells 43, 44, 45. Abatacept (CTLA‐4 Ig), which blocks co‐stimulation at the site of the APC by binding to CD80 and CD86, was included as a clinical benchmark 46 which has been approved for the treatment of RA 7, 47. Recently it was shown that CTLA‐4 might also operate through a CD28‐independent mechanism, mobilizing pathways in the APC involving nitric oxide (NO), regulatory T cells and transforming growth factor (TGF)‐β 48. Evaluation of the two strategies in mouse CIA was not considered, because FR104 does not bind mouse CD28. Moreover, considerable differences exist between murine and human CD28 expression on effector T cells and differences in co‐stimulatory interactions with CD28 have been reported between man and mice 10. In addition, selective mouse CD28 antagonists working with a similar mechanism of action as FR104 are not available.
The induction and pathogenesis of CIA in rhesus monkeys involves an interplay of humoral and cellular autoimmunity 40. Anti‐CII IgM antibodies have an essential pathogenic role in CIA initiation 29, 49, probably via mediating the initial autoimmune attack on joint cartilage 50. Our previously reported experience with CsA in this model 3 showed that the activation of autoreactive T cells is typically an early event in the model. This implies that the window of opportunity for treatment with T cell‐targeting therapies is narrow. In contrast to the situation in human autoimmune inflammatory diseases, where autoreactive T cells seem to be activated constitutively, T cell activation in the rhesus monkey CIA model seems a brisk and monophasic process. To maximize the detection of a relevant treatment effect, administration of FR104 and abatacept was therefore started at the time of CIA induction (psd 0). A similar approach was used for testing FR104 in the EAE model 39.
We report that treatment with FR104 induced profound changes of the humoral and cellular autoimmune reactions that drive CIA initiation. We observed a significant reduction in anti‐CII IgM and IgG serum antibody levels (Fig. 3). The reduction observed for CII‐specific IgM production is somewhat lower (75% for FR104 and 61% for abatacept) than CII‐specific IgG production (90% for FR104 and 74% for abatacept). The more robust effect of FR104 on the production of CII‐specific IgG may be explained by inhibition of helper T cells that promote antibody isotype switching. Our current data are compatible with observations by Mihara et al., that CTLA‐4 Ig treatment prevents B cell maturation resulting in a suppressed expansion of both IgG‐ and IgM‐producing autoreactive B cells 51.
Although it was shown previously that CD28 blockade with a single‐chain Fv antibody 45 or FR104 38 promoted regulatory functions in non‐human primate models of allograft rejection, we obtained no indication that FR104 had this effect in the CIA model. No differences in cellular phenotypes, indicative of regulatory T cell induction, were observed between placebo‐treated animals and FR104‐ or abatacept‐treated animals (Fig. 5). Interestingly, at the day of necropsy, which was approximately 3 weeks after the last treatment, CII‐induced proliferation of MNC from FR104‐treated animals was still substantially lower than in control animals, while proliferation in abatacept‐treated animals was comparable to placebo‐treated animals (Fig. 4). We did not test infection by specific pathogens during this study, but no overt infectious adverse event was noticed in a parallel study testing FR104 in a rhesus monkey EAE model 39.
This promising immunomodulatory potency of FR104 is translated into beneficial effects on the ensuing autoimmune inflammatory disease. All five placebo‐treated monkeys developed an inflammatory response on induction (CRP > 50 mg/l; Fig. 5j) and three of five developed clinically evident arthritis (60%). By contrast, only two of the seven monkeys treated with FR104 (30%) showed a subclinical response to the immunization, with minimal inflammatory reactions. The early treatment with abatacept had a comparable effect to FR104 on the inflammatory reaction and the development of outward clinical signs. Monkeys treated with FR104 or abatacept displayed only minimal histological features of arthritis in peripheral joints, such as synovitis or damage to cartilage and subchondral bone.
Taking the clinical and immunological data together, the conclusion is warranted that FR104 exerted a remarkable beneficial effect on the clinical and pathological aspects of the model. The effect of FR104 is comparable with the clinically approved benchmark drug abatacept. An additional value of FR104 was the more profound suppression of cellular autoimmune parameters compared to abatacept.
The current study demonstrates a strong potential of the selective CD28 inhibition strategy for treatment of RA.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Dot‐plots showing the cell gating strategy for flow cytometry analysis of whole blood from the rhesus monkey.
Fig. S2. A proliferation assay was used to demonstrate functional blocking by FR104. This was compared to proliferation in the presence of cytotoxic T lymphocyte antigen 4 immunoglobulin (CTLA‐4 Ig) and cyclosporin A (CsA). The data are presented as stimulation indices relative to medium alone. (a) Proliferation was measured for allogeneic responses of four major histocompatibility complex (MHC) class II mismatched peripheral blood mononuclear cell (PBMC) cultures in the presence of medium alone, FR104, CTLA‐4 Ig and CsA (r1 a r2 = PBMC of rhesus monkey 1 responds to irradiated PBMC of rhesus monkey 2). (b) CII‐specific proliferation was measured for two animals that were immunized with CII (rhesus 1, 2) in the presence of medium alone, FR104, CTLA‐4 Ig and CsA.
Fig. S3. FR104 serum levels during the experiment. (a) FR104 levels were measured after the first dosing and (b) during the entire evaluation period through levels of FR104 were measured before dosing.
Fig. S4. Dot‐plots showing the cell gating strategy for flow cytometry analysis of regulatory T cell population.
Table S1. CII‐specific proliferation presented as counts per minute and calculated stimulation index. Phorbol myristate acetate (PMA)/I was taken along as a positive control.
Acknowledgements
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007‐2013) under grant agreement no. 281493: Tolerance Restoration In Autoimmune Diseases (TRIAD) by selective manipulation of the CD28 co‐stimulatory pathway.
Disclosure
B. V., N. P. and C. M. are shareholders or employees of Effimune, a biotech company developing FR104. The other authors declare that they have no disclosures.
Author contributions
M. V. designed the CIA study and was responsible for the data acquisition and data interpretation of the in‐vitro experiments and in‐vivo efficacy study and drafted and revised the manuscript. E. B. assisted in the execution of the CIA study and managed the day‐to‐day logistics of the experiment. Y. K. established the cross reactivity profile. B. H. participated in the CIA study design and in writing the manuscript. B. V. participated in the CIA study design, data interpretation, revised the manuscript and funded the study. C. M. and N. P. participated in data interpretation and manuscript revision and performed ELISA and anti‐drug antibody assays.
References
- 1. Bankhurst AD, Husby G, Williams RC Jr. Predominance of T cells in the lymphocytic infiltrates of synovial tissues in rheumatoid arthritis. Arthritis Rheum 1976; 19:555–62. [DOI] [PubMed] [Google Scholar]
- 2. Kremer JM. Toward a better understanding of methotrexate. Arthritis Rheum 2004; 50:1370–82. [DOI] [PubMed] [Google Scholar]
- 3. Bakker NP, Van Besouw N, Groenestein R, Jonker M, Hart LA. The anti‐arthritic and immunosuppressive effects of cyclosporin A on collagen‐induced arthritis in the rhesus monkey. Clin Exp Immunol 1993; 93:318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Landewe RB, Goei The HS, van Rijthoven AW, Breedveld FC, Dijkmans BA. A randomized, double‐blind, 24‐week controlled study of low‐dose cyclosporine versus chloroquine for early rheumatoid arthritis. Arthritis Rheum 1994; 37:637–43. [DOI] [PubMed] [Google Scholar]
- 5. Cohen S, Cannon GW, Schiff M et al Two‐year, blinded, randomized, controlled trial of treatment of active rheumatoid arthritis with leflunomide compared with methotrexate. Utilization of leflunomide in the Treatment of Rheumatoid Arthritis Trial Investigator Group. Arthritis Rheum 2001; 44:1984–92. [DOI] [PubMed] [Google Scholar]
- 6. Smolen JS, Kalden JR, Scott DL et al Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: a double‐blind, randomised, multicentre trial. European Leflunomide Study Group. Lancet 1999; 353:259–66. [DOI] [PubMed] [Google Scholar]
- 7. Kremer JM, Westhovens R, Leon M et al Treatment of rheumatoid arthritis by selective inhibition of T‐cell activation with fusion protein CTLA4Ig. N Engl J Med 2003; 349:1907–15. [DOI] [PubMed] [Google Scholar]
- 8. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987; 30:1205–13. [DOI] [PubMed] [Google Scholar]
- 9. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996; 14:233–58. [DOI] [PubMed] [Google Scholar]
- 10. Yao S, Zhu Y, Zhu G et al B7‐h2 is a costimulatory ligand for CD28 in human. Immunity 2011; 34:729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol 2010; 22:333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCoy KD, Le Gros G. The role of CTLA‐4 in the regulation of T cell immune responses. Immunol Cell Biol 1999; 77:1–10. [DOI] [PubMed] [Google Scholar]
- 13. Freeman GJ, Long AJ, Iwai Y et al Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death‐1 ligand 1 interacts specifically with the B7‐1 costimulatory molecule to inhibit T cell responses. Immunity 2007; 27:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poirier N, Blancho G, Vanhove B. CD28‐specific immunomodulating antibodies: what can be learned from experimental models? Am J Transplant 2012; 12:1682–90. [DOI] [PubMed] [Google Scholar]
- 16. Cho ML, Yoon CH, Hwang SY et al Effector function of type II collagen‐stimulated T cells from rheumatoid arthritis patients: cross‐talk between T cells and synovial fibroblasts. Arthritis Rheum 2004; 50:776–84. [DOI] [PubMed] [Google Scholar]
- 17. Kim WU, Cho ML, Jung YO et al Type II collagen autoimmunity in rheumatoid arthritis. Am J Med Sci 2004; 327:202–11. [DOI] [PubMed] [Google Scholar]
- 18. Londei M, Savill CM, Verhoef A et al Persistence of collagen type II‐specific T‐cell clones in the synovial membrane of a patient with rheumatoid arthritis. Proc Natl Acad Sci USA 1989; 86:636–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klareskog L, Ronnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol 2008; 26:651–75. [DOI] [PubMed] [Google Scholar]
- 20. Shi J, Knevel R, Suwannalai P et al Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci USA 2011; 108:17372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clague RB, Shaw MJ, Holt PJ. Incidence and correlation between serum IgG and IgM antibodies to native type II collagen in patients with chronic inflammatory arthritis. Ann Rheum Dis 1981; 40:6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klareskog L, Johnell O, Hulth A, Holmdahl R, Rubin K. Reactivity of monoclonal anti‐type II collagen antibodies with cartilage and synovial tissue in rheumatoid arthritis and osteoarthritis. Arthritis Rheum 1986; 29:730–8. [DOI] [PubMed] [Google Scholar]
- 23. Raza K, Mullazehi M, Salmon M, Buckley CD, Ronnelid J. Anti‐collagen type II antibodies in patients with very early synovitis. Ann Rheum Dis 2008; 67:1354–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lindh I, Snir O, Lonnblom E et al Type II collagen antibody response is enriched in the synovial fluid of rheumatoid joints and directed to the same major epitopes as in collagen induced arthritis in primates and mice. Arthritis Res Ther 2014; 16:R143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silver PB, Hathcock KS, Chan CC, Wiggert B, Caspi RR. Blockade of costimulation through B7/CD28 inhibits experimental autoimmune uveoretinitis, but does not induce long‐term tolerance. J Immunol 2000; 165:5041–7. [DOI] [PubMed] [Google Scholar]
- 26. Perrin PJ, June CH, Maldonado JH, Ratts RB, Racke MK. Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J Immunol 1999; 163:1704–10. [PubMed] [Google Scholar]
- 27. Lenschow DJ, Zeng Y, Thistlethwaite JR et al Long‐term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science 1992; 257:789–92. [DOI] [PubMed] [Google Scholar]
- 28. Kirk AD, Harlan DM, Armstrong NN et al CTLA4‐Ig and anti‐CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA 1997; 94:8789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bakker NP, van Erck MG, Otting N et al Resistance to collagen‐induced arthritis in a nonhuman primate species maps to the major histocompatibility complex class I region. J Exp Med 1992; 175:933–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Otting N, Heijmans CM, Noort RC et al Unparalleled complexity of the MHC class I region in rhesus macaques. Proc Natl Acad Sci USA 2005; 102:1626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doxiadis GG, de Groot N, Otting N et al Haplotype diversity generated by ancient recombination‐like events in the MHC of Indian rhesus macaques. Immunogenetics 2013; 65:569–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vierboom MPM, Breedveld E, 't Hart BA. Preclinical evaluation of anti‐rheumatic drugs in a non‐human primate model of arthritic disease. Drug Discov Today Dis Models 2008; 5:89–95. [Google Scholar]
- 33. Woo J, Vierboom MP, Kwon H et al PDL241, a novel humanized monoclonal antibody, reveals CD319 as a therapeutic target for rheumatoid arthritis. Arthritis Res Ther 2013; 15:R207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pettit AR, Ji H, von Stechow D et al TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol 2001; 159:1689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vierboom MP, Breedveld E, Kondova I, 't Hart BA. Collagen‐induced arthritis in common marmosets: a new nonhuman primate model for chronic arthritis. Arthritis Res Ther 2010; 12:R200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vierboom MP, Jonker M, Bontrop RE, 't Hart B. Modeling human arthritic diseases in nonhuman primates. Arthritis Res Ther 2005; 7:145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Poirier N, Mary C, Le Bas‐Bernardet S et al Advantages of Papio anubis for preclinical testing of immunotoxicity of candidate therapeutic antagonist antibodies targeting CD28. MAbs 2014; 6:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Poirier N, Dilek N, Mary C et al FR104, an antagonist anti‐CD28 monovalent fab' antibody, prevents alloimmunization and allows calcineurin inhibitor minimization in nonhuman primate renal allograft. Am J Transplant 2015; 15:88–100. [DOI] [PubMed] [Google Scholar]
- 39. Haanstra KG, Dijkman K, Bashir N et al Selective blockade of CD28‐mediated T cell costimulation protects rhesus monkeys against acute fatal experimental autoimmune encephalomyelitis. J Immunol 2015; 194:1454–66. [DOI] [PubMed] [Google Scholar]
- 40. Bakker NP, van Erck MG, Botman CA, Jonker M, 't Hart BA. Collagen‐induced arthritis in an outbred group of rhesus monkeys comprising responder and nonresponder animals. Relationship between the course of arthritis and collagen‐specific immunity. Arthritis Rheum 1991; 34:616–24. [DOI] [PubMed] [Google Scholar]
- 41. Kraan MC, Versendaal H, Jonker M et al Asymptomatic synovitis precedes clinically manifest arthritis. Arthritis Rheum 1998; 41:1481–8. [DOI] [PubMed] [Google Scholar]
- 42. 't Hart BA, Bank RA, Brok H et al Collagen‐induced arthritis in rhesus monkeys: evaluation of markers for inflammation and joint degradation. Br J Rheumatol 1998; 37:314–23. [DOI] [PubMed] [Google Scholar]
- 43. Friedline RH, Brown DS, Nguyen H et al CD4+ regulatory T cells require CTLA‐4 for the maintenance of systemic tolerance. J Exp Med 2009; 206:421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA‐4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci USA 2010; 107:1524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Poirier N, Azimzadeh AM, Zhang T et al Inducing CTLA‐4‐dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci Transl Med 2010; 2:17ra0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dilek N, Poirier N, Hulin P et al Targeting CD28, CTLA‐4 and PD‐L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T‐cells. PLOS ONE 2013; 8:e83139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choy EH, Kavanaugh AF, Jones SA. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol 2013; 9:154–63. [DOI] [PubMed] [Google Scholar]
- 48. Deppong CM, Bricker TL, Rannals BD, Van Rooijen N, Hsieh CS, Green JM. CTLA4Ig inhibits effector T cells through regulatory T cells and TGF‐beta. J Immunol 2013; 191:3082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. 't Hart BA, Bakker NP, Jonker M, Bontrop RE. Resistance to collagen‐induced arthritis in rats and rhesus monkeys after immunization with attenuated type II collagen. Eur J Immunol 1993; 23:1588–94. [DOI] [PubMed] [Google Scholar]
- 50. Jasin HE, Noyori K, Takagi T, Taurog JD. Characteristics of anti‐type II collagen antibody binding to articular cartilage. Arthritis Rheum 1993; 36:651–9. [DOI] [PubMed] [Google Scholar]
- 51. Mihara M, Tan I, Chuzhin Y et al CTLA4Ig inhibits T cell‐dependent B‐cell maturation in murine systemic lupus erythematosus. J Clin Invest 2000; 106:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Dot‐plots showing the cell gating strategy for flow cytometry analysis of whole blood from the rhesus monkey.
Fig. S2. A proliferation assay was used to demonstrate functional blocking by FR104. This was compared to proliferation in the presence of cytotoxic T lymphocyte antigen 4 immunoglobulin (CTLA‐4 Ig) and cyclosporin A (CsA). The data are presented as stimulation indices relative to medium alone. (a) Proliferation was measured for allogeneic responses of four major histocompatibility complex (MHC) class II mismatched peripheral blood mononuclear cell (PBMC) cultures in the presence of medium alone, FR104, CTLA‐4 Ig and CsA (r1 a r2 = PBMC of rhesus monkey 1 responds to irradiated PBMC of rhesus monkey 2). (b) CII‐specific proliferation was measured for two animals that were immunized with CII (rhesus 1, 2) in the presence of medium alone, FR104, CTLA‐4 Ig and CsA.
Fig. S3. FR104 serum levels during the experiment. (a) FR104 levels were measured after the first dosing and (b) during the entire evaluation period through levels of FR104 were measured before dosing.
Fig. S4. Dot‐plots showing the cell gating strategy for flow cytometry analysis of regulatory T cell population.
Table S1. CII‐specific proliferation presented as counts per minute and calculated stimulation index. Phorbol myristate acetate (PMA)/I was taken along as a positive control.