

Summary
Rheumatoid arthritis (RA) may promote endothelial dysfunction. This phenomenon requires further investigation, especially in collagen‐induced arthritis (CIA), as it is considered the experimental model most similar to RA. The objectives of this study were to identify CIA‐induced changes in noradrenaline (NE) and acetylcholine (ACh) responses in mice aortas that may suggest endothelial dysfunction in these animals. Moreover, we characterize CIA‐induced modifications in inducible nitric oxide synthase (iNOS) expression in the aortas and cardiac and renal tissues taken from these mice that may be related to possible endothelial dysfunction. Male DBA/1J mice were immunized with 100 μg of emulsified bovine collagen type II (CII) plus complete Freund's adjuvant. Twenty‐one days later, these animals received a boost of an additional 100 μg plus incomplete Freund's adjuvant. Fifteen days after the onset of the disease, aortic rings from CIA and control mice were challenged with NE and ACh in an organ bath. In these animals, iNOS was detected through immunohistochemical analysis of aorta, heart and kidneys. Plasma nitrite concentration was determined using the Griess reaction. CIA did not change NE or ACh responses in mice aorta but apparently increased the iNOS expression not only in aorta, but also in cardiac and renal microcirculation. In parallel, CIA reduced nitrite plasma concentration. In mice, CIA appears to increase the presence of iNOS in aorta, as well as in heart and in kidney microcirculation. This iNOS increase occurs apparently in parallel to a reduction of the bioavailability of NO. This phenomenon does not appear to change NE or ACh responses in aorta.
Keywords: aorta, endothelium, experimental arthritis, oxidative stress, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease with unknown aetiology, which affects mainly synovial joints and periarticular structures. However, as the average life expectancy of RA patients is lower than that of the general population, it appears that manifestations of the disease may go beyond the pain and disability related to joint injury 1, 2, 3, 4. The correlation between systemic inflammation and increased cardiovascular morbidity and mortality in RA patients is well established in the literature 3, 4, 5, 6, but the pathophysiological mechanisms involved in these disturbances remain unclear. Some evidence has suggested that systemic release of proinflammatory cytokines from chronic articular inflammation promotes inflammation in blood vessels, leading to functional 7, 8, 9, 10, 11, 12, 13 and structural 12 modifications in both endothelial and smooth muscle layers. Based on these findings, some authors have proposed that endothelial dysfunction is actually an early manifestation of RA, and may be influenced by the intensity and/or duration of the joint inflammatory process as well as by the action of anti‐rheumatic drugs 14, 15.
In an attempt to understand more clearly the pathophysiological mechanisms that lead to the aforementioned endothelial dysfunction, several models of experimental arthritis have been developed. These experimental models share the objective of simulating with the highest possible reliability the inflammatory processes that characterize RA in humans. In this manner, previous studies have shown endothelial dysfunction related to arthritis in the adjuvant‐induced arthritis (AIA) model 7, 8, 10, 11, 12, 13, 16, 17, 18, 19, 20, 21. Actually, a significant reduction in endothelial function was observed, associated with increased oxidative stress due to increased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase expression and uncoupling of endothelial nitric oxide synthase (eNOS) in aorta taken from AIA rat 7, 8. It was also proposed that the increased activity of inducible nitric oxide synthases (iNOS), as well as the elevated levels of circulating myeloperoxidase, are involved in the endothelial dysfunction, thereby resulting in attenuation of acetylcholine‐induced flow in the forearms of patients with RA 6. According to these authors, this increase in iNOS consumes tetrahydrobiopterin co‐factor (BH4), leading to eNOS uncoupling. Uncoupled, eNOS produces superoxide anion ( ) instead of nitric oxide (NO). Moreover, this reacts with NO to produce peroxynitrite (ONOO−), a free radical against which there is no natural antioxidant defence. This process leads to a reduction of endothelial NO bioavailability in parallel with increasing concentrations of ONOO− 6, 7. Furthermore, ONOO− can oxidize BH4 into dihydrobiopterin (BH2), reducing the bioavailability of this co‐factor, which exacerbates eNOS uncoupling 22 and contributes to endothelial dysfunction. Indeed, iNOS may be considered a key enzyme in this pathophysiological mechanism.
However, evidence of vascular impairment related to collagen‐induced arthritis (CIA) – an experimental model that reproduces the inflammatory mechanisms of RA with greater accuracy than other experimental models used for this purpose 16, 18, 21, 23, 24, 25 – is scarce in the literature. CIA‐related endothelial dysfunction was demonstrated in mice aorta 26 as well as in sheep coronaries and digital arteries 27. Conversely, Reynolds et al. 25 failed to show CIA‐induced endothelial dysfunction in mice aorta at the moment of onset of the earliest signs of joint inflammation. This contradiction merits further investigation in order to establish bridges between the mechanism of endothelial dysfunction described in animal models and in RA patients. Furthermore, this investigation should be conducted in both macrovasculature and microvasculature, as the impact of RA on endothelial function may differ between these vascular beds 28. Moreover, it was observed in AIA animals that the course of microvascular endothelial dysfunction does not exactly mirror the macrovascular endothelial dysfunction 29.
Thus, the aim of the present study was to identify changes in noradrenaline (NE) and acetylcholine (ACh) responses induced by CIA in mice aortas that may suggest endothelial dysfunction in these animals. Moreover, the present study also sought to characterize CIA‐induced changes in iNOS expression in the aorta, cardiac and renal tissues of these animals; changes which may be related to endothelial dysfunction.
Materials and methods
Animals
Eighty male DBA/1J mice (10 ± 2 weeks old; 18–22 g), sourced from the Experimental Biotherium of Marília Medical School, were housed in temperature‐controlled rooms (22 ± 2°C) under a 12‐h light/dark cycle, with free access to food and water. All procedures were carried out with the approval of the Ethics Committee on Animal Use of Marília Medical School (protocol no. 829/12). All experiments were performed in accordance with international guidelines for the care and use of laboratory animals.
Induction and assessment of CIA
Animals received 100 μg bovine type II collagen (CII; 4 mg/ml; Sigma‐Aldrich, St Louis, MO, USA) dissolved in 25 μl of 5 × 10−2mol/l acetic acid and emulsified with an equal volume of complete Freund's adjuvant (CFA; 10 mg/ml of Mycobacterium tuberculosis; Difco, Detroit, MI, USA) by intradermal injection into the tail (day 0). On day 21 each animal received a similar treatment (boost), differing only in that incomplete Freund's adjuvant (IFA; Sigma‐Aldrich) replaced CFA. The control group (sham‐immunized) received the same CFA and IFA injection regimens, only without type II collagen administration. The onset of the earliest signs of CIA was characterized by mild erythema and/or paw swelling, usually observed 26–33 days post‐immunization. Once these early signs of inflammation were identified the animals were followed for a period of 15 days, until the end of the experimental protocol. On day 15 of the inflammatory process (i.e. days 41–48 post‐immunization), a scoring system was implemented to quantify the subjective evaluation of arthritic severity 24. Each paw was rated individually for its degree of inflammation on a scale of 0–4, making 16 the highest possible total score for a single animal. The rating system for severity of inflammation was defined as follows: 0 (no evidence of erythema or swelling); 1 (erythema and mild swelling confined to the tarsals or ankle joint); 2 (erythema and mild swelling extending from the ankle to the tarsals); 3 (erythema and moderate swelling extending from the ankle to metatarsal joints); and 4 (erythema and severe swelling encompass the ankle, foot and digits or ankylosis of the limb). At the end of this assessment, the animals were euthanized in a CO2 chamber in order to obtain the aortas, joints, kidneys, hearts and plasma needed. We excluded animals that showed no inflammatory signs 33 days post‐immunization. In the present study, 80% of the animals immunized with collagen developed arthritis, thereby permitting their inclusion into the CIA group.
Histopathological assessment of arthritis
Joints from these animals were removed, fixed in 2% glutaraldehyde and 4% paraformaldehyde in Sorensen phosphate buffer 10−1 mol/l pH 7·0, and subjected to descaling solution (NaCl 2·16 g; acetic acid 30 ml; formaldehyde 30 ml; distilled water 240 ml). Sections (5 μm) were stained with haematoxylin and eosin (H&E), toluidine blue and Masson's trichrome. A subjective score of 0–3 was employed to perform the histopathological assessment of these joints, wherein 0 represented an absence of inflammation, 1 = mild, 2 = moderate and 3 = severe. Severity was determined using the following parameters: intensity of the inflammatory exudate (neutrophils and lymphocytes), impairment of the inflammatory exudate (perivascular and synovial) and presence of pyoarthritis and initial fibroplasia.
Study of vascular responsiveness
Segments of thoracic aortas were dissected carefully and cleaned of perivascular adipose tissues. From this segment of thoracic aorta three rings were obtained (3–5 mm). These rings (two with intact endothelium and the other one without endothelium, removed mechanically) were then placed into 2 ml organ baths containing Krebs–Henseleit solution (composition in mmol/l: NaCl 130; KCl 4·7; CaCl2 1·6; KH2PO4 1·2; MgSO4 1·2; NaHCO3 15; glucose 11·1). The solution was adjusted to a pH of 7·4 and bubbled continuously at 37°C with a mixture of 95% O2 and 5% CO2. In the organ bath, the rings were arranged between two steel hooks inserted into the lumen, with one attached to a stationary support and the other connected to an isometric force transducer. Tension was monitored and recorded using a Powerlab 8/30 data‐acquisition system (AD Instruments, Castle Hill, NSW, Australia). Prior to administering drugs, rings were equilibrated for 60 min at a resting tension of 0·5 g.
All preparations were precontracted with 10−5 mol/l phenylephrine and then challenged with 10−5 mol/l ACh to verify endothelial integrity. Preparations that had their endothelium removed mechanically did not show 10−5 mol/l ACh‐induced relaxation. Later, intact preparations were challenged with cumulative concentrations of NE (10−10−10−4 mol/l) or with ACh (10−10−10−4 mol/l) after 10−5 mol/l NE‐induced precontraction. In parallel, the endothelium‐denuded preparation was challenged with cumulative concentrations of NE (10−10−10−4 mol/l). All drugs were added directly to the organ bath. The responses evoked (in g) by the cumulative addition of the aforementioned vasoactive agents into the organ bath were plotted to obtain concentration–response curves. Non‐linear regressions (variable slope) for these curves revealed the Rmax (maximal response; highest point of each concentration–response curve) and the pEC50 (negative logarithm of the concentration that evoked 50% of the maximal response). The pEC50 is indicative of the system sensitivity to the studied drug.
Immunohistochemical analysis
Paraffin‐embedded sections (3 μm) of aorta, heart and renal tissue collected from six euthanized animals/group were incubated for 20 min at 96°C with sodium citrate. Later, these sections were treated with 3% hydrogen peroxide diluted in methanol for 30 min. Subsequently, these sections were incubated overnight at 4°C with monoclonal anti‐NOS2 antibody (SC‐7271; Santa Cruz Biotechnology, Santa Cruz, CA, USA) (1 : 50). Non‐specific protein binding was blocked by simultaneous incubation with 1% bovine serum albumin (BSA) in phosphate‐buffered saline (PBS). Negative controls were incubated with an equivalent concentration of 1% PBS–BSA instead of primary antibody (control of reaction). Subsequently, the sections were incubated for 30 min with a biotinylated secondary antibody, followed by avidin‐conjugated horseradish peroxidase (959943B Histotain SP kit; Invitrogen, Camarillo, CA, USA). The sections were then enhanced with diaminobenzidine (DAB) chromogenic substrate (002014 liquid DAB substrate ki; Invitrogen) at room temperature. The sections were counterstained with haematoxylin, dehydrated and mounted. The analysis of the fragments was performed with Zeiss Axioskop 2 (Zeiss, Jena, Germany). All fields of the kidney, heart and aorta fragments were analysed using the densitometric method. For this, 20 different points were analysed in each field studied to obtain an average related to the intensity of immunoreactivity. The values were obtained as arbitrary units (a.u.) using AxioVision software (Carl Zeiss). An observer blinded to the treatment groups evaluated all the slides at random.
Determination of plasma nitrite concentration
The analysis of nitrite in plasma by assay based on the Griess reaction was performed according to a previously described protocol, with some modifications 30. On this occasion, 150 μl of plasma were deproteinized with 30 μl of sulphosalicylic acid (C7H6O6S; 35%) and maintained for 30 min at room temperature. Thereafter, samples were submitted to centrifugation (11,200g for 15 min) and the supernatant was collected for nitrite determination. Before continuing the analysis the supernatants (100 μl) were neutralized with 150 μl of ammonium chloride (NH4Cl; 5%) and 30 μl of sodium hydroxide (NaOH; 5%). The determination of chromophore compound generated by sample chemical reaction required: mixture of sodium phosphate buffer 20 mM; mixture of co‐factors (final concentration NADPH 100 mM and FAD 5 mM) and nitrate reductase enzyme 0·1 U/ml. The samples were then incubated for a period of 3 h in a microplate reader at 37°C. After this period, Griess reagent sulphanilamide (C6H8N2O2S – 1%) and phosphoric acid (H3PO4; 5%) were added, followed by Griess reagent naphtylethylenediamine (C12H16Cl2N2; 0·1%). Samples were read at 540 nm. The nitrite values were calculated for each sample and compared to ΔA540 nm of a sodium nitrite (NaNO2) standard solution of known concentrations (20 mM), which was tested in parallel. Standard regression coefficients were calculated from the calibration curve.
Statistical analysis
The data were expressed as mean ± standard deviation (s.d.). Control and CIA groups were compared by Student's t‐test. Only the animal weights throughout the experimental protocol were compared by one‐way analysis of variance (anova) followed by Bonferroni's post‐test. P < 0.05 was considered statistically significant.
Results
Characterization of CIA
No significant difference in body weight was observed between the CIA and control groups, neither on the day of the first immunization (22·4 ± 2·7; n = 11 and 20·8 ± 1·8; n = 13, respectively) nor at the boost on day 21 (21·3 ± 1·0; n = 11 and 20·3 ± 2·3; n = 11, respectively). However, immediately before euthanasia, the CIA group had a significantly (P < 0·001) lower body weight (21·1 ± 1·8; n = 13) than the controls (25·3 ± 1·0; n = 11), as well as intense paw erythema and oedema (scored between 7 and 16). The paw weights, determined immediately after euthanasia, were also higher in CIA animals compared to those of the controls (Table 1). In the histopathological assessment, all CIA animals presented a maximum score of 3 on joint inflammation analysis, while control animals presented scores of 0.
Table 1.
Paw weight detected on the day of euthanasia.
| Weight (g) | Control (n = 9) | CIA (n = 13) |
|---|---|---|
| Right front paw | 3·02 ± 0·21 | 5·50 ± 1·15** |
| Left front paw | 2·85 ± 0·21 | 5·03 ± 1·48** |
| Right back paw | 6·70 ± 0·42 | 11·84 ± 2·09** |
| Left back paw | 6·74 ± 0·27 | 12·04 ± 2·23** |
**P < 0·001 versus control group. Values represent means ± standard deviation. CIA = collagen‐induced arthritis.
Vascular responsiveness
CIA did not alter NE responses significantly in either intact (Fig. 1a) or endothelium‐denuded aortas (Fig. 1b). Moreover, CIA also did not alter significantly ACh responses determined in mice aortas (Fig. 1c).
Figure 1.
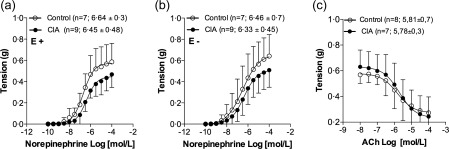
Concentration–response curves determined with norepinephrine (a) and acetylcholine (c) in intact (with endothelium; E+), as well as with norepinephrine (b) in denuded (without endothelium; E−) aorta preparations taken from control (○) and collagen‐induced arthritis (CIA) (•) animals. Values represent mean ± standard deviation (s.d.). The last point of each concentration–response curve is equivalent to Rmax response. The number of independent determinations made in aorta rings, taken from different animals, and the values of pEC50 are shown in parentheses.
Immunohistochemical analysis of iNOS
CIA increased iNOS immunolabelling significantly in aorta. In these preparations, CIA increased iNOS detection not only in the endothelium but also in the media layer (Fig. 2). The CIA also increased iNOS immunolabelling in the cardiac tissues, either in cardiomyocytes or in the endothelial layer of arterioles (diameter between 10 and 20 µm; Fig. 3). In the kidneys, iNOS immunolabelling was also increased in the glomeruli and interstitium, as well as in the endothelial layers of microvessels of the cortex (Fig. 4). In the medullary portion, a significant increase of iNOS immunolabelling was observed, mainly in the interstitium (Fig. 5).
Figure 2.
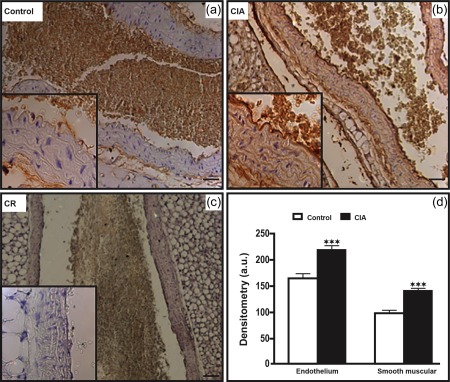
The aorta from arthritic animals presented enhancement of inducible nitric oxide synthase (iNOS) immunolabelling. (b) The collagen‐induced arthritis (CIA) group presents increased iNOS immunolabelling in endothelium and smooth muscle, compared to the control group (a). The details show enlargement of the highlighted areas in figures. (c) Control of reaction (CR). Counterstain: haematoxylin. Bars: 5 μm. (d) Densitometric analysis (arbitrary units, a.u.). Bars express mean ± standard deviation (n = 6; one‐way analysis of variance (anova) followed by Bonferroni's post‐test; ***P < 0·0001).
Figure 3.
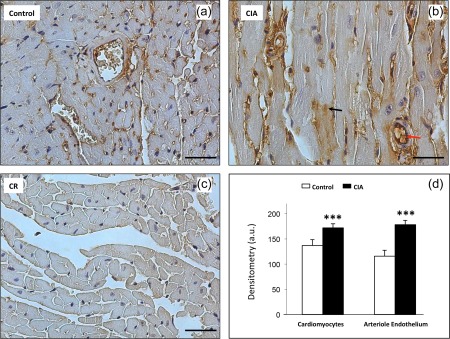
Enhancement of expression of inducible nitric oxide synthase (iNOS) in arthritic cardiac tissue. (b) The collagen‐induced arthritis (CIA) group presents increase in the expression of iNOS compared to the control group (a). Immunolabelling in cardiomyocytes (black arrow) and in the arteriole endothelium (red arrow). (c) Control of reaction (CR). Counterstain: haematoxylin. Bars: 20 μm. (d) Densitometric analysis (arbitrary units, a.u.). Bars express mean ± standard deviation (n = 6; one‐way analysis of variance (anova) followed by Bonferroni's post‐test; ***P < 0·0001).
Figure 4.
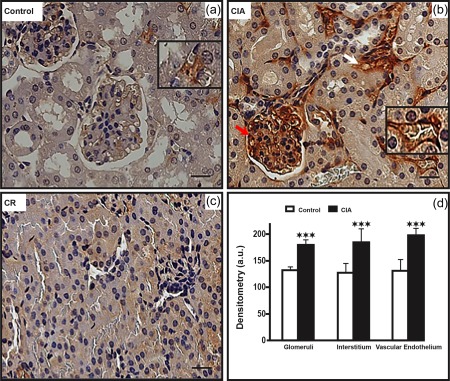
Increased inducible nitric oxide synthase (iNOS) immunolabelling in arthritic kidney cortex. (b) The collagen‐induced arthritis (CIA) group presents increased iNOS immunolabelling in glomeruli (red arrows), in the interstitium (white arrows) and in the vascular endothelium (detail), compared to the control group (a). (c) Control of reaction (CR). Counterstain: haematoxylin. Bars: 20 μm. (d) Densitometric analysis (arbitrary units, a.u.). Bars express mean ± standard deviation (n = 6; one‐way analysis of variance (anova) followed by Bonferroni's post‐test; ***P < 0·0001).
Figure 5.
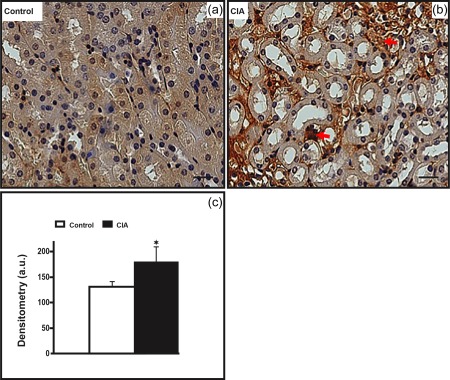
Increased inducible nitric oxide synthase (iNOS) immunolabelling in arthritic kidney medulla. (b) The collagen‐induced arthritis (CIA) group presents increased iNOS immunolabelling in the interstitium (red arrows), compared to the control group (a). Counterstain: haematoxylin. Bars: 20 μm. (c) Densitometric analysis (arbitrary units, a.u.). Bars express mean ± standard deviation (n = 6; one‐way analysis of variance (anova) followed by Bonferroni's post‐test; *P < 0·01).
Determination of plasma nitrite concentration
CIA reduced plasma nitrite concentrations significantly (Fig. 6).
Figure 6.
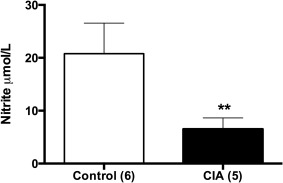
Plasma concentrations of nitrite in control and the collagen‐induced arthritis (CIA) group. Values are expressed as mean ± standard deviation (Student's t‐test; **P < 0·001). The numbers of independent determinations are shown in parentheses.
Discussion
Endothelial dysfunctions are described commonly in AIA 7, 8, 10, 11, 12, 13, 16, 17, 18, 19, 20, 21. However, this pathophysiological process has been scarcely studied in collagen‐induced models, which reproduce the inflammatory mechanisms of human RA with greater accuracy 16, 18, 21, 23, 24, 25. In the present study, CIA has not modified the NE or ACh responses in aortas of DBA/1J mice. As endothelial dysfunction normally increases NE‐induced vasoconstriction 31 and/or reduces the ACh‐induced relaxation 7, 8, 11, 12, 13 in isolated aortas, the data presented suggest a normal endothelial function in these CIA mice aortas. The present study, however, does not rule out possible modifications of NE or ACh responses in atherogenic‐prone sites of mice aorta. Indeed, it has been proposed that sites exposed to turbulent blood flow, such as areas of branching or high vessel curvature, are highly susceptible to the development of atherosclerotic lesions 32. In this regard, it has been demonstrated that turbulent shear stress may change the endothelial production of a large number of substances involved in either vascular tonus modulation or atherosclerotic lesions 33.
The absence of modification in NE or ACh responses observed in the present study cannot be justified by a failure in the development of the CIA‐related inflammatory process. In fact, joint inflammation was confirmed by the high paw inflammation scores obtained through the aforementioned scoring system for subjective evaluation of arthritic severity. Furthermore, the swelling in the CIA group was confirmed by the increases in paw weight. In addition, the weight gain observed between the boost and euthanasia was lower in CIA animals. Moreover, the histopathological assessment revealed the presence of pyoarthritis (through the intensity and the severity of the inflammatory exudate) and initial fibroplasia in CIA animals, but not in the controls. In fact, all the CIA animals presented scores of 3 for joint inflammation. These articular injuries are consistent with previously reported observations in joints of CIA mice, thereby corroborating the effectiveness of this experimental model 16, 18, 21, 23, 24, 25.
Histological analysis also revealed cartilage injuries and bone erosions. Indeed, these physiopathological features indicate that the CIA‐induced inflammatory process is fully established 34. Moreover, it was shown that the expression peak of interleukin IL‐2, IL‐6, macrophage inflammatory protein‐2 (MIP‐2) and IL‐1β in joints of CIA animals has ended 15 days after the onset of the earliest signs of inflammation 35. Further, the arthritis score reaches the plateau approximately 7 days after the onset of the earliest signs of inflammation 34. Collectively, these information indicate that the present study was performed in a subsequent phase to the maximum acute inflammatory period of CIA.
Also, no modifications of ACh responses in mice aorta were observed in an earlier phase of CIA at the moment of onset of the earliest signs of joint inflammation 25. Conversely, it was observed that CIA attenuated the ACh‐induced relaxation in mice aorta, probably through an impairment of mechanisms related to NO 26. These authors, however, assessed the ACh responses at a later phase of CIA in comparison to the present study (at day 56 post‐immunization versus at days 41–48 post‐immunization in the present study). Indeed, collectively, these studies indicate that endothelial dysfunction may establish itself over time. Moreover, increased concentrations of / in urine and plasma were observed 12 weeks after the first immunization (about 56 days after the onset of the earliest signs of arthritis) 23. Perhaps the aforementioned endothelial dysfunction may be in spontaneous regression in this later phase of CIA, thereby reinforcing that such mechanisms can be modified over time. Therefore, as proposed in the AIA model 29, the time allowed for CIA to develop should always be taken into consideration when analysing endothelial function within this experimental model.
Assuming that endothelial dysfunction is ongoing in these animals, we decided to investigate iNOS presence in the aorta as well as in the cardiac and renal tissues. We chose to focus our study on iNOS because, as stated previously, this enzyme may be involved in the genesis of endothelial dysfunction. In fact, the increased iNOS in the endothelium may consume BH4, leading to eNOS uncoupling. When uncoupled, eNOS produces instead of NO, leading to oxidative stress in these tissues. This process reduces endothelial NO bioavailability and, consequently, leads to endothelial dysfunction 6, 7. From this, it was posited that a reduction in nitric oxide bioavailability, attributed to increased generation of iNOS, was responsible for decreased nitrite plasma levels in rats exposed to restraint stress 31.
In the present study, CIA significantly augmented iNOS immunolabelling in aortas, either in the endothelium or in the smooth media layer. Is worth stressing that the immunohistochemical technique permits only semiquantitative analysis. Thus, although the obtained results suggest an increase of iNOS expression, they do not indicate accurately the magnitude of this phenomenon. In parallel, a significant reduction in nitrite ( ) plasma concentration was observed in the CIA group. These findings suggest that even though modifications in NE and ACh responses were not observed in aorta, a vascular inflammatory process already exists in these CIA‐submitted animals. In this regard, nitrite anions have been considered an important vascular storage pool and source of NO 36, 37. Therefore, a reduction in nitrite plasma concentration in the CIA group may indicate a reduction in NO bioavailability. However, it is necessary to emphasize that although NO is an important endothelium‐derived mediator, the endothelial function, mainly in arteries of resistance, involves other mechanisms beyond those NO‐related. In fact, elevations in plasma nitrite concentrations are not always accompanied by increases in vasodilator responses induced by ACh in the microcirculation 38. The assessment of NE and ACh responses in cardiac or renal microcirculation would allow for more precise conclusions. However, this assessment was not possible due to methodological limitations.
The data obtained suggest an increase of iNOS expression not only in cardiomyocytes but also in the endothelium layer of cardiac arterioles of resistance (diameter between 10 and 20 µm), taken from CIA mice. The data presented also suggest an increase of iNOS expression in renal tissues in either the cortical or medullary portion, including in its microcirculation, thereby suggesting a greater presence of iNOS in these tissues. Coincidentally, it has been affirmed that microcirculatory abnormalities are the first vascular manifestations of RA 39, 40. According to these authors, RA patients normally present functional vascular changes followed by progressive anatomical modifications in these vessels, characterized by an increase of the intima‐media thickness leading to increased cardiovascular disease. Therefore, the vascular manifestations observed in the present study appear to reflect the vascular injuries related to RA. These results also reinforce the need for particular care being given to cardiac and renal microcirculation in the treatment of patients with RA.
Conclusion
The results of this study suggest that, in mice, CIA induces an increase in iNOS expression in aorta, as well as in cardiac and renal microcirculation. These increases in iNOS expression occur in parallel to a reduction in nitrite plasma concentration, thereby suggesting CIA‐induced reduction of NO bioavailability. This change in iNOS expression, however, apparently does not attenuate endothelial function in aorta, at least not by approximately 15 days after the onset of the earliest signs of arthritis.
Disclosure
The authors declare that they have no disclosures.
Acknowledgements
The authors declare that they had no financial support for this study.
References
- 1. Goeldner I, Skare TL, Reason IT, Utiyama SR. Rheumatoid arthritis: a current view. J Bras Patol Med Lab 2011; 47:495–503. [Google Scholar]
- 2. Torigoe DY, Laurindo IM. Rheumatoid arthritis and cardiovascular disease. Rev Bras Reumatol 2006; 46 (Suppl. 1):60–6. [Google Scholar]
- 3. Gabriel SE. Cardiovascular morbidity and mortality in rheumatoid arthritis. Am J Med 2008; 121:S9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khan F, Galarraga B, Belch JJ. The role of endothelial function and its assessment in rheumatoid arthritis. Nat Rev Rheumatol 2010; 6:253–61. [DOI] [PubMed] [Google Scholar]
- 5. Yki‐Järvinen H, Bergholm R, Leirisalo‐Repo M. Increased inflammatory activity parallels increased basal nitric oxide production and blunted response to nitric oxide in vivo rheumatoid arthritis. Ann Rheum Dis 2003; 62:630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mäki‐Petäjä KM, Cheriyan J, Booth AD et al Inducible nitric oxide synthase is increased in patients with rheumatoid arthritis and contributes to endothelial dysfunction. Int J Cardiol 2008; 129:399–405. [DOI] [PubMed] [Google Scholar]
- 7. Haruna Y, Morita Y, Komai N et al Endothelial dysfunction in rat adjuvant‐induced arthritis: vascular superoxide production by NAD(P)H oxidase and uncoupled endothelial nitric oxide synthase. Arthritis Rheum 2006; 54:1847–55. [DOI] [PubMed] [Google Scholar]
- 8. Haruna Y, Morita Y, Yada T, Satoh M, Fox DA, Kashihara N. Fluvastatin reverses endothelial dysfunction and increased vascular oxidative stress in rat adjuvant‐induced arthritis. Arthritis Rheum 2007; 56:1827–35. [DOI] [PubMed] [Google Scholar]
- 9. Alkaitis MS, Crabtree MJ. Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr Heart Fail Rep 2012; 9:200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fontaine J, Herchuelz A, Famaey JP. A pharmacological analysis of the responses of isolated aorta from rats with adjuvant arthritis. Agents Actions 1984; 14:684–7. [DOI] [PubMed] [Google Scholar]
- 11. Fang ZY, Fontaine J, Unger P, Berkenboom G. Alterations of the endothelial function of isolated aortae in rats with adjuvant arthritis. Arch Int Pharmacodyn Ther 1991; 311:122–30. [PubMed] [Google Scholar]
- 12. Sakuta T, Morita Y, Satoh M, Fox DA, Kashihara N. Involvement of the renin‐angiotensin system in the development of vascular damage in a rat model of arthritis: effect of angiotensin receptor blockers. Arthritis Rheum 2010; 62:1319–28. [DOI] [PubMed] [Google Scholar]
- 13. Prati C, Berthelot A, Wendling D, Demougeot C. Endothelial dysfunction in rat adjuvant‐induced arthritis: up‐regulation of the vascular arginase pathway. Arthritis Rheum 2011; 63:2309–17. [DOI] [PubMed] [Google Scholar]
- 14. Foster W, Lip GYH, Raza K, Carruthers D, Blann AD. An observational study of endothelial function in early arthritis. Eur J Clin Invest 2012; 42:510–6. [DOI] [PubMed] [Google Scholar]
- 15. Galarraga B, Belch JJ, Pullar T, Ogston S, Khan F. Clinical improvement in RA is associated with healthier microvascular function in patients who respond to anti‐rheumatic therapy. J Rheumatol 2010; 37:521–8. [DOI] [PubMed] [Google Scholar]
- 16. Oliver SJ, Brahn E. Combination therapy in rheumatoid arthritis: the animal model perspective. J Rheumatol Suppl 1996; 44:56–60. [PubMed] [Google Scholar]
- 17. Cinar MG, Can C, Ulker S et al Effect of vitamin E on vascular responses of thoracic aorta in rat experimental arthritis. Gen Pharmacol 1998; 31:149–53. [DOI] [PubMed] [Google Scholar]
- 18. Bendele A. Animal models of rheumatoid arthritis. J Musculoskelet Neuronal Interact 2001; 1:377–85. [PubMed] [Google Scholar]
- 19. Can C, Cinar MG, Kosay S, Evinç A. Vascular endothelial dysfunction associated with elevated serum homocysteine levels in rat adjuvant arthritis: effects of vitamin E administration. Life Sci 2002; 71:401–10. [DOI] [PubMed] [Google Scholar]
- 20. Nozaki K, Goto H, Nakagawa T et al Effects of keishibukuryogan on vascular function in adjuvant‐induced arthritis rats. Biol Pharm Bull 2007; 30:1042–7. [DOI] [PubMed] [Google Scholar]
- 21. Hegen M, Keith JC Jr, Collins M, Nickerson‐Nutter CL. Utility of animal models for identification of potential therapeutics for rheumatoid arthritis. Ann Rheum Dis 2008; 67:1505–15. [DOI] [PubMed] [Google Scholar]
- 22. Vasquez‐Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J 2002; 362:733–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakaguchi Y, Shirahase H, Ichikawa A, Kanda M, Nozaki Y, Uehara Y. Effects of selective iNOS inhibition on type II collagen‐induced arthritis in mice. Life Sci 2004; 75:2257–67. [DOI] [PubMed] [Google Scholar]
- 24. Brand DD, Latham KA, Rosloniec EF. Collagen‐induced arthritis. Nat Protoc 2007; 2:1269–75. [DOI] [PubMed] [Google Scholar]
- 25. Reynolds SI, Williams AS, Williams H et al Contractile, but not endothelial, dysfunction in early inflammatory arthritis: a possible role for matrix metalloproteinase‐9. Br J Pharmacol 2012; 167:505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He M, Liang X, He L, W Wen, S Zhao, L Wen, Y Liu, JYJ Shyy, Z. Yuan Endothelial dysfunction in rheumatoid arthritis: the role of monocyte chemotactic protein‐1‐induced protein. Arterioscler Thromb Vasc Biol 2013; 33:1384–91. [DOI] [PubMed] [Google Scholar]
- 27. Dooley LM, Washington EA, Abdalmula A, Tudor EM, Kimpton WG, Bailey SR. Endothelial dysfunction in an ovine model of collagen‐induced arthritis. J Vasc Res 2014; 51:90–101. [DOI] [PubMed] [Google Scholar]
- 28. Sandoo A, Carroll D, Metsios GS, Kitas GD, Veldhuijzen van Zanten JJ. The association between microvascular and macrovascular endothelial function in patients with rheumatoid arthritis: a cross‐sectional study. Arthritis Res Ther 2011; 13:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Totoson P, Maguin‐Gaté K, Nappey M, Pratti C, Wendling D, Demougeot C. Microvascular abnormalities in adjuvant‐induced arthritis: relationship to macrovascular endothelial function and markers of endothelial activation. Arthritis Rheumatol 2015; 67:1203–13. [DOI] [PubMed] [Google Scholar]
- 30. Granger DL, Traintor RR, Boockvar KS, Hibbs JB Jr. Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol 1996; 268:142–51. [DOI] [PubMed] [Google Scholar]
- 31. Carda AP, Marchi KC, Rizzi E et al Acute restraint stress induces endothelial dysfunction: role of vasoconstrictor prostanoids and oxidative stress. Stress 2015; 18:1–11. [DOI] [PubMed] [Google Scholar]
- 32. Vanderlaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site‐selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol 2004; 24:12–22. [DOI] [PubMed] [Google Scholar]
- 33. Topper JN, Cai J, Falb D, Gimbrone MA Jr. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase‐2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up‐regulated by steady laminar shear stress. Proc Natl Acad Sci USA 1996; 93:10417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oliveira PG, Grespan R, Pinto LG, L Meurer, JC Brenol, R Roesler, G Schartsmann, FQ Cunha, RM Xavier. Protective effect of RC‐3095, an antagonist of the gastrin‐releasing peptide receptor, in experimental arthritis. Arthritis Rheum 2011; 63:2956–65. [DOI] [PubMed] [Google Scholar]
- 35. Thorton S, Duwel LE, Boivin GP, Ma Y, Hirsch R. Association of the course of collagen‐induced arthritis with distinct patterns of cytokine and chemokine messenger RNA expression. Arthritis Rheum 1999; 42:1109–18. [DOI] [PubMed] [Google Scholar]
- 36. Kosby K, Partovi KS, Crawford JH et al Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 2003; 9:1498–505. [DOI] [PubMed] [Google Scholar]
- 37. Li H, Cui H, Kundu TK, Alzawahra W, Zweier JL. Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem 2008; 283:17855–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huguenin GV, Moreira AS, Saint'pierre TD et al Effects of dietary supplementation with Brazil nuts on microvascular endothelial function in hypertensive and dyslipidemic patients: a randomized crossrover placebo‐controlled trial. Microcirculation 2015; 22:687–99. [DOI] [PubMed] [Google Scholar]
- 39. Foster W, Carruthers D, Lip GY, Blann AD. Inflammation and microvascular and macrovascular endothelial dysfunction in rheumatoid arthritis: effect of treatment. J Rheumatol 2010; 37:711–6. [DOI] [PubMed] [Google Scholar]
- 40. Sattar N, McInnes IB. Vascular comorbidity in rheumatoid arthritis: potential mechanisms and solutions. Curr Opin Rheumatol 2005; 17:286–92. [DOI] [PubMed] [Google Scholar]