

Summary
Atherosclerosis is an autoimmune inflammatory disease involving both innate and adaptive immune mechanisms. Immune tolerance induction may have therapeutic potential for the suppression of atherosclerosis. Current interest is directed towards mucosal tolerance induction, especially nasal tolerance. Previous studies have shown that heat shock protein 60 (HSP60) is recognized as an important autoantigen in atherosclerosis, and nasal or oral HSP60 can induce tolerance and ameliorate atherosclerosis by inducing several subsets of regulatory T cells (Tregs) such as latency‐associated peptide (LAP)+ and forkhead box transcription factor 3 (FoxP3)+ Tregs. However, little is known regarding the detailed mechanisms of nasal tolerance. Here, we again investigated the impact of nasal HSP60 on atherosclerosis and the mechanisms underlying the anti‐atherosclerosis responses. We found that nasal HSP60 caused a significant 33·6% reduction in plaque size at the aortic root in the early stages of atherosclerosis (P < 0·001). Notably, a significant increase in activated CD4+CD25+ glycoprotein A repetitions predominant (GARP)+ Tregs, type 1 Tregs (Tr1 cells), and CD4+CD25+FoxP3+ Tregs, as well as a marked decrease in the numbers of type 1 and 17 T helper cells was detected in the spleens and cervical lymph nodes of HSP60‐treated mice. Moreover, nasal HSP60 increases the production of transforming growth factor (TGF)‐β and interleukin (IL)‐10 and decreases the secretion of IFN‐γ and IL‐17. Interestingly, the atheroprotective role of nasal HSP60 treatment was abrogated partly by the neutralization of IL‐10. Our findings show that nasal administration of HSP60 can attenuate atherosclerotic formation by inducing GARP+ Tregs, Tr1 cells and FoxP3+ Tregs, and that these Tregs maintain immune homeostasis by secreting IL‐10 and TGF‐β.
Keywords: nasal tolerance, GARP, atherosclerosis, Regulatory T cells, inflammation
Introduction
The forkhead box transcription factor P3 (FoxP3) is particularly expressed in CD4+CD25+ Tregs, which are necessary and sufficient for the active suppression of immune responses against autoantigens and is essential for Treg development and function 1, 2, 3. However, several lines of evidence suggest that FoxP3 alone is not able to completely establish a stable regulatory phenotype, and its status as a genuine Treg marker has been questioned 4, 5, 6. Recently, glycoprotein‐A repetitions predominant (GARP or LRRC32), a transmembrane protein with a large extracellular domain that contains 20 leucine‐rich repeats, was found to be expressed specifically on the surface of activated human Tregs and to play a crucial role in Treg suppression 7, 8. Importantly, GARP has been identified as a receptor for latency‐associated peptide (LAP), which requires transforming growth factor (TGF)‐β for binding to GARP 9, 10. Furthermore, a positive feedback loop between GARP and FoxP3 is present in human Tregs 11. Emerging experimental evidence suggests that several subsets of Tregs, including FoxP3+ Tregs, LAP+ Tregs and type 1 Tregs (Tr1 cells), can inhibit autoimmunity in animal models of inflammatory and autoimmune diseases 12, 13, 14, particularly atherosclerosis 15, 16, through the down‐regulation of activated T cell responses. In a recent study, we found that the frequency and function of circulating CD4+CD25+GARP+ Tregs were down‐regulated in patients with acute coronary syndrome, suggesting that GARP may promote the protective function of Tregs in atherosclerotic disease 17.
The autoimmune concept of atherosclerosis was first presented by Wick et al. in 1992, and has since been proven in a large number of experimental and clinical studies 18; according to this idea, cells of the innate and adaptive immune system, especially activated CD4+ T cells, may be involved in the initiation and progression of atherosclerosis 19, 20, 21. HSP60, a member of the highly conserved heat shock protein (HSP) family, can be considered an important antigenic determinant of autoantibodies that participates in several autoimmune diseases, in particular atherosclerosis 22, 23, 24. Previous studies have shown enhanced HSP60 expression in human atherosclerotic plaques 25. Recently, Xu et al. and Zhang et al. reported that elevated titres of HSP60‐specific antibodies can be found in the serum of patients with atherosclerosis 26, 27. Additionally, activated CD4+ T cells that react specifically to self‐HSP60 peptides have been detected in carotid plaques of atherosclerosis patients, suggesting that HSP60‐reactive CD4+ T cells contribute to the autoimmune reactions in atherosclerosis 28. This autoimmune process may be related to an existing deficit in immunological tolerance and dampened by activated Tregs. Accumulating evidence has shown that mucosal administration of autoantigens such as HSPs, oxidized low‐density lipoprotein (ox‐LDL) and glycoproteins could induce immune tolerance, resulting in increased numbers of several subsets of Tregs and decreased atherosclerotic plaque area 28, 29, 30, 31, 32, 33.
Currently, two principal routes of mucosal tolerance induction are known: oral and nasal. Several previous studies have demonstrated that administering low doses of antigens orally induces many types of antigen‐specific Tregs, including CD4+CD25+FoxP3+ Tregs, T helper type 3 (Th3) cells, Tr1 cells and CD4+LAP+ Tregs, indicating that oral tolerance induction is an effective method for treating autoimmune diseases such as atherosclerotic disease 15, 33, 34. Simultaneously, nasal tolerance induction has been shown to be as effective as immune tolerance induced via the oral route in inhibiting autoimmune diseases; furthermore, the nasal route requires substantially lower doses of antigen than the oral route 35, 36. Recently, we found that nasal administration of low doses of ox‐LDL inhibits the development of atherosclerosis through a significant increase in CD4+CD25+FoxP3+ and CD4+LAP+ Tregs, which can suppress immune responses due to types 1, 2, and 17 T helper cells 33. In addition, our experimental groups also indicated that CD4+CD25+FoxP3+ and CD4+LAP+ Tregs induced by nasal HSP60 may be relevant to the suppression of atherosclerosis 32. However, as yet it remains uncertain whether nasal administration of HSP60 could induce other subsets of Tregs, such as CD4+CD25+GARP+ Tregs and Tr1 cells.
In the present study, we again show that nasal administration of HSP60 significantly attenuated early atherosclerosis in apolipoprotein E (ApoE)−/− mice and was associated with the induction of several types of Treg populations, including CD4+CD25+GARP+ Tregs, Tr1 cells and CD4+CD25+FoxP3+ Tregs, which suppressed T helper type 1 (Th1) and Th17 cell immune responses.
Methods
Animals
Six‐week‐old male ApoE−/− mice from a C57BL/6J background (Jackson Laboratory, Bar Harbor, ME, USA) were bred and maintained in the Animal Center at Beijing University. The mice were kept in a specific pathogen‐free facility (Tongji Medical College, Wuhan, China) and were given a western‐type diet containing 0·15% cholesterol and 21% fat. The mice were fed a normal chow diet during nasal HSP60 or phosphate‐buffered saline (PBS) treatment. All experiments were performed according to the guidelines for the Care and Use of Laboratory Animals (Science and Technology Department of Hubei Province, China, 2005). The protocol was approved by the Animal Care and Use Committee of Hubei Province.
Experimental groupings
Six‐ and 16‐week‐old male mice were nasally administered 15 μl of PBS (pH 7·4) or 3 μg of HSP60 dissolved in 15 μl PBS by a micropipette once daily for 5 consecutive days (n = 6 for each group). At 16 and 24 weeks of age, the mice were killed by cervical dislocation and the atherosclerotic plaques were evaluated. For the interleukin (IL)‐10 blocking experiment, 6‐week‐old mice (n = 6) were administered 3 μg of HSP60 nasally once daily for 5 consecutive days. Subsequently, these mice were injected intraperitoneally once weekly with 100 μg of neutralizing anti‐interleukin (IL)‐10 antibody from 7 to 16 weeks of age 33, 37, after which atherosclerotic plaques were evaluated. Control mice (n = 6) were treated with equal amounts of rat immunoglobulin (Ig)G.
Reagents and antibodies
Recombinant Helicobacter pylori HSP60 was obtained from Shanghai Linc‐Bio Science Co. Ltd (Shanghai, China). Anti‐CD4‐fluorescein isothiocyanate (FITC) monoclonal antibody (mAb) (Cat. no.11‐0041), anti‐CD25‐phycoerythrin (PE) mAb (Cat. no. 12‐0251), anti‐FoxP3‐allophycocyanin (APC) mAb (Cat. no. 88‐8118), anti‐interferon (IFN)‐γ‐PE mAb (Cat no. 12‐7311), anti‐IL‐4‐PE mAb (Cat. no. 12‐7041), anti‐IL‐17A‐PE mAb (Cat. no. 12‐7177), anti‐CD3 mAb (Cat no. 14‐0032), anti‐CD28 mAb (Cat. no. 14‐0281), anti‐GARP‐APC mAb (Cat. no. 17‐9891), anti‐IgG1‐APC mAb (Cat. no. 17‐4714) and anti‐IgG1‐PE mAb (Cat. no. 12‐4714) were purchased from eBioscience (San Diego, CA, USA). Anti‐IL‐10Rβ‐APC mAb (Cat. no. FAB5368A), anti‐IFN‐γR1‐PE mAb (Cat. no. FAB1026P), anti‐IL‐10 mAb (Cat. no. MAB417) and control rat IgG (Cat. no. 6‐001‐A) were obtained from R&D Systems (Minneapolis, MN, USA).
Serum total cholesterol levels
After clotting at room temperature, serum was obtained from experimental and control group mice via centrifugation at 1200 g for 10 min and stored at −80°C. Total cholesterol levels were measured as described in detail previously 33.
Atherosclerotic plaque analysis
Mice were killed at 16 and 24 weeks of age, and the aortic root plaques were assessed. After removal of the blood residue, the aortas were quickly dissected and removed from the higher region of the aortic sinus to the lower region of the left and right iliac artery. In addition, we retained the heart with the aortic sinus, as well as spleens and cervical lymph nodes (CLNs), which were routinely considered the nose‐draining lymphatic nodes 33, 38. For plaque analysis, the hearts with the aortic root were embedded in optimal cutting temperature (OCT) compound for frozen tissue sectioning. Six consecutive cryosections (10‐μm thickness) with three aortic valves were obtained from the aortic root of each mouse and stained with oil red O and haematoxylin for lipid visualization. All slices were collected on a Leica CM 1850 Cryostat (Leica Microsystems, Wetzlar, Germany). Plaque area was calculated using Image‐Pro Plus version 6·0 software (Media Cybernetics, Rockville, MD, USA).
Flow cytometry analysis
At the days 4 and 14 after the final nasal administration, splenocytes and CLN cells were harvested from HSP60‐treated mice (n = 6 for each group). Untreated mice (n = 6) were used as controls. The monocytes were suspended and cultured as described previously 33. For GARP induction, the cells (2 × 106/sample) were stimulated with soluble anti‐CD3 mAb (1·5 μg/ml) and anti‐CD28 mAb (2 μg/ml) at 37°C with 5% CO2 for 24 h, and the cells were subsequently harvested for staining.
Tregs were analysed as follows: (1) for the detection of CD4+CD25+GARP+ Tregs, the cells were stained with anti‐GARP‐APC, anti‐CD4‐FITC and anti‐CD25‐PE mAb, as described previously 17; (2) for the detection of CD4+ IFN‐γR+ IL‐10R+ Tregs (Tr1), the cells were stained with anti‐IFN‐γR1‐PE, anti‐IL‐10Rβ‐APC and anti‐CD4‐FITC mAb as described previously 39; and (3) for the detection of CD4+CD25+FoxP3+ Tregs, the cells were stained as described previously 33.
For the analysis of CD4+IFN‐γ+ (Th1), CD4+IL‐4+ (Th2) and CD4+IL‐17+ (Th17) T cells, the cells were stimulated with phorbol myristate acetate (PMA, 20 ng/ml) plus ionomycin (1 μg/ml; Alexis Biochemicals, San Diego, CA, USA) for 4 h in the presence of 2 μmol/ml monensin (Alexis Biochemicals). The incubator was set at 37°C under a 5% CO2 environment. After 4 h of culture, the monocytes were collected for staining with anti‐CD4‐FITC mAb according to the manufacturer's instructions. Fixation and permeabilization were necessary before staining with anti‐IFN‐γ‐PE, anti‐IL‐4‐PE or anti‐IL‐17‐PE mAb.
Isotype‐specific controls were given to enable correct compensation and to validate antibody specificity. All stained cells were analysed immediately by flow cytometry using a fluorescence activated cell sorter (FACS)Calibur (BD Biosciences), and all data were analysed using the FlowJo 7·6·1 software program (TreeStar Inc., Ashland, OR, USA).
Responder T cells and Treg co‐culture
On day 14 after the last nasal administration, CD4+ T cells were isolated from pooled splenocytes of HSP60‐ or PBS‐treated mice using a CD4+ T cell‐positive isolation Kit II (Miltenyi Biotec, Gladbach, Germany). The purified CD4+ T cells were stained with anti‐GARP‐APC, anti‐CD4‐FITC and anti‐CD25‐PE mAb for 30 min at 4°C. Responder T cells (Tresps, CD4+CD25−GARP− T cells) and CD4+CD25+GARP+ Tregs were isolated by FACS. The purity of the sorted population was > 93% using FACS analysis. The Tregs and Tresps were divided into two groups: (1) CD4+CD25+GARP+ Tregs (1 × 104 cells/well) were stimulated with concanavalin A (ConA) in vitro for 72 h, and the supernatants were collected for detecting the anti‐inflammatory cytokines IL‐10 and TGF‐β1; (2) purified CD4+CD25+GARP+ Tregs were co‐cultured with CD4+CD25−GARP− T cells (5 × 103 cells/well) at different ratios (Tregs/Tresps ratios: 1 : 1, 1 : 2, 1 : 4 and 1 : 8) as described previously 17. The inhibitory effects were measured using an MTT assay. MTT (20 μl of 5 mg/ml) was added to each well 4 h prior to harvest. The supernatant in each well was removed, and 150 μl of DMSO was added and stirred for 10 min. The absorbance (A) values were determined at 570 nm on a microplate reader. Dulbecco's modified Eagle's medium (DMEM)‐F12 culture medium was used as blank control in the normal control group and for zero adjustment of the microplate reader. All measurements were performed in triplicate. The rate of cell proliferation was calculated as follows: cell proliferation rate = (A value in test group – A value in normal control group)/A value in normal control group × 100%.
Enzyme‐linked immunosorbent assay (ELISA)
At the days 4 and 14 and weeks 8 and 16 after the last administration, splenocytes were isolated and cultured with ConA. Untreated or PBS‐treated mice were used as controls. The IFN‐γ, IL‐4, IL‐17, TGF‐β1 and IL‐10 levels in the supernatants were quantified using ELISA kits (eBioscience). In addition, as mentioned above, the levels of IL‐10 and TGF‐β1 in the CD4+CD25+GARP+ Treg culture supernatant were measured by ELISA. All procedures were conducted following the manufacturer's instructions. All samples were measured in duplicate.
Real‐time reverse transcription–polymerase chain reaction (RT–PCR)
Total RNA from splenocytes and the descending aortas was prepared using RNAiso Plus (Takara Biotechnology, Shiga, Japan). cDNA was transcribed from purified RNA using a RNA PCR kit (Takara Biotechnology). Real‐time PCR was performed as described previously 33. The primers are indicated in Table 1.
Table 1.
Sequences of primers for real‐time reverse transcription–polymerase chain reaction (RT–PCR)
| Molecule | Sequence (5'–3') |
|---|---|
| IFN‐γ sense | CGGCACAGTCATTGAAAGCCTA |
| IFN‐γ anti‐sense | GTTGCTGATGGCCTGATTGTC |
| IL‐4 sense | TCTCGAATGTACCAGGAGCCATATC |
| IL‐4 anti‐sense | AGCACCTTGGAAGCCCTACAGA |
| IL‐10 sense | GACCAGCTGGACAACATACTGCTAA |
| IL‐10 anti‐sense | GATAAGGCTTGGCAACCCAAGTAA |
| IL‐17 sense | TCTCTGATGCTGTTGCTGCT |
| IL‐17 anti‐sense | CGTGGAACGGTTGAGGTAGT |
| TGF‐β sense | GTGTGGAGCAACATGTGGAACTCTA |
| TGF‐β anti‐sense | TTGGTTCAGCCACTGCCGTA |
| CD25 sense | CTGATCCCATGTGCCAGGAA |
| CD25 anti‐sense | AGGGCTTTGAATGTGGCATTG |
| Foxp3 sense | CTCATGATAGTGCCTGTGTCCTCAA |
| Foxp3 anti‐sense | AGGGCCAGCATAGGTGCAAG |
| GARP sense | ACCTCCACAGCAATGTCCTC |
| GARP anti‐sense | TGCTGTTGCAGCTCAAGTCT |
| VCAM‐1 sense | CCTCACTTGCAGCACTACGGGCT |
| VCAM‐1 anti‐sense | TTTTCCAATATCCTCAATGACGGG |
| MCP‐1 sense | TTCCTCCACCACCATGCAG |
| MCP‐1 anti‐sense | CCAGCCGGCAACTGTGA |
| GAPDH sense | ACCACAGTCCATGCCATCAC |
| GAPDH anti‐sense | TCCACCACCCTGTTGCTGTA |
Statistical analysis
All statistical analyses were performed using spss version 17·0. The results are expressed as the mean ± standard deviation (s.d.). One‐way analysis of variance (anova) was used for multiple comparisons, followed by the Newman–Keuls test. Differences were considered significant at P < 0·05.
Results
Nasal HSP60 inhibits early atherosclerosis in ApoE−/− mice
To determine the influence of nasal induction of HSP60 tolerance on atherosclerosis initiation, atherosclerosis was induced after the nasal administration of PBS (group I) or HSP60 (group II) for 5 consecutive days to ApoE−/− mice. Mice were placed subsequently on a western‐type diet for 10 weeks, and then atherosclerotic plaque formation was analysed in oil red O and haematoxylin‐stained cryosections of the aortic root. Interestingly, the atherosclerotic plaque size in the aortic root was reduced significantly (33·6% reduction) in mice treated with HSP60 compared with PBS‐treated mice (272 645·6 ± 30 346·6 μm2 versus 410 687.7 ± 38 708·4 μm2, respectively; P < 0·001; Fig. 1). In addition, the influence of nasal HSP60 on atherosclerosis progression was evaluated. Six‐week‐old ApoE−/− mice were fed a western‐type diet for 10 weeks to induce atherosclerotic plaque formation in the aortic root. Then, these mice were administered nasally with PBS (group III) or HSP60 (group IV) for 5 consecutive days and subsequently fed a western‐type diet for another 7 weeks. At 24 weeks, the mice were euthanized and atherosclerotic plaques were calculated. However, only a mild but not statistically significant reduction was found in group IV compared with group III (Supporting information, Fig. S1a,b,d). To develop an effective approach for inhibiting atherosclerosis progression, we carried out another animal experiment. On the basis of group IV, mice were treated nasally with 3 μg of HSP60 (group V) once weekly from 18 to 24 weeks and then euthanized. Unfortunately, only a trend of an 11·3% decrease in atherosclerotic plaque size was found in group V compared with group III, and no significant differences were observed (Supporting information, Fig.S1c,b,d). As expected, nasal HSP60 did not change body weight or total serum cholesterol levels significantly, in agreement with our previous findings (Supporting information, Table S1) 32, 33.
Figure 1.
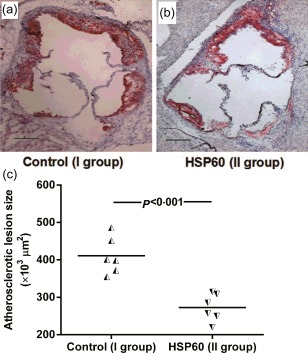
Nasal heat shock protein (HSP)60 inhibits atherosclerotic lesion formation. (a) and (b) representative photomicrographs of oil red O‐ and haematoxylin‐stained aortic root sections from the phosphate‐buffered saline (PBS)‐treated group (n = 6) and the HSP60‐treated group (n = 6), respectively. (c) The data from two groups are shown: the half black regular triangles represent animals from the PBS‐treated group, and the half black inverted triangles represent animals from the HSP60‐treated group. A black bar represents 200 μm. The horizontal bars represent the means.
Nasal HSP60 induces CD4+CD25+GARP+ Tregs in spleens and CLNs
We investigated whether nasal HSP60 atheroprotection was dependent on the induction of CD4+CD25+GARP+ Tregs, which were detected on days 4 and 14 after the final nasal treatment, consistent with our previous studies 32, 33. As shown in Fig. 2, we found that the frequency of CD4+CD25+GARP+ Tregs present in untreated mice was 0·77 ± 0·12% in spleens and 1·07 ± 0·14% in CLNs. Surprisingly, the frequency of CD4+CD25+GARP+ Tregs increased markedly to 1·37 ± 0·14% in the spleens and 2·90 ± 0·32% in the CLNs of HSP60‐treated mice on day 4 and to 2·28 ± 0.20% in spleens and 2·07 ± 0·22% in CLNs on day 14 when compared with untreated mice (Fig. 2f,h,j,k; all P < 0·05). Furthermore, absolute numbers of CD4+CD25+GARP+ Tregs were also increased in the spleens and CLNs of HSP60‐treated mice (Fig. 2l, all P < 0·05). Increasing evidence has implicated GARP as a specific marker of activated Tregs 8, 9, 10. Therefore, we next measured the frequency of activated CD4+CD25+GARP+ Tregs after 24 h of T cell receptor (TCR) stimulation with anti‐CD3 mAb and anti‐CD28 mAb, which were used to activate immunocytes because of its high specificity and properties of physiological stimulation. Our data showed that the frequency of activated CD4+CD25+GARP+ Tregs was significantly higher than observed under resting conditions in all the groups examined. Interestingly, there was a significant increase in the frequency of activated CD4+CD25+GARP+ Tregs in the spleens and CLNs of HSP60‐treated mice on day 4 (HSP60‐treated mice versus untreated mice; 6·08 ± 0·54% versus 4·01 ± 0·38% in spleens, P < 0·05; 13·37 ± 1·39% versus 5·90 ± 0·40% in CLNs, P < 0·05; Fig. 2g, i–k) and day 14 compared with untreated mice (HSP60‐treated mice versus untreated mice; 9·74 ± 0·92% versus 4·01 ± 0·38% in spleens, P < 0·05; 8·89 ± 0·97% versus 5·90 ± 0·40% in CLNs, P < 0·05; Fig. 2g, i–k).
Figure 2.
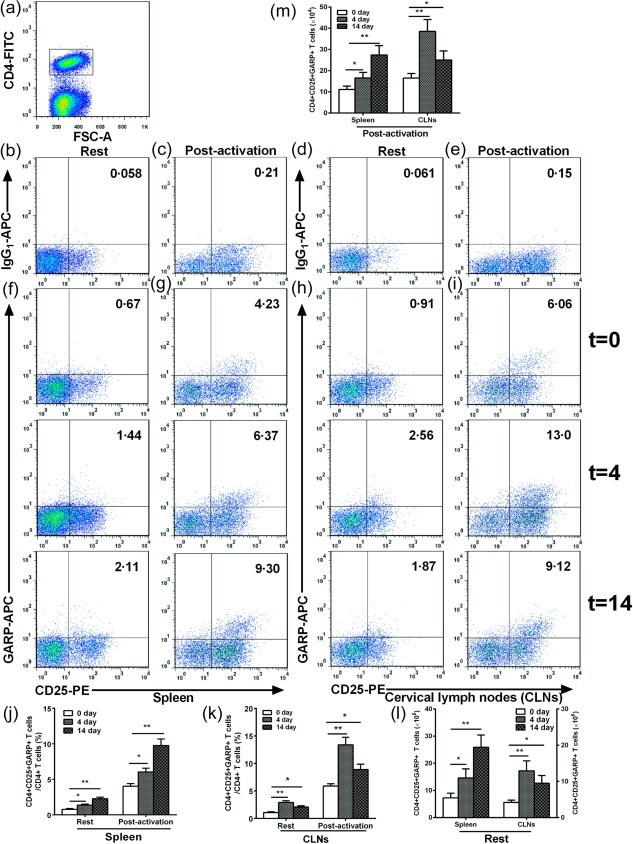
Effects of nasal heat shock protein (HSP)60 on CD4+CD25+GARP+ regulatory T cells (Tregs) in spleens and cervical lymph nodes (CLNs). Apolipoprotein E (ApoE)−/− mice were nasally administered HSP60 for 5 days and were killed 4 and 14 days after the final nasal treatment. Untreated mice (t = 0) were considered controls. For glycoprotein A repetitions predominant (GARP) activation, the cells (2 × 106/sample) were stimulated with soluble anti‐CD3 monoclonal antibody (mAb) (1·5 μg/ml) and anti‐CD28 mAb (2 μg/ml) at 37°C with 5% CO2 for 24 h. (a) CD4+ T cell subsets were gated. (b–e) Representative results for isotype controls in spleens and CLNs estimated by fluorescence activated cell sorter (FACS) analysis. (f–i) Representative results for unstimulated and stimulated CD4+CD25+GARP+ Tregs in spleens and CLNs estimated by FACS analysis. (j,k) The graphs represent the percentage of unstimulated and stimulated CD4+CD25+GARP+ Tregs in spleens and CLNs. (l,m) The graphs represent the absolute number of unstimulated and stimulated CD4+CD25+GARP+ Tregs in spleens and CLNs. *P < 0·05, **P < 0·01. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments.
Furthermore, we also investigated whether nasal HSP60 affects the suppressive function of activated CD4+CD25+GARP+ Tregs. Our data indicated that the suppressive function of activated CD4+CD25+GARP+ Tregs was not remarkably altered by nasal HSP60 using in‐vitro suppression assays (Supporting information, Fig. S2). In addition, we examined anti‐inflammatory cytokine production from CD4+CD25+GARP+ Tregs in response to ConA, which was a polyclonal stimulant and exhibited a more powerful effect than anti‐CD3 mAb and anti‐CD28 mAb. The most exciting implications are that a significant up‐regulation of TGF‐β secreted by CD4+CD25+GARP+ Tregs was observed in HSP60‐treated mice compared with PBS‐treated mice (147·7 ± 10·4 pg/ml versus 83·1 ± 10·4 pg/ml, P < 0·01; Fig. 3). Nevertheless, the levels of IL‐10 were similar in the supernatants of CD4+CD25+GARP+ Tregs between HSP60‐treated and PBS‐treated mice (Fig. 3).
Figure 3.
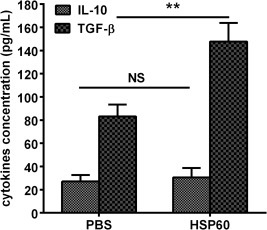
Effects of nasal heat shock protein (HSP)60 on transforming growth factor (TGF)‐β and interleukin (IL)‐10 production in CD4+CD25+ glycoprotein A repetitions predominant (GARP)+ regulatory T cells (Tregs). CD4+CD25+GARP+ Tregs isolated from spleens on the day 14 after the final nasal HSP60 and phosphate‐buffered saline (PBS) treatments were stimulated with concanavalin A (ConA) in vitro for 72 h. TGF‐β and IL‐10 production was detected in the supernatants. **P < 0·01. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments.
Nasal HSP60 induces Tr1 and CD4+CD25+FoxP3+ Tregs in spleens and CLNs
Next, we evaluated the effect of nasal induction of HSP60 tolerance on Tr1 and CD4+CD25+FoxP3+ Tregs. When compared with untreated mice (0·48 ± 0·05% in spleens, 0·88 ± 0·09% in CLNs; Fig. 4d), the frequency of Tr1 cells was up‐regulated significantly to 1·02 ± 0·16% and 2·01 ± 0·17% in spleens (all P < 0·01; Fig. 4f,h) and 2·48 ± 0·20% and 1·55 ± 0·14% in CLNs (all P < 0·01; Fig. 4g,h) on days 4 and 14 after the final nasal treatment, respectively. Interestingly, absolute numbers of Tr1 cells were also up‐regulated in the spleens and CLNs of HSP60‐treated mice, respectively (Fig. 4i, all P < 0·01). Similar to our previous report 32, 33, we also found a significant increase in the frequency and number of CD4+CD25+FoxP3+ Tregs in the spleens and CLNs of HSP60‐treated mice on days 4 and 14 compared to untreated mice, respectively (data not shown).
Figure 4.
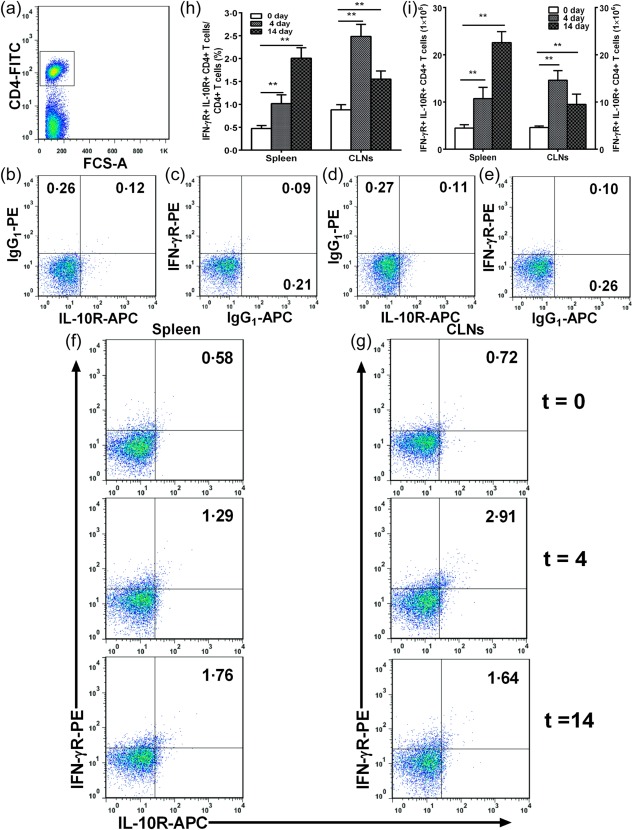
Effects of nasal heat shock protein (HSP)60 on CD4+interleukin (IL)−10R+interferon (IFN)‐γR+ regulatory T cells (Tregs) (Tr1) in spleens and cervical lymph nodes (CLNs). Apolipoprotein E (ApoE)−/− mice were administered nasally HSP60 for 5 days and were killed 4 and 14 days after the final nasal treatment. Untreated mice (t = 0) were considered controls. (a) CD4+ T cell subsets were gated. (b–e) Representative results for isotype controls in spleens and CLNs estimated by fluorescence activated cell sorter (FACS) analysis. (f,g) Representative results for type 1 Tregs (Tr1) cells in spleens and CLNs estimated by fluorescence activated cell sorter (FACS) analysis. (h) The graphs represent the percentage of Tr1 cells in spleens and CLNs. (i) The graphs represent the absolute number of Tr1 cells in spleens and CLNs. **P < 0·01. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments.
Nasal HSP60 suppresses Th1 and Th17 cells in spleens and CLNs
Furthermore, we examined whether nasal HSP60 affected Th1, Th2 and Th17 cells in spleens and CLNs. We observed a significant reduction in the frequency of Th1 cells in the spleens and CLNs of HSP60‐treated mice on days 4 and 14 compared with untreated mice (untreated mice versus HSP60‐treated mice on day 4 and versus HSP60‐treated mice on day 14; 14·4 ± 1·13% versus 10·9 ± 1·02% and versus 8·11 ± 0·74% in spleens, respectively, all P < 0·001; 13·5 ± 1·05% versus 8·56 ± 0·64% and versus 9·91 ± 0·76% in CLNs, respectively, all P < 0·01; Fig. 5e,h; Fig. 6e,h). Meanwhile, we also found that the frequency of Th17 cells in the spleens and CLNs of HSP60‐treated mice on days 4 and 14 was lower than in those of untreated mice (untreated mice versus HSP60‐treated mice on day 4 and versus HSP60‐treated mice on day 14; 1·68 ± 0·13% versus 1·1 ± 0·09% and versus 0·67 ± 0·08% in spleens, respectively, all P < 0·001; 1·36 ± 0·09% versus 0·69 ± 0·09% and versus 0·89 ± 0·08% in CLNs, respectively, all P < 0·001; Fig. 5g,h, Fig. 6g,h). Additionally, our data showed that absolute numbers of Th1 and Th17 were decreased in the spleens and CLNs of HSP60‐treated mice, respectively (Fig. 5i, Fig. 6i; all P < 0·01). Unexpectedly, there were no significant differences in the frequency and number of Th2 cells in spleens and CLNs of HSP60‐treated mice on days 4 and 14 compared with untreated mice (Fig. 5f,h,i, Fig. 6g–i).
Figure 5.
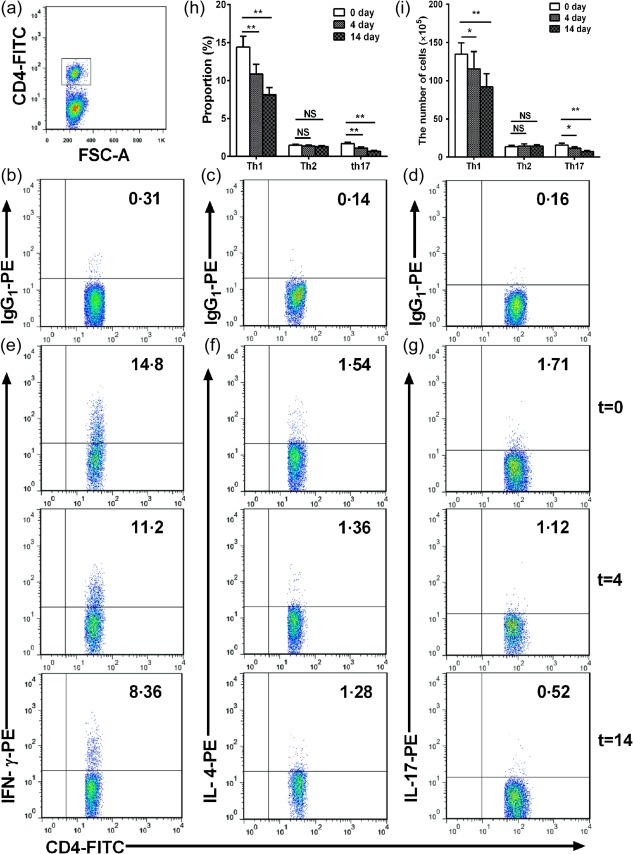
Effects of nasal heat shock protein (HSP)60 on T helper type 1 (Th1), Th2 and Th17 cells in spleens. Apolipoprotein E (ApoE)−/− mice were administered HSP60 nasally for 5 days and were killed 4 and 14 days after the final nasal treatment. Untreated mice (t = 0) were considered controls. (a) CD4+ T cell subsets were gated. (b,d) Representative results for isotype controls in spleens estimated by fluorescence activated cell sorter (FACS) analysis. (e–g) Representative results for Th1 cells, Th2 cells and Th17 cells, respectively, in spleens estimated by FACS analysis. (h) The graphs represent the percentage of Th1, Th2 and Th17 cells in spleens, respectively. (i) The graphs represent the absolute number of Th1, Th2 and Th17 cells in spleens, respectively. *P < 0·05, **P < 0·01, and n.s. P > 0·05. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments.
Figure 6.
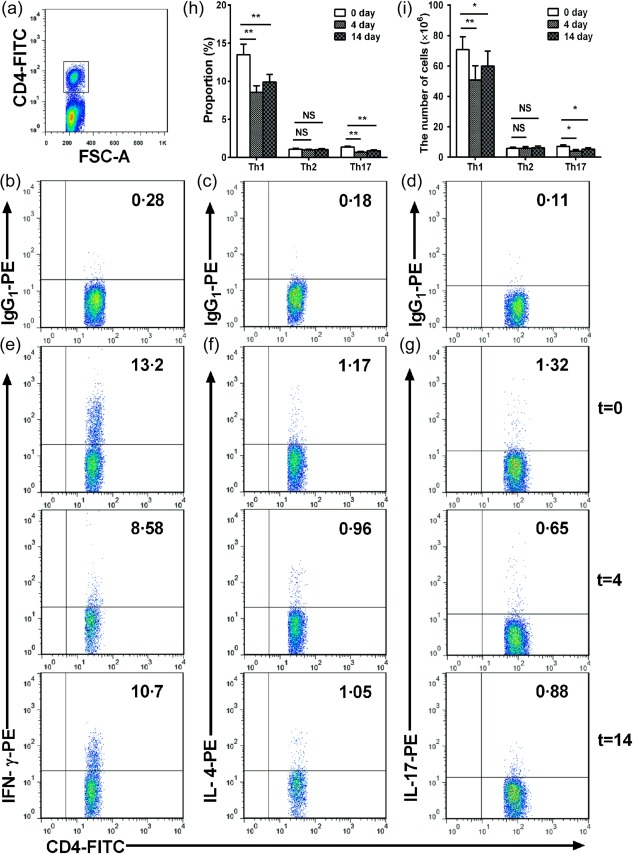
Effects of nasal heat shock protein (HSP)60 on T helper type 1 (Th1), Th2 and Th17 cells in cervical lymph nodes (CLNs). Apolipoprotein E (ApoE)−/− mice were administered HSP60 nasally for 5 days and were killed 4 and 14 days after the final nasal treatment. Untreated mice (t = 0) were considered controls. (a) CD4+ T cell subsets were gated. (b–d) Representative results for isotype controls in spleens estimated by fluorescence activated cell sorter (FACS) analysis. (e,g) Representative results for Th1 cells, Th2 cells and Th17 cells, respectively, in CLNs estimated by FACS analysis. (h), The graphs represent the percentage of Th1, Th2 and Th17 cells in CLNs, respectively. (i), The graphs represent the absolute number of Th1, Th2 and Th17 cells in CLNs, respectively. *P < 0·05, **P < 0·01 and n.s. P > 0·05. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments.
Nasal HSP60 increases TGF‐β and IL‐10 secretion and decreases IFN‐γ and IL‐17 production in splenocytes
To determine the effect of the various Tregs induced by nasal HSP60 on the cytokine production profile from lymphocytes, we examined the cytokine levels in the supernatants of ConA‐stimulated splenocytes on days 4 and 14 after the final nasal treatment using ELISA. There was a significant increase in TGF‐β levels and an obvious decrease in the levels of IFN‐γ and IL‐17 in HSP60‐treated mice on days 4 and 14 compared with untreated mice, respectively (Table 2, all P < 0·001). Intriguingly, when compared with untreated mice, a marked up‐regulation in IL‐10 production was observed in HSP60‐treated mice on day 14 (Table 2, P < 0·05), but not on day 4. Moreover, we also examined cytokine production by splenocytes at 16 and 24 weeks, and a significant increase in TGF‐β and IL‐10 levels and a marked decrease in IFN‐γ and IL‐17 production was found in group II compared with group I (Table 2, all P < 0·05). However, there were no significant differences in the levels of TGF‐β, IL‐10, IFN‐γ and IL‐17 between group III and groups IV or V (Table 2). Unfortunately, the levels of IL‐4 were below the threshold of detection of the assay in all experiments.
Table 2.
Cytokine production by concanavalin (ConA)‐stimulated splenocytes
| Days after treatment | ||||||||
|---|---|---|---|---|---|---|---|---|
| 8 weeks | 16 weeks | |||||||
| 0 day (n = 8) | 4 day (n = 6) | 14 day n = 7) | Group I(n = 6) | Group II(n = 6) | Group III(n = 6) | Group IV(n = 6) | Group V(n = 6) | |
| TGF‐β1 | 207·5 ± 19·5 | 380·3 ± 27·9* | 448·3 ± 32·2* | 192·8 ± 18·2 | 307·6 ± 26·1† | 174·6 ± 16·4 | 187·1 ± 19 | 203·1 ± 19·7 |
| IL‐10 | 45·1 ± 11·7 | 51 ± 10·9 | 65·6 ± 11·3‡ | 57·8 ± 9·9 | 81·1 ± 13·5§ | 73·7 ± 13·1 | 79·2 ± 16·1 | 85·1 ± 12·9 |
| IFN‐γ | 288·1 ± 19·9 | 267·5 ± 18·9* | 251·5 ± 17·8* | 318·5 ± 24·1 | 203·2 ± 19·4† | 371·8 ± 36·2 | 382·5 ± 35·2 | 353·2 ± 23·3 |
| IL‐4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IL‐17 | 29·6 ± 2·6 | 25·8 ± 1·7‡ | 20·9 ± 1·6* | 41·6 ± 6·4 | 30·1 ± 4·7§ | 45·3 ± 8·6 | 43·2 ± 6·7 | 39·6 ± 7·4 |
* P < 0·001 versus untreated mice;
† P < 0·001 versus group I
‡ P < 0·05 versus untreated mice
§ P < 0·05 versus Group I. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments. Effects of nasal heat shock protein (HSP)60 on cytokine production in splenocytes. Apolipoprotein E (ApoE)−/− mice were administered phosphate‐buffered saline (PBS) or HSP60 nasally for 5 days; untreated or PBS‐treated mice were considered controls. Splenocytes collected from mice that were killed 4 and 14 days after the final nasal treatment, and at 16 and 24 weeks of age were stimulated with ConA in vitro for 72 h. Interferon (IFN)‐γ, interleukin (IL)‐4, IL‐17, transforming growth factor (TGF)‐β and IL‐10 production in the supernatants was measured by enzyme‐linked immunosorbent assay (ELISA).
Next, we determined the levels of cytokine mRNA expression by unstimulated splenocytes using real‐time RT–PCR. Interestingly, a marked decrease in IFN‐γ and IL‐17 mRNA levels in HSP60‐treated mice on days 4 (Fig. 7a, P < 0·05) and 14 (Fig. 7a, P < 0·01) were found than in untreated mice. Of note, there was a significant increase in TGF‐β and IL‐10 mRNA levels on day 14 and in mRNA TGF‐β levels on day 4; however, the IL‐4 mRNA levels were unchanged. Similarly, we observed increased TGF‐β and IL‐10 mRNA levels and decreased IFN‐γ and IL‐17 mRNA levels in group II compared with group I (Fig. 7b, all P < 0·05). However, no differences were noticed in IL‐4 mRNA levels between groups I and II. Additionally, up‐regulated TGF‐β mRNA levels were observed in groups IV and V compared with group III (Fig. 7c, P < 0·05). Unexpectedly, IFN‐γ, IL‐4, IL‐17, and IL‐10 mRNA levels did not differ among groups III, IV and V.
Figure 7.
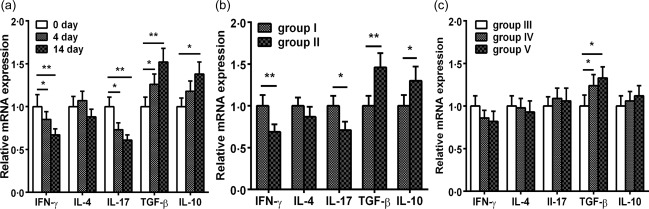
Effects of nasal heat shock protein (HSP)60 on the relative expression of cytokine mRNA in splenocytes. Apolipoprotein E (ApoE)−/− mice were administered phosphate‐buffered saline (PBS) or HSP60 nasally for 5 days; untreated or PBS‐treated mice were considered controls. (a–c) Relative cytokine mRNA levels of interferon (IFN)‐γ, interleukin (IL)‐4, IL‐17, transforming growth factor (TGF)‐β and IL‐10 in unstimulated splenocytes collected from mice that were killed 4 and 14 days after the final nasal treatment, and at 16 and 24 weeks of age were measured by real‐time reverse transcription–polymerase chain reaction (RT–PCR). Fold change relative to PBS‐treated mice is shown. *P < 0·05, **P < 0·01. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments.
The mRNA expression of Treg markers and inflammatory markers in atherosclerotic lesions
To reveal whether nasal HSP60 affects the accumulation of Tregs and inflammatory cells in atherosclerotic lesions, we performed real‐time RT–PCR analysis of TGF‐β, CD25, FoxP3, IFN‐γ, GARP and the adhesion molecules vascular cell adhesion protein 1 (VCAM‐1) and monocyte chemotactic protein 1 (MCP‐1) in thoracic aortas. Our data showed that TGF‐β and FoxP3 mRNA expression were increased markedly, and the expression of IFN‐γ, VCAM‐1 and MCP‐1 mRNA was reduced significantly in atherosclerotic lesions in group II compared with group I (Fig. 8a, all P < 0·05). Unexpectedly, CD25 and GARP mRNA expression was similar in groups I and II. Notably, TGF‐β and FoxP3 mRNA expression were increased markedly in group V compared with group III (Fig. 8b, P < 0·05). In accordance with expectation, the expression levels of TGF‐β and FoxP3 mRNA were unchanged in group IV compared with group III (Fig. 8b). Additionally, we found that the mRNA expression levels of the other cytokines did not differ significantly among groups III, IV and V (Fig. 8b).
Figure 8.
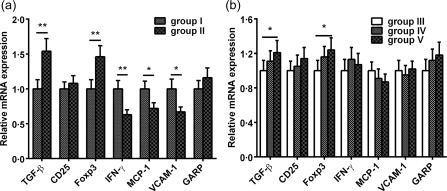
Effects of nasal heat shock protein (HSP)60 on the relative mRNA expression of regulatory T cell markers and inflammatory markers in atherosclerotic plaques. (a,b) Total RNA was extracted from the aortas of 16‐ and 24‐week‐old phosphate‐buffered saline (PBS)‐ or HSP60‐treated mice. mRNA expression of regulatory T cell markers [transforming growth factor (TGF)‐β, CD25, forkhead box transcription factor (FoxP3) and glycoprotein A repetitions predominant (GARP)] and inflammatory markers interferon (IFN)‐γ, monocyte chemotactic protein (MCP)‐1, vascular cell adhesion molecule (VCAM)‐1 was determined quantitatively by real‐time reverse transcription–polymerase chain reaction (RT–PCR). Fold‐change relative to PBS‐treated mice is shown. *P < 0·05, **P < 0·01. The data are expressed as the mean ± standard error of the mean (s.e.m.) and are representative of at least three independent experiments.
Neutralizing anti‐IL‐10 antibody partly blocks the atheroprotective effect of nasal HSP60
Our finding showed that nasal HSP60 increased TGF‐β and IL‐10 production significantly. Our previous study found that a neutralizing anti‐TGF‐β‐antibody increased atherosclerotic lesion formation significantly in HSP60‐treated mice, suggesting that the atheroprotective effect of nasal HSP60 is partly dependent upon TGF‐β 32. However, whether IL‐10 participates directly in inhibiting lesion formation following nasal HSP60 treatment remains uncertain. Therefore, an in‐vivo IL‐10 neutralization study was carried out using an anti‐IL‐10 antibody. As mentioned above, HSP60 was administered nasally to 6‐week‐old mice before the injection of neutralizing anti‐IL‐10‐antibody (group VI) or control rat IgG (group VII). Our data revealed a significant increase (24·8%) in atherosclerotic lesion formation in group VI compared with group VII (Fig. 9; 353 437·2 ± 33 609·9 μm2 versus 265 816·4 ± 26 705·3 μm2; P = 0·003), indicating that inhibiting IL‐10 markedly increased atherosclerotic plaque formation.
Figure 9.
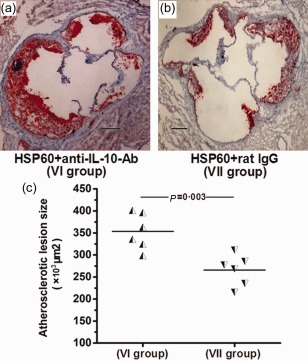
Anti‐atherosclerotic effect is partly interleukin (IL)‐10 dependent. (a,b) Representative photomicrographs of oil red O‐ and haematoxylin‐stained aortic root sections from the heat shock protein (HSP)60+ anti‐IL‐10‐antibody‐treated group (VI, n = 6) and the HSP60+ rat immunoglobulin (Ig)G‐treated group (VII, n = 6). (c) The data from two groups are shown: the half black regular triangles represent animals from group VI and the half black inverted triangles represent animals from group VII. A black bar represents 200 μm. The horizontal bars represent the means.
Discussion
Emerging clinical and experimental facts have demonstrated that HSP60 plays an important role in atherosclerosis initiation and progression. Elevated plasma levels of soluble HSP60 and anti‐HSP60 autoantibodies have been found in atherosclerotic patients and experimental atherosclerotic mice 26, 27, 40. Autologous HSP60 can induce the generation of autoreactive HSP60‐specific T cells and anti‐HSP60 autoantibodies in ApoE−/− or LDLr−/− mice 41. Furthermore, anti‐HSP60 autoantibodies contribute to the development of atherosclerosis by accelerating endothelial damage and macrophage lysis 42, 43. In addition, HSP60‐specific T cells have been observed in human atherosclerotic plaques and in atherosclerotic lesions in ApoE−/− mice, suggesting that HSP60‐specific T cells are associated with early atherosclerotic events and terminating the HSP60‐specific autoimmune response is known to be protective in atherosclerosis 28, 44, 45.
Previous studies have demonstrated clearly that the mucosal administration of autoantigens can be used to induce peripheral antigen‐specific immune tolerance and to down‐regulate antigen‐specific T cells, eventually inhibiting excessive immune responses and helping to maintain self‐tolerance to prevent autoimmune diseases 29, 30, 31, 32, 33, 36. Activated CD4+ T cells that are reactive to several antigens, including HSP60 and ox‐LDL, mainly secrete a Th1‐pattern of cytokines in atherosclerotic lesions, leading to an imbalance between Th1 and the Tregs specific for self‐ and non‐self‐antigens 46. Restoring the balance between Th1 cells and Tregs may be effective in ameliorating atherosclerosis, suggesting that Tregs play a crucial role in controlling the initiation and progression of atherosclerosis 47. Substantial studies have shown that HSP60 delivered via the oral and nasal route can induce immune tolerance successfully and increase the number of Tregs, which inhibit effector T cell responses by producing anti‐inflammatory cytokines and attenuate the development of atherosclerosis 29, 32. The experimental results from our laboratory have revealed that the adoptive transfer of HSP60‐specific Tregs to recombination‐activating gene (RAG)−/− low‐density lipoprotein receptor (LDLr)−/− mice can prevent the development of atherosclerosis 48. Consequently, the above research suggested that the induction of mucosal tolerance and the subsequent activation of Tregs may be a novel and promising treatment approach for attenuating atherosclerosis.
In the present study, we again found that nasal tolerance induction to a low dose of HSP60 suppressed early atherosclerosis significantly. This result is in line with previous studies from our laboratory and others, showing that nasal or oral tolerance induction to HSP60/65, β2‐glycoprotein I and ox‐LDL can reduce early atherosclerotic lesion formation 29, 30, 31, 32, 33, 34, 49. In addition, this result is also supported by several lines of clinical observations, which indicate a more prominent role of HSP60‐specific cellular immunity in the early stages of atherosclerosis 45, 50. However, our results are in agreement with studies showing that mucosal tolerance to self‐antigens is not expected to inhibit advanced atherosclerosis 29, 31. Hence, we attempted to perform another experiment in group V, as mentioned above. Disappointingly, our data showed that the enhanced immunization once weekly led to only a mild but not significant reduction in lesion size during the progression of atherosclerosis. Nevertheless, our previous study demonstrated that an appropriate enhanced immune response to ox‐LDL can contribute to attenuating atherosclerosis progression 33. This discrepancy may be explained by the different types of antigens used in the above two studies. Therefore, further studies are required to identify a more effective method for reducing atherosclerosis progression via nasal HSP60.
It is well known that natural CD4+CD25+FoxP3+ Tregs, which are the main population of Treg subsets, exert suppressive effects in atherogenesis in mice 29, 30, 47. In addition to naturally occurring Tregs, however, recent reports have shown that other types of Tregs, including Tr1, Th3 and CD4+LAP+ T cells, may be responsible for anti‐atherosclerotic effects through the production of anti‐inflammatory cytokines such as IL‐10 and TGF‐β 32, 33, 34. Recently, GARP regulates the bioavailability and activation of TGF‐β by binding to LAP on the cell surface 9, 10, 51. Importantly, Wang et al. have demonstrated that αVβ6 and αVβ8 integrins are favoured to activate TGF‐β from the GARP–proTGF‐β1 complex, and cell‐surface GARP contributed to this activation, suggesting that the GARP‐proTGF‐β1 complex could serve as a source of activated TGF‐β1 52. Kalathil et al. has demonstrated recently that the number of GARP+FoxP3+ Tregs was increased significantly in patients with advanced hepatocellular carcinoma, and GARP expression was utilized to identify antigen‐specific Tregs with high suppressive potential 53. Moreover, our clinical investigations have shown that the frequency and function of GARP+ Tregs were impaired in patients with acute coronary syndrome (ACS) 17, 54, 55. These clinical findings suggested that GARP+ Tregs may be involved in immune modulation of autoimmune and inflammatory diseases by producing immunosuppressive cytokines such as TGF‐β. In the present study, our findings showed that nasal HSP60 increased the number of CD4+CD25+GARP+ Tregs significantly, in particular activated CD4+CD25+GARP+ Tregs, in spleens and CLNs on 4 days after nasal HSP60 treatment. More importantly, the influence lasted for at least 14 days. Although the suppressive function of CD4+CD25+GARP+ Tregs remained unchanged nasal HSP60 led to a significant increase in TGF‐β, which was secreted by CD4+CD25+GARP+ Tregs in the spleens. This result is also supported by our clinical observations, which indicated that the TGF‐β1 levels in the supernatants of cultured GARP+ Tregs were reduced in ACS patients 17. Similarly, we found a significant up‐regulation of CD4+CD25+FoxP3+ Tregs in the spleens and CLNs of HSP60‐treated mice. To the best of our knowledge, this is the first report demonstrating a possible role for CD4+CD25+GARP+ Tregs in the development of atherosclerosis in mice; however, further studies are required to offer more direct experimental support for the association between increased CD4+CD25+GARP+ Tregs number or function and decreased atherosclerosis. In addition, whether or not CD4+CD25+GARP+ Tregs can represent a new regulatory CD4+ T cell phenotype that differs from other Treg subtypes remains to be investigated.
Several studies have indicated that IL‐10‐secreting Tr1 cells have immunosuppressive properties, and these cells have been shown to play key roles in the development of Th1‐mediated autoimmune diseases 56, 57. A decreased frequency of IL‐10‐producing Tr1 cells was observed in the inflamed synovium and peripheral blood of patients with rheumatoid arthritis, suggesting that a deficiency in Tr1 cells may play an important role in the loss of self‐tolerance in autoimmune diseases 14, 58. Recently, Mallet et al. showed that adoptive transfer of Tr1 cells to ApoE−/− mice suppressed pathogenic Th1‐mediated responses and reduced the development of atherosclerosis 16. Interestingly, several studies have demonstrated that oral administration of self‐antigens induces Tr1 cells that can inhibit multiple sclerosis and diabetes in animal models 59. However, whether nasal administration of autoantigens can induce Tr1 cells is not entirely clear. In the current study, we demonstrated that nasal administration of HSP60 can induce a marked increase in Tr1 cells in spleens and CLNs. This result is consistent with a study by Kilngenberg et al., who reported that intranasal immunization with ApoB‐100 attenuated atherosclerotic plaque formation by inducing an increased number of IL‐10‐producing Tr1 cells 34. As the same time, we also found a significant decrease in Th1 and Th17 cells in the spleens and CLNs of HSP60‐treated mice. These results demonstrated that nasal HSP60 can induce a shift from T helper cells towards Tregs in spleens and CLNs.
A substantial body of evidence indicates that TGF‐β and IL‐10 suppress autoimmune and inflammatory diseases, and IFN‐γ and IL‐17 are detrimental to many immunoinflammatory diseases 16, 61, 62. TGF‐β maintains the suppressor function of Tregs and inhibits the proliferation, activation and differentiation of CD4+ T cells towards Th1, Th2, and Th17 in the periphery 63. Previous studies have demonstrated that TGF‐β or IL‐10 deficiency increases atherosclerotic lesion formation in ApoE−/− mice 64, 65. In this study, our data showed that nasal HSP60 increases TGF‐β and IL‐10 production and decreases the secretion of IFN‐γ and IL‐17 in splenocytes. Simultaneously, we also observed a significant up‐regulation of TGF‐β and IL‐10 mRNA levels and a marked down‐regulation of IFN‐γ and IL‐17 mRNA levels in splenocytes in HSP60‐treated mice. In addition, increased TGF‐β and FoxP3 mRNA expression and decreased IFN‐γ, VCAM‐1 and MCP‐1 mRNA expression were found in early lesions in HSP60‐treated mice. Quite serendipitously, we did not observe a significant increase in GARP mRNA expression in lesions in HSP60‐treated mice, possibly because there are other latent TGF‐β‐binding proteins in the plaques. Although IL‐4 levels were unchanged in all groups, the differential effects of other inflammatory cytokines that were implicated previously in atherogenesis were again confirmed.
We and others have demonstrated previously that neutralizing anti‐TGF‐β antibodies reverse the suppressive function of Tregs in animal models of several autoimmune and inflammatory diseases, especially atherosclerosis 15, 32, 33, 66. In this study, in addition to a significant increase in TGF‐β‐producing Tregs, a noticeable up‐regulation of IL‐10‐secreting Tr1 cells was also observed in HSP60‐treated mice. It remains unknown, however, whether anti‐IL‐10 antibody treatment abrogates the suppressive effect of Tregs. We therefore performed a neutralizing anti‐IL‐10 antibody study and found that anti‐IL‐10 antibody treatment partly abolished the anti‐atherosclerotic effects of nasal HSP60. This result is in concordance with several earlier studies describing that the suppressive effect of Tr1 cell clones on CD4+ T cells was abrogated by neutralizing anti‐IL‐10 monoclonal antibodies 67, 68. Although we did not rule out the possible beneficial effect of nasal HSP60 on the humoral immune response due to a lack of detection of HSP60‐specific antibodies, our study results support the possibility that nasal HSP60 inhibits atherosclerosis through an enhanced cellular immune response, including the induction of several types of Tregs and the production of TGF‐β and IL‐10.
In conclusion, we demonstrated that nasal tolerance induction to HSP60 inhibits early atherosclerotic lesion formation by inducing CD4+CD25+GARP+ Tregs, Tr1 cells and CD4+CD25+FoxP3+ Tregs, which produce large amounts of TGF‐β and IL‐10. Although further work is necessary to investigate the mechanism by which nasal HSP60 treatment increases Tregs and by which GARP regulates the activation of TGF‐β, our results again confirm the fact that the induction of Tregs by nasal tolerance induction to HSP60 could serve as a promising therapeutic method for treating atherosclerotic diseases.
Disclosure
The authors have no disclosures.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Nasal heat shock protein (HSP)60 fails to inhibit atherosclerotic lesion progression. (a–c) Representative photomicrographs of oil red O‐ and haematoxylin‐stained aortic root sections from the control‐treated group (III, n = 6), the HSP60‐treated group (IV, n = 6) and the HSP60‐treated enhancement group (V, n = 6). (d) The data from three groups are shown: the half black regular triangles, the half black inverted triangles and the black inverted triangles represent animals from groups III, IV and V. A black bar represents 200 μm. The horizontal bars represent the means.
Fig. S2. Effects of nasal heat shock protein (HSP)60 on the suppressive function of CD4+CD25+ glycoprotein A repetitions predominant (GARP)+ regulatory T cells (Tregs). CD4+CD25+GARP+ Tregs and CD4+CD25–GARP– T cells were isolated from spleens 14 days after the final nasal HSP60 treatment. CD4+CD25+GARP+ Tregs from phosphate‐buffered saline (PBS)‐treated or HSP60‐treated mice were co‐cultured with CD4+CD25–GARP– T cells at different ratios in the presence of soluble anti‐CD3‐antibody and anti‐CD28‐antibody. Cell proliferation was assessed using the MTT assay. The rate of cell proliferation was calculated as follows: cell proliferation rate = (A value in test group – A value in normal control group)/A value in normal control group × 100%. The data are representative of at least three independent experiments.
Table S1. Apolipoprotein E (ApoE)−/− mice were nasally administered phosphate‐buffered saline (PBS) or heat shock protein (HSP)60, as described in the Methods section. The data are expressed as the mean ± standard error of the mean (s.e.m.).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81070237, 81270354, and 81300213)
References
- 1. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4:330–6. [DOI] [PubMed] [Google Scholar]
- 2. Williams LM, Rudensky AY. Maintenance of the Foxp3‐dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol 2007; 8:277–84. [DOI] [PubMed] [Google Scholar]
- 3. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30:531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allan SE, Crome SQ, Crellin NK et al Activation‐induced Foxp3 in human t effector cells does not suppress proliferation or cytokine production. Int Immunol 2007; 19:345–54. [DOI] [PubMed] [Google Scholar]
- 5. Wang J, Ioan‐Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of Foxp3 in human activated nonregulatory CD4+ T cells. Eur J Immunol 2007; 37:129–38. [DOI] [PubMed] [Google Scholar]
- 6. Roncarolo MG, Gregori S. Is FOXP3 a bona fide marker for human regulatory T cells? Eur J Immunol 2008; 38:925–7. [DOI] [PubMed] [Google Scholar]
- 7. Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLOS ONE 2008; 3:e2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci USA 2009; 106:13439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (lrrc32) is essential for the surface expression of latent TGF‐beta on platelets and activated Foxp3+ regulatory T cells. Proc Natl Acad Sci USA 2009; 106:13445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF‐beta on the surface of activated human Treg. Eur J Immunol 2009; 39:3315–22. [DOI] [PubMed] [Google Scholar]
- 11. Probst‐Kepper M, Geffers R, Kroger A et al GARP: a key receptor controlling foxp3 in human regulatory T cells. J Cell Mol Med 2009; 13:3343–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stephens LA, Gray D, Anderton SM. CD4+CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor cross reactivity. Proc Natl Acad Sci USA 2005; 102:17418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu HY, Center EM, Tsokos GC, Weiner HL. Suppression of murine SLE by oral anti‐CD3: inducible CD4+CD25‐LAP+ regulatory T cells control the expansion of IL‐17+ follicular helper T cells. Lupus 2009; 18:586–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yudoh K, Matsuno H, Nakazawa F, Yonezawa T, Kimura T. Reduced expression of the regulatory CD4+ T cell subset is related to Th1/Th2 balance and disease severity in rheumatoid arthritis. Arthritis Rheum 2000; 43:617–27. [DOI] [PubMed] [Google Scholar]
- 15. Sasaki N, Yamashita T, Takeda M et al Oral anti‐CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation 2009; 120:1996–2005. [DOI] [PubMed] [Google Scholar]
- 16. Mallat Z, Gojova A, Brun V et al Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E‐knockout mice. Circulation 2003; 108:1232–7. [DOI] [PubMed] [Google Scholar]
- 17. Meng K, Zhang W, Zhong Y et al Impairment of circulating CD4+CD25+GARP+ regulatory T cells in patients with acute coronary syndrome. Cell Physiol Biochem 2014; 33:621–32. [DOI] [PubMed] [Google Scholar]
- 18. Wick G, Kleindienst R, Dietrich H, Xu Q. Is atherosclerosis an autoimmune disease? Trends Food Sci Technol 1992; 3:114–9. [Google Scholar]
- 19. Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol 2009; 31:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol 2009; 27:165–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wick G, Jakic B, Buszko M, Wick MC, Grundtman C. The role of heat shock proteins in atherosclerosis. Nat Rev Cardiol 2014; 11:516–29. [DOI] [PubMed] [Google Scholar]
- 22. Alard JE, Dueymes M, Youinou P, Jamin C. Modulation of endothelial cell damage by anti‐Hsp60 autoantibodies in systemic autoimmune diseases. Autoimmun Rev 2007; 6:438–43. [DOI] [PubMed] [Google Scholar]
- 23. Cappello F, Conway de Macario E, Di Felice V, Zummo G, Macario AJ. Chlamydia trachomatis infection and anti‐Hsp60 immunity: the two sides of the coin. PLOS Pathog 2009; 5:e1000552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jang EJ, Jung KY, Hwang E, Jang YJ. Characterization of human anti‐heat shock protein 60 monoclonal autoantibody Fab fragments in atherosclerosis: genetic and functional analysis. Mol Immunol 2013; 54:338–46. [DOI] [PubMed] [Google Scholar]
- 25. Kleindienst R, Xu Q, Willeit J, Waldenberger FR, Weimann S, Wick G. Immunology of atherosclerosis. Demonstration of heat shock protein 60 expression and T lymphocytes bearing alpha/beta or gamma/delta receptor in human atherosclerotic lesions. Am J Pathol 1993; 142:1927–37. [PMC free article] [PubMed] [Google Scholar]
- 26. Xu Q, Schett G, Perschinka H et al Serum soluble heat shock protein 60 is elevated in subjects with atherosclerosis in a general population. Circulation 2000; 102:14–20. [DOI] [PubMed] [Google Scholar]
- 27. Zhang X, He M, Cheng L et al Elevated heat shock protein 60 levels are associated with higher risk of coronary heart disease in Chinese. Circulation 2008; 118:2687–93. [DOI] [PubMed] [Google Scholar]
- 28. Almanzar G, Öllinger R, Leuenberger J et al Autoreactive HSP60 epitope‐specific T‐cells in early human atherosclerotic lesions. J Autoimmun 2012; 39:441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Puijvelde GH, van Es T, van Wanrooij EJ et al Induction of oral tolerance to HSP60 or an HSP60‐peptide activates T cell regulation and reduces atherosclerosis. Arterioscler Thromb Vasc Biol 2007; 27:2677–83. [DOI] [PubMed] [Google Scholar]
- 30. van Puijvelde GH, Hauer AD, de Vos P et al Induction of oral tolerance to oxidized low‐density lipoprotein ameliorates atherosclerosis. Circulation 2006; 114:1968–76. [DOI] [PubMed] [Google Scholar]
- 31. George J, Yacov N, Breitbart E et al Suppression of early atherosclerosis in LDL‐receptor deficient mice by oral tolerance with beta 2‐glycoprotein I. Cardiovasc Res 2004; 62:603–9. [DOI] [PubMed] [Google Scholar]
- 32. Li H, Ding Y, Yi G, Zeng Q, Yang W. Establishment of nasal tolerance to heat shock protein‐60 alleviates atherosclerosis by inducing TGF‐β‐dependent regulatory T cells. J Huazhong Univ Sci Technolog Med Sci 2012; 32:24–30. [DOI] [PubMed] [Google Scholar]
- 33. Zhong Y, Wang X, Ji Q et al CD4+LAP+ and CD4+CD25+Foxp3+ regulatory T cells induced by nasal oxidized low‐density lipoprotein suppress effector T cells response and attenuate atherosclerosis in ApoE−/− mice. J Clin Immunol 2012; 32:1104–17. [DOI] [PubMed] [Google Scholar]
- 34. Klingenberg R, Lebens M, Hermansson A et al Intranasal immunization with an apolipoprotein B‐100 fusion protein induces antigen‐specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol 2010; 30:946–52. [DOI] [PubMed] [Google Scholar]
- 35. Ma CG, Zhang GX, Xiao BG, Link J, Olsson T, Link H. Suppression of experimental autoimmune myasthenia gravis by nasal administration of acetylcholine receptor. J Neuroimmunol 1995; 58:51–60. [DOI] [PubMed] [Google Scholar]
- 36. Maron R, Sukhova G, Faria AM et al Mucosal administration of heat shock protein‐65 decreases atherosclerosis and inflammation in aortic arch of low‐density lipoprotein receptor‐deficient mice. Circulation 2002; 106:1708–15. [DOI] [PubMed] [Google Scholar]
- 37. Lu H, Wagner WM, Gad E et al Treatment failure of a TLR‐7 agonist occurs due to self‐regulation of acute inflammation and can be overcome by IL‐10 blockade. J Immunol 2010; 184:5360–7. [DOI] [PubMed] [Google Scholar]
- 38. Wolvers DA, Coenen‐de Roo CJ, Mebius RE et al Intranasally induced immunological tolerance is determined by Characteristics of the draining lymph nodes: studies with OVA and human cartilage gp‐39. J Immunol 1999; 162:1994–8. [PubMed] [Google Scholar]
- 39. Ding Q, Lu L, Wang B et al B7H1‐Ig fusion protein activates the CD4+ IFN‐gamma receptor+ type 1 T regulatory subset through IFN‐gamma‐secreting Th1 cells. J Immunol 2006; 177:3606–14. [DOI] [PubMed] [Google Scholar]
- 40. Mandal K, Foteinos G, Jahangiri M, Xu Q. Role of antiheat shock protein 60 autoantibodies in atherosclerosis. Lupus 2005; 14:742–6. [DOI] [PubMed] [Google Scholar]
- 41. Grundtman C, Kreutmayer SB, Almanzar G, Wick MC, Wick G. Heat shock protein 60 and immune inflammatory responses in atherosclerosis. Arterioscler Thromb Vasc Biol 2011; 31:960–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Foteinos G, Afzal AR, Mandal K, Jahangiri M, Xu Q. Anti‐heat shock protein 60 autoantibodies induce atherosclerosis in apolipoprotein E‐deficient mice via endothelial damage. Circulation 2005; 112:1206–13. [DOI] [PubMed] [Google Scholar]
- 43. Schett G, Metzler B, Mayr M et al Macrophage‐lysis mediated by autoantibodies to heat shock protein 65/60. Atherosclerosis 1997; 128:27–38. [DOI] [PubMed] [Google Scholar]
- 44. Curry AJ, Portig I, Goodall JC, Kirkpatrick PJ, Gaston JS. T lymphocyte lines isolated from atheromatous plaque contain cells capable of responding to Chlamydia antigens. Clin Exp Immunol 2000; 121:261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knoflach M, Kiechl S, Mayrl B et al T‐cell reactivity against HSP60 relates to early but not advanced atherosclerosis. Atherosclerosis 2007; 195:333–8. [DOI] [PubMed] [Google Scholar]
- 46. Mallat Z, Ait‐Oufella H, Tedgui A. Regulatory T cell responses: potential role in the control of atherosclerosis. Curr Opin Lipidol 2005; 16:518–24. [DOI] [PubMed] [Google Scholar]
- 47. Ait‐Oufella H, Salomon BL, Potteaux S et al Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med 2006; 12:178–80. [DOI] [PubMed] [Google Scholar]
- 48. Yang K, Li D, Luo M, Hu Y. Generation of HSP60‐specific regulatory T cell and effect on atherosclerosis. Cell Immunol 2006; 243:90–5. [DOI] [PubMed] [Google Scholar]
- 49. Xiong Q, Li J, Jin L, Liu J, Li T. Nasal immunization with heat shock protein 65 attenuates atherosclerosis and reduces serum lipids in cholesterol‐fed wild‐type rabbits probably through different mechanisms. Immunol Lett 2009; 125:40–5. [DOI] [PubMed] [Google Scholar]
- 50. Knoflach M, Kiechl S, Penz D et al Cardiovascular risk factors and atherosclerosis in young women: atherosclerosis risk factors in female youngsters (ARFY study). Stroke 2009; 40:1063–9. [DOI] [PubMed] [Google Scholar]
- 51. Gauthy E, Cuende J, Stockis J et al GARP is regulated by miRNAs and controls latent TGF‐β1 production by human regulatory T cells. PLOS ONE 2013; 8:e76186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGF‐β. Mol Biol Cell 2012; 23:1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+) CTLA‐4(+)Foxp3(+) T regulatory cells and myeloid‐derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T‐cell functionality. Cancer Res 2013; 73:2435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhu ZF, Meng K, Zhong YC et al Impaired circulating CD4+ LAP+ regulatory T cells in patients with acute coronary syndrome and its mechanistic study. PLOS ONE 2014; 9:e88775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu Y, Zhao X, Zhong Y et al Heme oxygenase‐1 restores impaired GARPCD4+CD25+ regulatory T cells from patients with acute coronary syndrome by upregulating LAP and GARP expression on activated T lymphocytes. Cell Physiol Biochem 2015; 35:553–70. [DOI] [PubMed] [Google Scholar]
- 56. Groux H, O'Garra A, Bigler M et al A CD4+ T‐cell subset inhibits antigen‐specific T‐cell responses and prevents colitis. Nature 1997; 389:737–42. [DOI] [PubMed] [Google Scholar]
- 57. Lou W, Wang C, Wang Y, Han D, Zhang L. Enhancement of the frequency and function of IL‐10‐secreting type I T regulatory cells after 1 year of cluster allergen‐specific immunotherapy. Int Arch Allergy Immunol 2012; 159:391–8. [DOI] [PubMed] [Google Scholar]
- 58. Han D, Wang C, Lou W, Gu Y, Wang Y, Zhang L. Allergen‐specific IL‐10‐secreting type I T regulatory cells, but not CD4(+)CD25(+)Foxp3(+) T cells, are decreased in peripheral blood of patients with persistent allergic rhinitis. Clin Immunol 2010; 136:292–301. [DOI] [PubMed] [Google Scholar]
- 59. Battaglia M, Gianfrani C, Gregori S, Roncarolo MG. IL‐10‐producing T regulatory type 1 cells and oral tolerance. Ann NY Acad Sci 2004; 1029:142–53. [DOI] [PubMed] [Google Scholar]
- 60. Bettini M, Vignali DA. Regulatory T cells and inhibitory cytokines in autoimmunity. Curr Opin Immunol 2009; 21:612–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shabgah AG, Fattahi E, Shahneh FZ. Interleukin‐17 in human inflammatory diseases. Postepy Dermatol Alergol 2014; 31:256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rizza P, Moretti F, Belardelli F. Recent advances on the immunomodulatory effects of IFN‐alpha: implications for cancer immunotherapy and autoimmunity. Autoimmunity 2010; 43:204–9. [DOI] [PubMed] [Google Scholar]
- 63. Mantel PY, Schmidt‐Weber CB. Transforming growth factor‐beta: recent advances on its role in immune tolerance. Methods Mol Biol 2011; 677:303–38. [DOI] [PubMed] [Google Scholar]
- 64. Mallat Z, Gojova A, Marchiol‐Fournigault C et al Inhibition of transforming growth factor‐beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res 2001; 89:930–4. [DOI] [PubMed] [Google Scholar]
- 65. Caligiuri G, Rudling M, Ollivier V et al Interleukin‐10 deficiency increases atherosclerosis, thrombosis, and low‐density lipoproteins in apolipoprotein E knockout mice. Mol Med 2003; 9:10–7. [PMC free article] [PubMed] [Google Scholar]
- 66. Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor‐beta but not interleukin 4 in the suppression of T helper type 1‐mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med 1996; 183:2669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin‐10 induces a long‐term antigen‐specific anergic state in human CD4+ T cells. J Exp Med 1996; 184:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hunter MM, Wang A, Hirota CL, McKay DM. Neutralizing anti‐IL‐10 antibody blocks the protective effect of tapeworm infection in a murine model of chemically induced colitis. J Immunol 2005; 174:7368–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Nasal heat shock protein (HSP)60 fails to inhibit atherosclerotic lesion progression. (a–c) Representative photomicrographs of oil red O‐ and haematoxylin‐stained aortic root sections from the control‐treated group (III, n = 6), the HSP60‐treated group (IV, n = 6) and the HSP60‐treated enhancement group (V, n = 6). (d) The data from three groups are shown: the half black regular triangles, the half black inverted triangles and the black inverted triangles represent animals from groups III, IV and V. A black bar represents 200 μm. The horizontal bars represent the means.
Fig. S2. Effects of nasal heat shock protein (HSP)60 on the suppressive function of CD4+CD25+ glycoprotein A repetitions predominant (GARP)+ regulatory T cells (Tregs). CD4+CD25+GARP+ Tregs and CD4+CD25–GARP– T cells were isolated from spleens 14 days after the final nasal HSP60 treatment. CD4+CD25+GARP+ Tregs from phosphate‐buffered saline (PBS)‐treated or HSP60‐treated mice were co‐cultured with CD4+CD25–GARP– T cells at different ratios in the presence of soluble anti‐CD3‐antibody and anti‐CD28‐antibody. Cell proliferation was assessed using the MTT assay. The rate of cell proliferation was calculated as follows: cell proliferation rate = (A value in test group – A value in normal control group)/A value in normal control group × 100%. The data are representative of at least three independent experiments.
Table S1. Apolipoprotein E (ApoE)−/− mice were nasally administered phosphate‐buffered saline (PBS) or heat shock protein (HSP)60, as described in the Methods section. The data are expressed as the mean ± standard error of the mean (s.e.m.).