

Abstract
Metallo-β-lactamases (MBLs) hydrolyze virtually all β-lactam antibiotics, including penicillins, cephalosporins, and carbapenems. The worldwide emergence of antibiotic-resistant bacteria harboring MBLs poses an increasing clinical threat. The MBL German imipenemase-1 (GIM-1) possesses an active site that is narrower and more hydrophobic than the active sites of other MBLs. The GIM-1 active-site groove is shaped by the presence of the aromatic side chains of tryptophan at residue 228 and tyrosine at residue 233, positions where other MBLs harbor hydrophilic residues. To investigate the importance of these two residues, eight site-directed mutants of GIM-1, W228R/A/Y/S and Y233N/A/I/S, were generated and characterized using enzyme kinetics, thermostability assays, and determination of the MICs of representative β-lactams. The structures of selected mutants were obtained by X-ray crystallography, and their interactions with β-lactam substrates were modeled in silico. Steady-state kinetics revealed that both positions are important to GIM-1 activity but that the effects of individual mutations vary depending on the β-lactam substrate. Activity against type 1 substrates bearing electron-donating C-3/C-4 substituents (cefoxitin, meropenem) could be enhanced by mutations at position 228, whereas hydrolysis of type 2 substrates (benzylpenicillin, ampicillin, ceftazidime, imipenem) with methyl or positively charged substituents was favored by mutations at position 233. The crystal structures showed that mutations at position 228 or the Y233A variant alters the conformation of GIM-1 loop L1 rather than that of loop L3, on which the mutations are located. Taken together, these data show that point mutations at both positions 228 and 233 can influence the catalytic properties and the structure of GIM-1.
INTRODUCTION
Members of the carbapenem class of β-lactams are among the most important antimicrobial agents available for the treatment of serious bacterial infections (1). However, their use against Gram-negative bacteria is now threatened by the dissemination of carbapenemases, β-lactamases that hydrolyze the amide bond of the β-lactam ring, thereby inactivating carbapenems and other β-lactams (2). To date, only a few effective carbapenemase inhibitors have been available for clinical use. Enzymes with carbapenemase activity have been identified in multiple β-lactamase classes. β-Lactamases are divided into four classes, of which classes A, C, and D are serine enzymes that catalyze hydrolysis of the β-lactam through a serine-bound acyl intermediate, whereas class B β-lactamases, or metallo-β-lactamases (MBLs), coordinate one or two zinc ions in the active site, which are essential for their enzymatic activity (1–3). The class B MBLs can be divided into four subclasses, according to their primary structure: B1a (e.g., VIM, IMP, DIM, and SPM), B1b (NDM), B2 (e.g., CphA), and B3 (e.g., L1 and AIM) (3). Currently, no clinically effective MBL inhibitor has been approved for use (4). Avibactam, the only clinically approved effective carbapenemase inhibitor, has activity against class A carbapenemases and some class D carbapenemases (5).
The German imipenemase-1 (GIM-1) MBL was first identified in clinical isolates of Pseudomonas aeruginosa in Germany in 2002 (6). Recently, GIM-1 has been identified in other bacterial species, such as Serratia marcescens (7), Enterobacter cloacae (8), and Acinetobacter pittii (9), indicating that it is transmitted on mobile genetic elements (6, 7). A new variant of GIM-1 (GIM-2) with a single mutation, A290G, was recently discovered (10). GIM-1 is categorized as a subclass B1a enzyme and shows the closest resemblance to DIM-1 (55% amino acid sequence identity) (8) and a more distant relationship to enzymes such as IMP-6 (44% identity), VIM-7 (31% identity), and SPM-1 (28% identity) (6).
The GIM-1 sequence contains the characteristic canonical MBL 116HXHXD120 motif (according to the standard numbering scheme for class B β-lactamases [11]) with a serine at the first X and a glutamic acid at the second X, which are infrequently found in other MBLs (6). The GIM-1 active site contains aromatic side chains of tryptophan at residue 228 (W228) and tyrosine at residue 233, positions where hydrophilic residues are normally present in other MBLs. These residues create a substrate-binding groove narrower than that found in other MBLs (12). Crystal structures indicate that the GIM-1 active site is flanked by two flexible loops, the L1 loop (residues 57 to 68) and the L3 loop (residues 223 to 242). The conformations of these loops are defined by hydrogen-bonding patterns involving tryptophan at position 228 (12). The capacity of these loops to undergo conformational rearrangement could explain why GIM-1 can hydrolyze a range of β-lactam antibiotics, despite the narrow active site (13). Previous studies have concluded that mutations in loop L3 outside the active site can contribute to the tuning of MBL catalytic activity (14, 15). The structure of a complex of VIM-2 and a mercaptocarboxylate inhibitor suggests that the residues at positions 228 and 233 play a role in the binding of substrates and inhibitors (16). In other MBLs, mutations at position 233 have been shown to affect substrate specificity, suggesting that this position is involved in substrate binding and/or substrate release during catalysis (15, 17, 18). Consistent with this conclusion, in NDM-1 the N233 side chain amide is hydrogen bonded to hydrolyzed cefuroxime (19).
In this study, four mutants with site-directed mutations of GIM-1 at positions 228 (W228R/A/Y/S) and 233 (Y233N/A/I/S) were constructed, and their properties were investigated. The selection of residue substitutions was based on the sequences of other MBLs, shown in the sequence alignment in Fig. 1, and the properties of individual amino acids. Thus, tryptophan 228 was mutated to arginine (R), as in VIM-2; tyrosine (Y), as in L1; and serine (S), as in VIM-1. The tyrosine at position 233 was mutated to asparagine (N), which is present in IMP-1 and ∼67% of all known MBLs (17); isoleucine (I), since it is a smaller amino acid but still hydrophobic; and serine (S), to investigate the effect of a polar amino acid. Both W228 and Y233 were also mutated to alanine (A) to investigate whether the narrow groove of GIM-1 could be opened and the enzyme made more receptive to various substrates (Fig. 2). The effects of the mutations on the activity of GIM-1 compared to that of recombinant wild-type GIM-1 were investigated by measuring β-lactam hydrolysis under steady-state conditions, MICs, and thermal stabilities. The structures of five GIM-1 mutants were solved by X-ray crystallography to obtain structural explanations for the observed effects. Taken together, our results indicate that positions 228 and 233 are important to the activity of GIM-1 and that substitutions at these positions can have an impact on the catalytic efficiency against various substrates.
FIG 1.

Alignment of the GIM-1 sequence with the sequences of other important MBLs. The large boxes show the sequence alignment of the loop regions of GIM-1, IMP-1, VIM-2, VIM-7, and NDM-1, and secondary structure annotations retrieved from GIM-1 (PDB accession number 2YNW) and NDM-1 (PDB accession number 4RL0) are shown in the top and bottom rows, respectively. Arrows, β strands; TTT and TT, strict α and β turns, respectively; spirals, α helices. Blue boxes indicate conserved positions, wherein red backgrounds indicate identical residues and red letters indicate similar residues. Aligned sequences were rendered by use of the ESPript (v3.0) (41).
FIG 2.

Molecular structures of β-lactam substrates. Type 1 substrates have electron donor-containing R2 groups, like cefoxitin and meropenem, and type 2 substrates have positive charged R2 groups, like ceftazidime and imipenem, or axial methyl R2 groups, like ampicillin and benzylpenicillin.
MATERIALS AND METHODS
Construction of recombinant GIM-1 W228 and Y233 mutants.
Residues Q19 to D250 of the blaGIM-1 gene (GenBank accession no. Q704V1; Q19 is the same as Q37 according to the class B β-lactamase numbering scheme [20]) were cloned into the pDEST14 vector as previously described (12) using an N-terminal six-His tag and the tobacco etch virus (TEV) (14) cleavage site, resulting in a construct comprising residues A18 to D250, where A18 remains from the TEV cleavage site. The eight mutants, W228R/A/Y/S and Y233N/A/I/S, were constructed using the pDEST14 construct containing blaGIM-1 as the template, a QuikChange site-directed mutagenesis kit (Agilent Bioscience), and eight different primer pairs specific for the mutations (Table 1). Parental DNA was digested using DpnI, and the remaining DNA was transformed into Escherichia coli XL1-Blue cells (Stratagene). Luria-Bertani (LB) agar plates containing 100 μg/ml of ampicillin (Sigma-Aldrich) were used for clone selection, and DNA sequencing (BigDye Terminator cycle sequencing kit, v3.1; Applied Biosystems) with the primers listed in Table 1 was performed to confirm the mutations.
TABLE 1.
Primers used for GIM-1 cloning, site-directed mutagenesis, and sequencing
| Primer name | Forward primer |
Reverse primer |
||
|---|---|---|---|---|
| Sequencea (5′–3′) | Tmb (°C) | Sequencea (5′–3′) | Tmb (°C) | |
| GIM-1 W228R | AGG AGT CAT GAG cGc GAA GGC TTA GGT | 62.8 | ACC TAA GCC TTC gCg CTC ATG ACT CCT | 62.8 |
| GIM-1 W228A | AGG AGT CAT GAG gcG GAA GGC TTA GGT | 62.8 | ACC TAA GCC TTC Cgc CTC ATG ACT CCT | 62.8 |
| GIM-1 W228Y | GTG AGG AGT CAT GAG Tac GAA GGC TTA GGT TAC | 64.4 | GTA ACC TAA GCC TTC gtA CTC ATG ACT CCT CAC | 64.4 |
| GIM-1 W228S | GTG AGG AGT CAT GAG aGc GAA GGC TTA GGT TAC | 65.6 | GTA ACC TAA GCC TTC gCt CTC ATG ACT CCT CAC | 65.6 |
| GIM-1 Y233N | GAA GGC TTA GGT aAC GTA GGC GAC GCC | 64.3 | GGC GTC GCC TAC GTt ACC TAA GCC TTC | 64.3 |
| GIM-1 Y233A | GAA GGC TTA GGT gcg GTA GGC GAC GCC | 67.3 | GGC GTC GCC TAC cgc ACC TAA GCC TTC | 67.3 |
| GIM-1 Y233I | GAA GGC TTA GGT att GTA GGC GAC GCC | 62.8 | GGC GTC GCC TAC aat ACC TAA GCC TTC | 62.8 |
| GIM-1 Y233S | GAA GGC TTA GGT agC GTA GGC GAC GCC | 65.8 | GGC GTC GCC TAC Gct ACC TAA GCC TTC | 65.8 |
| T7 promoter | TAA TAC GAC TCA CTA TAG GG | 47.7 | GCT AGT TAT TGC TCA GCG G | 51.1 |
| M13 | GTA AAA CGA CGG CCA GT | 47.1 | GGA AAC AGC TAT GAC CAT G | 48.9 |
| GIM-1-RBS | AGG AGG GTC TTG ATG AAA AAT GTA TTA GTG TTTc | 69.0 | TTA ATC AGC CGA CGC TTC AGC | 68.0 |
The mutated nucleotides are given in lowercase letters and underlined.
The melting temperature was calculated at http://insilico.ehu.es/tm.php.
The ribosomal binding site (RBS) is bold, and the ATG start codon is italicized.
Expression and purification of GIM-1 and the W228 and Y233 mutants.
The constructed plasmids were transformed into in-house-modified E. coli BL21 Star(DE3)pLysS (Invitrogen) cells containing the pRARE plasmid (Novagen), allowing expression of genes encoding tRNAs for rare codons (21). The precultures were grown overnight in LB containing 100 μg/ml ampicillin (Sigma-Aldrich) and 34 μg/ml chloramphenicol (Sigma-Aldrich). Twenty milliliters of preculture was inoculated into 2 liters of Terrific broth (TB) containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol and grown at 37°C to reach an optical density at 600 nm (OD600) between 0.5 and 1.0. Isopropyl-β-d-thiogalactopyranoside (IPTG; 0.4 mM; Sigma-Aldrich) was added to induce expression. The cultures were incubated overnight at 20°C before the cells were collected by centrifugation (8,900 × g, 30 min, 4°C). The cell pellets were resuspended in buffer A (50 mM HEPES [or Tris-HCl], pH 7.2, 100 μM ZnCl2, 150 mM NaCl) and lysed by sonication. The supernatants were collected by centrifugation (25,400 × g, 40 min, 4°C). Recombinant GIM-1 and the mutants were affinity purified using a 5-ml or 1-ml His-Trap HP column (GE Healthcare) in buffer A, washed with 5% buffer B (50 mM HEPES, pH 7.2, 100 μM ZnCl2, 150 mM NaCl, 500 mM imidazole), and eluted across a gradient (5 to 100%) of buffer B (22). The presence of GIM-1 in peak fractions was verified using SDS-PAGE (Bio-Rad) (22). In-house-made His-tagged TEV protease (14) was added to GIM-1-containing fractions in a 1:100 mg ratio of TEV protease/protein, before dialysis overnight at 4°C using 10-kDa-molecular-mass cutoff SnakeSkin dialysis tubing (Pierce) in buffer C (50 mM HEPES, pH 7.2, 250 mM NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol). A second His-Trap purification was performed as described above to remove uncleaved protein and TEV protease. The flowthrough fractions containing the enzymes were estimated to be ∼95% pure from the SDS-polyacrylamide gel analysis and were pooled and dialyzed against buffer A before being concentrated by ultrafiltration using a 10-kDa-molecular-mass cutoff Ultra centrifugal filter unit (Amicon).
Enzyme kinetics.
The hydrolysis of ampicillin (Δε235 = −820 M−1 cm−1), benzylpenicillin (Δε235 = −775 M−1 cm−1), cefoxitin (Δε260 = −7,700 M−1 cm−1), ceftazidime (Δε260 = −9,000 M−1 cm−1), meropenem (Δε300 = −6,500 M−1 cm−1), and imipenem (Δε300 = −9,000 M−1 cm−1) (23) by recombinant GIM-1 and the mutants was measured using a SpectraMax M2e spectrophotometer (Molecular Devices) in assay buffer D (50 mM HEPES, pH 7.2, 100 μM ZnCl2, 1 mg/ml bovine serum albumin [BSA]). BSA was added to the buffer in order to prevent protein unfolding and loss of activity due to a low protein concentration (23, 24). All reactions were performed at 25°C in UV-transparent 96-well plates or, for the penicillins, half-well plates (Corning) with a total reaction volume of 100 μl for all antibiotics except imipenem, which for selected mutants was analyzed using a total volume of 50 μl. The final enzyme concentration was 10 nM. The steady-state kinetic parameters Km (in micromolar) and kcat (in seconds−1) were determined by fitting initial velocities (measured in triplicate and averaged) to the Michaelis-Menten equation in GraphPad Prism (v5.0) software (GraphPad).
Determination of MICs.
For determination of the MICs, the full-length GIM-1 gene, including the native leader sequence, was expressed with an artificial E. coli optimized ribosomal binding site (RBS) 6 nucleotides upstream of the ATG start codon. The RBS was introduced to regulate protein expression. The GIM-1 gene (GenBank accession no. Q704V1 [6]) was amplified using a GIM-1- and RBS-specific primer pair (Table 1) in a Phusion PCR system (NEB). The purified PCR product was cloned into the pET26b vector and used as a template for generating the W228R/A/Y/S and Y233N/A/I/S mutants as described above. The GIM-1 sequences from pET26b were subsequently amplified by PCR using the GIM-1- and RBS-specific primer pair, cloned into the pCRBluntII-TOPO vector (Invitrogen), and transformed into E. coli TOP10 cells (Invitrogen). Clones were selected on LB plates containing 25 μg/ml kanamycin (Sigma). Correct cloning of the plasmids was verified by sequencing. The MICs of the β-lactams used in the kinetic experiments and some additional substrates for GIM-1 and the mutant expression clones were determined using gradient strips (Liofilchem) according to the manufacturer's instruction.
Constitutive expression was tested for wild-type GIM-1 and each of the mutants in E. coli TOP10. Cells were grown in 15 ml LB supplemented with 25 μg/ml kanamycin at 37°C to an OD600 of 0.7 and compared to untransformed E. coli TOP10 cells. Whole cell extracts were analyzed on SDS-polyacrylamide gels.
Crystallization, X-ray data collection, structure determination, and refinement of GIM-1 W228R/A/Y/S and Y233N/A/I/S.
Crystallization trials were set up for all mutants; however, only the four GIM-1 W228 mutants and the Y233A mutant were successfully crystallized. The crystals were obtained through the sitting-drop method using MRC 96-well plates (Molecular Dimensions) and a Phoenix DT crystallization robot (Rigaku) with 60 μl reservoir solution and drops made of 500 nl reservoir solution and 500 nl protein. Five millimolar β-mercaptoethanol was added to the protein storage buffer to avoid the oxidation of Cys221 (12, 25). Crystals grew in approximately 2 days at 25°C and were harvested after 1 to 11 weeks. Crystals of GIM-1 W228R (from protein at 9.8 mg/ml) and W228A (11.8 mg/ml) were grown in 19.8% polyethylene glycol (PEG) 10000 and 0.1 M sodium cacodylate, pH 6.5. GIM-1 W228Y (10 mg/ml) crystallized from a reservoir solution of 24% PEG 5000 monomethyl ether (PEG 5K MME), 0.1 M sodium acetate, pH 5.5, and 0.12 potassium thiocyanate. GIM-1 W228S (11.9 mg/ml) crystallized from a reservoir solution of 26.8% PEG MME 2K, 0.1 M Tris, pH 7.0, 0.11 M potassium phosphate monobasic (KH2PO4), and 2.5% glycerol. GIM-1 Y233A (13.9 mg/ml) was crystallized from 28% PEG 4000, 0.1 M HEPES, pH 7.0, and 0.23 M potassium chloride. All crystals were cryoprotected with 32% PEG 5K MME, 0.1 M succinate, pH 7, and 5% glycerol and then flash frozen in liquid nitrogen.
Diffraction data were collected at beamlines ID29 and BM30 of the European Synchrotron Radiation Facility, Grenoble, France. The GIM-1 mutant structures were solved by molecular replacement in Phaser crystallographic software (26), using the structure of wild-type GIM-1 (PDB accession number 2YNT.A [12]) as a search model. The structures were refined in the Phenix software suite (27, 28) with manual rebuilding in the WinCoot program (28).
Structural in silico analysis of hydrolyzed ampicillin binding to GIM-1 and GIM-1 Y233N.
To structurally investigate the differences between wild-type GIM-1 and the GIM-1 Y233N mutant, the binding mode of the hydrolyzed β-lactam substrate ampicillin was investigated through linear interaction approximation and molecular mechanics with generalized Born and surface area solvation (MM/GBSA). The model-building and binding energy calculations were performed with Maestro (v9.9) software (Schrödinger LLC, New York, NY). The crystal structure of GIM-1 (PDB accession number 2YNW.A [12]) was used to create the in silico model of the Y233N mutant. All crystallographic water molecules were removed, except for the metal-bridging water between the zinc ions, since this water molecule is present in the crystal structure of NDM-1 with hydrolyzed ampicillin (PDB accession number 3Q6X.A [29]). Missing side chains and hydrogen atoms were built and added to using Protein Preparation Wizard (v2.9) software (suite 2014-3; Schrödinger LLC, New York, NY). Both GIM-1 and the Y233N mutant were subjected to restrained energy minimization. The hydrolyzed ampicillin ligand was extracted from the NDM-1 crystal structure (PDB accession number 3Q6X.A [29]) through secondary-structure matching of NDM-1 and GIM-1 in Coot (30). During performance of the Liaison (v5.9) program (Schrödinger LLC, New York, NY), flexibility in the active site was allowed to obtain more reliable complex structures. In our Liaison minimization, a portion of the enzyme was assigned to be flexible by use of the medium (12/16) option. The receptor region extending 12 Å from the ampicillin ligand was flexible, that extending from 12 to 16 Å was restrained, and that extending beyond 16 Å was fixed. Minimizations of the complexes were performed using the truncated Newton algorithm and the OPLS_2005 force field with 1,000 steps of minimization. The Prime (v3.2) (Schrödinger LLC, New York, NY) MM/GBSA was used for calculating the energy of ampicillin binding to GIM-1 and GIM-1 Y233N. The binding free energies (ΔGbinds) were computed according to equation 1, where the minimized energies of the free substrate (E_substrate) and the free enzyme (E_enzyme) were subtracted from the minimized energy of the enzyme-substrate complex (E_enzyme-substrate complex).
| (1) |
Fluorescence-based protein thermal stability assay.
The thermal stability of recombinant GIM-1 and the mutants was determined using a fluorescence-based thermal stability assay in an MJ minicycler (Bio-Rad) (31). The assay volume used was 25 μl, which included 1.75 μM enzyme, 22.5× SYPRO Orange solution from a 5,000× stock solution (Sigma-Aldrich), and buffer E (50 mM HEPES, pH 7.2). Thermostability was also tested in the presence of 150 mM NaCl. The temperature gradient was from 10 to 70°C with an increase of 1°C per minute. The melting temperatures (Tms) were determined to be the inflection point of the melting transition found from the first derivative. All experiments were performed in triplicate.
Protein structure accession numbers.
Coordinate and structure factor files for the GIM-1 W228R (PDB accession number 5ACP), W228A (PDB accession number 5ACQ), W228Y (PDB accession number 5ACR), W228S (PDB accession number 5ACT), and Y233A (PDB accession number 5ACS) mutants were deposited in the Protein Data Bank (PDB).
RESULTS AND DISCUSSION
In class B1 MBLs, such as GIM-1, the L1 and L3 loops help to define the active-site groove. Alterations to residues within these regions have been demonstrated to affect the binding and hydrolysis of a range of β-lactam substrates by several class B1 MBLs (14, 32). In GIM-1, residues W228 and Y233 are part of the L3 loop that faces the L1 loop (residues 57 to 68) across the active-site groove, and these aromatic residues distinguish GIM-1 from other class B1 MBLs (12). The W228 side chain participates in alternative hydrogen bonds within the protein, constricting the active-site groove in the proposed binding site for the R2 substituents of cephalosporin substrates (12). Residue 233 is implicated in the binding and/or hydrolysis of β-lactam substrates (17, 25, 34), and in many MBLs this position is occupied by asparagine. In VIM-2 the asparagine is shown to interact with a mercaptocarboxylate inhibitor (16), whereas in NDM-1 it binds the hydrolyzed cefuroxime substrate (19).
In this study, the impact of W228 and Y233 substitutions on the activity and structure of GIM-1 was investigated through the generation of eight different mutants with single site-directed mutations: W228R/A/Y/S and Y233N/A/I/S.
Enzymatic activity of GIM-1 and the W228R/A/Y/S and Y233N/A/I/S mutants.
To investigate the enzymatic properties of the eight mutants that were generated in this study, kinetic analyses were carried out. The substrates for these analyses were selected to include two substrates from each main group of β-lactam antibiotics, penicillins, cephalosporins, and carbapenems (Fig. 2). The steady-state kinetic parameters for GIM-1 and the mutants with the six β-lactam substrates, ampicillin, benzylpenicillin, cefoxitin, ceftazidime, meropenem, and imipenem, are presented in Table 2. The catalytic efficiency (kcat/Km) ratios for the GIM-1 mutants compared to the ratio for wild-type GIM-1 are shown in Fig. 3. The data indicate that the two residues W228 and Y233 contribute to the substrate specificity of GIM-1. In general, the mutations resulted in a reduction in catalytic efficiency compared to that of wild-type GIM-1. In most cases, the effects of the mutations were manifested as changes in Km values rather than changes in kcat values, indicating that the substitutions at these positions primarily affect association steps rather than catalytic steps in the reaction. Overall, the deleterious effects of substitution were greater at position 233 than at position 228, with the Y233N mutant presenting the greatest reductions in catalytic efficiency, since the kcat/Km value was reduced by more than an order of magnitude for four of the six substrates tested.
TABLE 2.
Steady-state enzyme kinetics of GIM-1 and mutants with six β-lactam antibioticsa
| Antibiotic group and antiboticb | GIM-1 wild type |
GIM-1 W228R |
GIM-1 W228A |
GIM-1 W228Y |
GIM-1 W228S |
GIM-1 Y233N |
GIM-1 Y233A |
GIM-1 Y233I |
GIM-1 Y233S |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | Km (μM) | kcat (s−1) | kcat/Km (s−1/mM) | |
| Penicillins | |||||||||||||||||||||||||||
| AMP (type 2) | 147 ± 22 | 36 ± 1 | 247 | 280 ± 39 | 45 ± 2 | 159 | 216 ± 21 | 36 ± 1 | 166 | 412 ± 108 | 53 ± 5 | 128 | 310 ± 30 | 68 ± 2 | 220 | 104 ± 15 | 4.7 ± 0.2 | 45 | 320 ± 37 | 135 ± 5 | 420 | 400 ± 50 | 65 ± 3 | 160 | 500 ± 80 | 30 ± 2 | 60 |
| PEN (type 2) | 73 ± 13 | 46 ± 2 | 630 | 330 ± 30 | 96 ± 3 | 291 | 330 ± 28 | 95 ± 3 | 288 | 270 ± 30 | 95 ± 4 | 358 | 896 ± 153 | 190 ± 15 | 212 | 47 ± 18 | 5.8 ± 0.6 | 123 | 265 ± 30 | 213 ± 8 | 804 | 286 ± 61 | 92 ± 6 | 322 | 152 ± 25 | 24 ± 1 | 158 |
| Cephalosporins | |||||||||||||||||||||||||||
| FOX (type 1) | 57 ± 13 | 0.7 ± 0.1 | 12 | 37 ± 5 | 0.5 ± 0.014 | 14 | 5.7 ± 0.7 | 0.6 ± 0.01 | 113 | 33 ± 6 | 0.5 ± 0.1 | 15 | 9 ± 1 | 0.7 ± 0.014 | 80 | 190 ± 30 | 0.7 ± 0.04 | 3.7 | 160 ± 12 | 0.9 ± 0.02 | 5.6 | 980 ± 200 | 1.1 ± 0.1 | 1 | 780 ± 120 | 0.9 ± 0.1 | 1 |
| CAZ (type 2) | 12 ± 3 | 0.9 ± 0.1 | 75 | 22 ± 5 | 0.2 ± 0.023 | 9 | 17 ± 3 | 0.8 ± 0.048 | 47 | 18 ± 3 | 0.6 ± 0.04 | 33 | 10 ± 2 | 0.7 ± 0.1 | 70 | 350 ± 60 | 1.8 ± 0.2 | 5 | 28 ± 4 | 2.0 ± 0.5 | 71 | 40 ± 10 | 1.5 ± 0.2 | 38 | 76 ± 6 | 0.7 ± 0.1 | 9 |
| Carbapenems | |||||||||||||||||||||||||||
| MEM (type 1) | 20 ± 1 | 6.0 ± 0.1 | 307 | 40 ± 4 | 11.0 ± 0.3 | 289 | 27 ± 2 | 23.8 ± 0.4 | 884 | 29 ± 2 | 6.4 ± 0.1 | 224 | 27 ± 2 | 27 ± 1 | 1014 | 119 ± 7 | 1.70 ± 0.04 | 14 | 52.8 ± 3.3 | 16.0 ± 0.4 | 303 | 1242 ± 133 | 7.9 ± 0.4 | 6 | 51 ± 4 | 2.06 ± 0.05 | 40 |
| IPM (type 2) | 295 ± 16 | 15.0 ± 0.3 | 49 | 645 ± 36 | 17.0 ± 0.5 | 26 | 950 ± 110 | 32 ± 2 | 34 | 2900 ± 480 | 39 ± 4 | 13 | 950 ± 90 | 44 ± 1 | 46 | 5400 ± 800 | 13 ± 1 | 2 | 1600 ± 140 | 36 ± 2 | 23 | 510 ± 390 | 5.0 ± 0.3 | 10 | 413 ± 14 | 10.0 ± 0.2 | 24 |
Data in bold are discussed in the text.
AMP, ampicillin; PEN, benzylpenicillin; FOX, cefoxitin; CAZ, ceftazidime; MEM, meropenem; IPM, imipenem. The type of substrate according to the suggestions of Oelschlaeger et al. (34) is indicated in parentheses.
FIG 3.
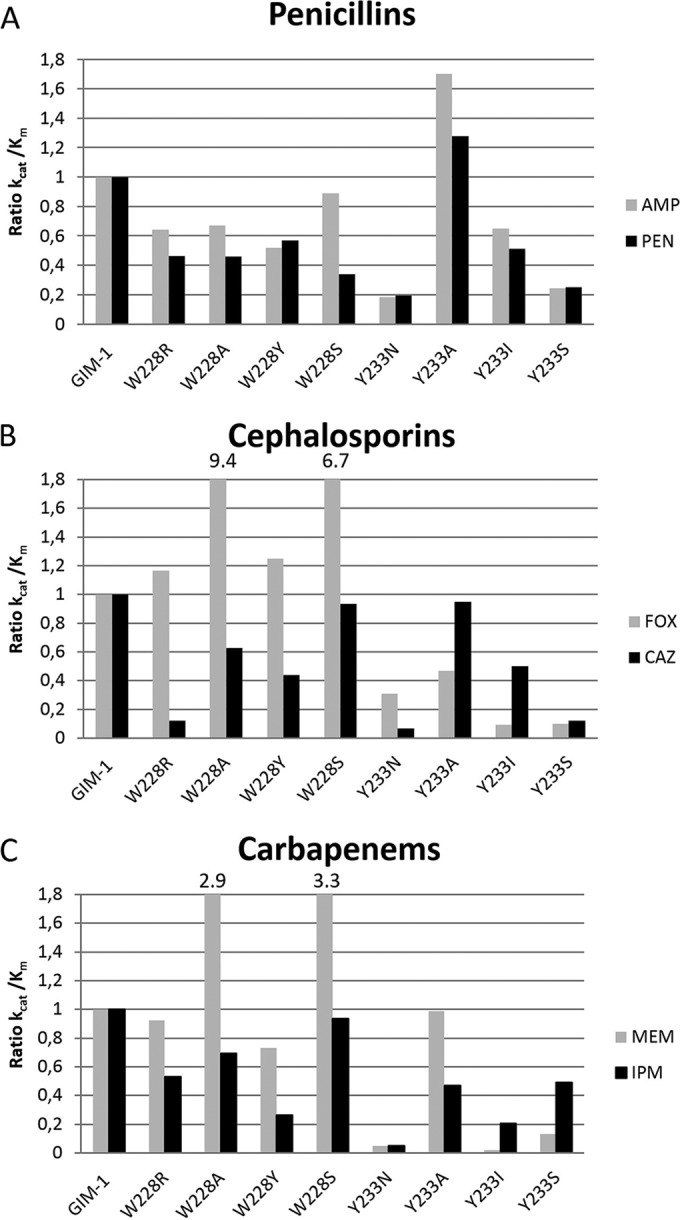
Ratio of catalytic efficiencies (kcat/Km ratios) for wild-type GIM-1 and the GIM-1 mutants toward six β-lactam antibiotics. The kcat/Km ratio for wild-type GIM-1 was set to 1, and the values for the mutants were calculated relative to this. The six β-lactam antibiotics tested were the penicillins ampicillin (AMP) and benzylpenicillin (PEN) (A), the cephalosporins cefoxitin (FOX) and ceftazidime (CAZ) (B), and the carbapenems meropenem (MEM) and imipenem (IPM) (C).
Oelschlaeger et al. (34) have suggested a division of β-lactam substrates into two types according to their structures. Type 1 substrates (cefoxitin and meropenem) have electron donors at their R2 positions, whereas type 2 substrates have axial methyl groups (ampicillin and benzylpenicillin) or a positively charged R2 side chain (ceftazidime and imipenem) (Fig. 2) (34). For some mutations, particularly those at position 228, differential effects were observed for type 1 and type 2 substrates. All four substitutions at residue 228 resulted in a modest reduction in activity against both penicillins tested. However, in the case of both the cephalosporins and carbapenems, the kcat/Km values for the hydrolysis of type 1 substrates (cefoxitin, meropenem) increased for the W228A and W228S mutants compared to the value for the wild type, while those for the hydrolysis of type 2 substrates (ceftazidime, imipenem) were little changed. Conversely, while the effects of substitution at position 233 were usually deleterious (with the exception of penicillin hydrolysis by the Y233A mutant), for mutations other than Y233N, efficiencies were, in general, less profoundly affected against type 2 substrates than against type 1 substrates. These effects were the most marked for the Y233I mutant, where, in comparison to the kcat/Km value for the wild type, the kcat/Km value for the Y233I mutant was reduced by 12- and 50-fold for the type 1 substrates cefoxitin and meropenem, respectively, but only 2- and 5-fold for the type 2 substrates ceftazidime and imipenem, respectively. Taken together, our data show that while the effects on activity against individual substrates were often modest, substitutions at these positions influenced specificity; in the case of mutations at position 228, the mutants were found to have more efficient activity against type 1 substrates, whereas mutants with changes at position 233 were found to have less efficient activity against type 2 substrates.
MIC determination.
To investigate the effects of mutations in an in vivo model system, MICs for recombinant wild-type GIM-1 and its mutants in E. coli were determined. The same substrates used in the kinetic experiments together with some additional β-lactams were tested in MIC assays. PCR products encoding full-length wild-type GIM-1, the GIM-1 mutants, and a ribosomal binding site 6 nucleotides upstream of the start codon were cloned into the pCRBluntII-TOPO vector and transformed into E. coli TOP10 cells.
Purified GIM-1 and empty E. coli TOP10 cells (Fig. 4, first two lanes) were included as positive and negative controls, respectively. E. coli TOP10 cells without GIM-1 did not show strong expression of E. coli proteins with molecular masses similar to the molecular mass of purified GIM-1 in the second lane of Fig. 4. Tests on constitutive (uninduced) whole-cell extracts established that expression levels did not differ substantially among the various mutants (Fig. 4). For all substrates with the exception of imipenem, the presence of GIM-1 or its mutants increased the MICs significantly (defined as a more than one 2-fold dilution step difference) compared to the MICs obtained with E. coli TOP10 cells without GIM-1 (Table 3). However, no clear correlation between the MIC data and the kinetic data could be observed, mainly because the MIC values with the majority of substrates did not differ substantially between wild-type GIM-1 and the mutants. Surprisingly, these findings extended to the Y233N mutant, which behaved like the wild type with all antibiotics tested, even though the in vitro experiments (Table 2) showed that the catalytic activity was the one most strongly reduced among the eight mutants tested. The exceptions where a significant difference in MICs (a ≥4-fold decrease in the MICs for the mutants compared to those for the wild type) could be observed were the MICs of piperacillin for the W228R mutant, cefepime for the W228S mutant, and meropenem and ertapenem for the Y233I mutant. Most strikingly, while expression of wild-type GIM-1 elevated the cefoxitin MIC above 256 mg/liter, this value decreased by at least 4-fold for all mutants except the W228Y and Y233N mutants.
FIG 4.

Constitutive expression levels of GIM-1 (not induced) and mutants in E. coli TOP10. (Left) Protein extracts from empty TOP10 cells and purified wild-type GIM-1 (theoretical mass of 25.63 kDa); (right) whole-cell (TOP10) extracts of wild-type GIM-1 and mutants obtained from LB cultures grown to an OD600 of 0.7, as labeled at the top. The box and arrow show the position of recombinant GIM-1. The numbers in the Marker lane are molecular masses (in kilodaltons).
TABLE 3.
MICs for recombinant full-length GIM-1 and mutants cloned in pCRBluntII-TOPO in E. coli TOP10 cells
| Isolate | MICa (mg/liter) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Penicillins |
Cephalosporins |
Monobactam |
Carbapenems |
|||||||||
| AMP | PIP | PTc | CAZ | CTX | CXM | FOX | FEP | ATM | MEM | IPM | ETP | |
| TOP10 | 4 | 4 | 8 | 0.5 | 0.125 | 8 | 4 | 0.0625 | 0.125 | 0.0625 | 1 | 0.008 |
| GIM-1 | ≥256 | 64 | ≥256 | 16 | 32 | ≥256 | ≥256 | 0.25 | 0.125 | 0.25 | 2 | 0.25 |
| W228R | ≥256 | 16 | 32 | 4 | 8 | ≥256 | 8 | 0.125 | 0.125 | 0.25 | 2 | 0.5 |
| W228A | ≥256 | 32 | 64 | 4 | 16 | ≥256 | 8 | 0.125 | 0.125 | 0.5 | 1 | 0.5 |
| W228Y | ≥256 | ≥256 | 64 | 8 | 16 | ≥256 | 64 | 0.25 | 0.125 | 0.5 | 1 | 0.5 |
| W228S | ≥256 | 32 | 64 | 8 | 16 | ≥256 | 8 | 0.0625 | 0.125 | 0.25 | 1 | 0.25 |
| Y233N | ≥256 | 128 | ≥256 | 16 | 32 | ≥256 | ≥256 | 0.5 | 0.125 | 0.125 | 4 | 0.125 |
| Y233A | ≥256 | ≥256 | ≥256 | 16 | 16 | ≥256 | 16 | 0.25 | 0.125 | 0.125 | 2 | 0.25 |
| Y233S | ≥256 | 64 | ≥256 | 16 | 16 | ≥256 | 8 | 0.25 | 0.125 | 0.25 | 1 | 0.25 |
| Y233I | ≥256 | 32 | 128 | 16 | 8 | ≥256 | 8 | 0.125 | 0.125 | 0.0625 | 1 | 0.0625 |
AMP, ampicillin; PIP, piperacillin; PTc, piperacillin-tazobactam; CAZ, ceftazidime; CTX, cefotaxime; CXM, cefuroxime; FOX, cefoxitin; FEP, cefepime; ATM; aztreonam; MEM, meropenem; IPM, imipenem; ETP, ertapenem.
As stated above, the correlation between kinetic and MIC data was variable. Changes in MICs reflected changes in catalytic efficiency in a few cases, e.g., a reduced ceftazidime MIC for the W228R mutant, for which kcat/Km fell by a factor of 8, and a reduced meropenem MIC for the Y233I mutant, for which kcat/Km fell by a factor of 50. Still, in most cases we did not observe substantial differences in the MICs between wild-type GIM-1 and its mutants. This is consistent with the relatively modest effects (≤1 order of magnitude) of most of these mutations on the overall catalytic efficiency. Further, the periplasmic environment where MBLs exert their hydrolytic activity against β-lactams is different from the in vitro environment applied in the kinetic assay; i.e., the availability of zinc ions in the periplasm has been shown to be limited (35). In addition, Gonzalez et al. have recently shown for SPM-1 that host-specific enzyme-substrate interactions are important for substrate selectivity and that the effect of specific residues on substrate selectivity is observed only in the periplasmic environment (36). Since the levels of expression of GIM-1 and the various GIM-1 mutants were similar in E. coli, this indicates that the effect of the mutations is dependent on the environment.
Analysis of W228R, W228A, W228Y, W228S, and Y233A mutant crystal structures.
In order to obtain information on the effects of mutations on the active site, crystallization trials were set up, and where crystals were obtained, molecular replacement with the wild-type GIM-1 crystal structure (PDB accession number 2YNT.A) was used to solve the mutant structures. Crystals were obtained for five of the eight GIM-1 mutants, all of which crystallized in a P21 crystal form, with two molecules being found in the asymmetric unit under conditions similar to those used for the wild-type enzyme (PDB accession number 2YNU [12]). All five mutant structures were obtained at a high resolution (1.46 to 1.98 Å) with good R factors (12.8 to 20.2%) and Rfree values (16.7 to 24.4%) and favorable geometries (Table 4). In general, the B values were lower for chain A, indicating more disorder for chain B (Table 4). All mutants showed similar packing, with the extended L1 loop (residues 57 to 68) of one chain protruding into the active site of the other. Analysis of Wilson B factors (Table 4) showed that the structure of the Y233A mutant was the most ordered, consistent with the quality of the observed electron density maps. Consequently, the structure of the Y233A mutant was used as the main reference structure in structural comparisons. Root mean square deviations (RMSDs) for CA atoms after matching of the secondary structure (30) of chain A of the Y233A mutant with the secondary structures of the other mutants were 0.28 to 0.76 Å, with the greatest divergence being observed between chain B of the W228Y mutant and chain B of the W228S mutant (RMSD, 0.975 Å). The differences were located in the L1 loop (residues 57 to 68). Similarly, differences between the structures of the various mutants and the structure of wild-type GIM-1 (Fig. 5) showed that the differences in the main-chain architecture were largely confined to loop L1 and that the mutations had little effect upon the orientation of loop L3. Thus, mutations at positions 228 and 233 can be considered to exert predominantly local, rather than global, effects upon the GIM-1 structure.
TABLE 4.
X-ray data collection and crystallographic refinement statistics for GIM-1 mutants
| Parametera | Value(s) forb: |
||||
|---|---|---|---|---|---|
| GIM-1 W228R | GIM-1W228A | GIM-1W228Y | GIM-1W228S | GIM-1Y233A | |
| PDB accession no. | 5ACP | 5ACQ | 5ACR | 5ACT | 5ACS |
| X-ray source | ID29 | ID29 | BM30 | ID29 | ID29 |
| Data collection statistics | |||||
| Space group | P21 | P21 | P21 | P21 | P21 |
| Unit cell dimensions | |||||
| a (Å) | 38.76 | 38.55 | 38.53 | 38.64 | 38.43 |
| b (Å) | 133.62 | 131.80 | 129.73 | 133.01 | 131.22 |
| c (Å) | 40.70 | 40.58 | 40.40 | 40.86 | 40.84 |
| β (°) | 95.26 | 95.51 | 96.50 | 95.28 | 94.83 |
| Resolution (Å) | 41–1.98 (2.05–1.98) | 44–1.70 (1.76–1.70) | 25–1.90 (2.0–1.9) | 45–1.81 (1.87–1.81) | 44–1.46 (1.54–1.46) |
| Wavelength (Å) | 1.2400 | 1.2400 | 0.979720 | 1.2400 | 1.2400 |
| No. of unique reflections | 28,430 (2,827) | 42,445 (4,023) | 30,606 (4,374) | 36,990 (3,628) | 68,700 (9,733) |
| Multiplicity | 2.5 (3.6) | 2.9 (2.8) | 3.5 (3.3) | 3.5 (3.5) | 3.8 (3.7) |
| Completeness (%) | 99.2 (99.4) | 96.2 (92.5) | 98.9 (97.5) | 99.1 (99.3) | 98.4 (95.8) |
| Mean (〈I〉/〈σI〉) | 13.0 (2.0) | 8.7 (2.2) | 6.5 (2.3) | 14.0 (2.1) | 12.2 (2.2) |
| Rsym (%) | 4.0 (48.6) | 6.8 (40.3) | 7.4 (46.2) | 3.8 (48.0) | 4.3 (45.8) |
| Wilson B factor (Å2) | 38.9 | 30.5 | 25.4 | 31.3 | 17.4 |
| Refinement statistics | |||||
| Resolution range (Å) | 25–1.98 | 25–1.70 | 25–1.90 | 25–1.81 | 25–1.46 |
| Rfactor (all reflections) (%) | 20.17 | 18.78 | 20.17 | 17.94 | 12.80 |
| Rfreec (%) | 23.36 | 22.70 | 24.42 | 22.95 | 16.71 |
| RMSD | |||||
| Bond length (Å) | 0.004 | 0.012 | 0.008 | 0.013 | 0.006 |
| Bond angle (°) | 0.85 | 1.38 | 1.27 | 1.39 | 1.00 |
| Average B factor (Å2) | |||||
| All atoms | 59.9 | 49.3 | 43.1 | 45.5 | 23.4 |
| Chain A/B | 48.7/71.5 | 38.6/60.4 | 31.4/54.9 | 39.4/51.9 | 21.5/24.1 |
| Solvent | 52.7 | 46.3 | 40.6 | 42.5 | 31.9 |
| Occupancy for Zn1/Zn2d | 2.0/0.2 | 1.2/0.3 | 2.0/0.0 | 2.0/0.0 | 2.0/2.0 |
I, intensity of a reflection; 〈I〉, average intensity; Rsym = [ΣhΣi|Ii(h) − 〈I(h)〉|]/[ΣhΣI I(h)], where Ii(h) is the ith measurement of reflection h and 〈I(h)〉 is the weighted mean of all measurements of h.
Values in parentheses are for the highest-resolution shell.
Five percent of the reflections were used in the Rfree calculations.
The total occupancy for Zn1 and Zn2 is 2.0, since there are two chains (A and B) in the asymmetric unit.
FIG 5.

Observed electron density maps and interactions for residue 228 or 233. 2Fo-Fc maps are displayed for W228R at 1.0σ (a), W228A at 1.2σ (b), W228Y at 1.0σ (c), W228S at 1.0σ (d), and Y233A at 1.0σ (e). (f) Superposition of the wild-type GIM-1 structure (PDB accession number 2YNT.A) and the structures of the W228R, W228A, W228Y, W228S, and Y233A mutants showing selected residues. Red spheres, water molecules; orange spheres, zinc ions. The red dotted lines (a, d) indicate hydrogen bonds, and the red arrow (d) shows the movement of the H263 residue.
The active sites of the various mutant structures are shown in Fig. 5. In the structures of all four mutants with a mutation at position 228 (the W228R, W228A, W228Y, and W228S mutants), the active-site residue Cys221 was oxidized and 1.5 to 2.0 zinc ions per crystallographic asymmetric unit (i.e., 0.75 to 1.0 zinc ion per GIM-1 molecule) were observed, despite the addition of 5 mM β-mercaptoethanol as a reducing agent in all crystallization trials. However, in the W228A and W228R mutant structures, some electron density was observed at the position of the second zinc ion (Zn2, Asp-Cys-His), enabling this ion to be modeled at a low occupancy. In contrast, the Y233A mutant structure clearly showed a reduced C221 residue and four zinc ions in the asymmetric unit, indicating that both zinc sites were fully occupied.
In wild-type GIM-1, W228 is notable for the involvement of the N atom of the indole side chain in interactions either with residue D68 at the base of loop L1, thus forming a bridge across the active site between loops L1 and L3, or with the backbone carbonyl of H263, one of the three zinc ligands in the Zn2 site (Asp-Cys-His), with W228 thus acting as a second-shell zinc ligand. H263 is also held in position by additional hydrogen bonds between its backbone carbonyl and a bound water molecule positioned close to the S225 backbone amide and the protonated ND1 of the imidazole side chain and the backbone carbonyl of D68. In addition, the adjacent G264 NH contacts the D68 carboxylate side chain via a crystallographic water molecule. Substitutions at position 228 perturb these interactions.
For the W228R mutant (Fig. 5a), the arginine side chain is positioned similarly to the W228 indole but makes only weak interactions with the H263 carbonyl (4.2 Å to R228 NE) and the D68 carboxylate side chain (3.9 Å to D68 OD1). While a shift in the backbone conformation around D68 brings its OD2 atom into hydrogen-bonding distance (3.1 Å to G264 NH) of the main chain around H263, the associated movement of the D68 carbonyl results in a repositioning of the H263 side chain in order to maintain an H-bonding interaction and, consequently, a shift in the position of Zn2 and an increase in the Zn-Zn distance from 3.49 Å in the wild type (PDB accession number 2YNW) to 4.27 Å. In this mutant the most obvious effect is a reorientation of the side chain of Y64 on the L1 loop, creating extra space in the active site in the predicted binding pocket for β-lactam R2 substituents.
Similar effects were observed for W228A (Fig. 5b), although in this case Zn2 did not reposition to track the H263 side chain, and the result was an increase in the H263 NE2-Zn2 distance from 2.14 Å to 2.47 Å. Equally, for the W228Y mutant (Fig. 5c), while the H263 backbone conformation was maintained by interactions of the H263 carbonyl with the S225 amide (via a water molecule) and of the G264 amide with the D68 side chain, the position of the H263 side chain again shifted to maintain contact with the D68 carbonyl. However, in the W228A mutant structure no electron density was observed for Zn2, and as a consequence, the position of this metal ion could not be modeled.
The most dramatic effects were observed for the W228S mutant. Although the backbone conformations around residues 68 and 263 were very similar to those in the other three W228 mutant structures, in this instance the H263 side chain was rotated out of the active site and ND1 contacted the side chain carboxylate of D68 (Fig. 5d). It is also notable that, in the absence of Zn2 from this structure, the Zn2 ligand D120 was partially disordered. However, in all four W228 mutant structures, the positions of other residues, such as R224 and Y233, that are likely to be involved in interactions with the substrate were little affected compared to their positions in the wild-type enzyme, emphasizing that the effects of the mutations at position 228 are local rather than global in their extent. However, residue Y233 was found in the same orientation in all four W228 mutants, as shown in Fig. 5f. The Y233A crystal structure was very similar to the four W228 mutant structures, but the L1 loop was slightly more closed (closer to the L3 loop) even if the L1 loop was even more closed in wild-type GIM-1 (PDB accession number 2YNT.A) (Fig. 5e and f).
Compared to the effects of mutations at W228 on the structures of the mutants, the structure of the Y233A mutant (Fig. 5e) differed less profoundly from that of the wild-type enzyme. Notably, both zinc ions were present in the active site and a hydrogen bond was observed between the W228 indole N and the main-chain carbonyl of H263. However, unlike the wild-type enzyme, direct hydrogen bonds were observed both between the D68 side chain and the G264 amide and between the D68 carbonyl and H263 ND1. As a consequence, the zinc-zinc distance increased to 4.06 and 3.80 Å in chains A and B, respectively. Taken together, the crystal structures indicate that mutations at both positions 228 and 233 can perturb the local architecture of the GIM-1 active site.
In silico modeling of hydrolyzed ampicillin binding to GIM-1 and GIM-1 Y233N.
As we were unable to obtain a crystal structure of the GIM-1 Y233N mutant and this was the variant for which the most profound changes in activity were observed, in silico models of GIM-1 and GIM-1 Y233N bound to hydrolyzed ampicillin were constructed (Fig. 6) in order to investigate the interactions between them and to undertake binding affinity calculations. The ability of the Schrödinger Liaison module to account for flexibility within the active-site regions of GIM-1 and GIM-1 Y233N, particularly the L1 and L3 loops, permitted positioning of hydrolyzed ampicillin itself within a reasonable distance of both zinc ions (2.07 Å to Zn1 and 1.93 Å to Zn2). The position of the hydrolyzed β-lactam varied little between the wild-type and mutant enzymes. However, due to the mutation at position 233, the interactions made by the C-7 carboxylate group of hydrolyzed ampicillin differed between the two structures. In wild-type GIM-1, one oxygen atom of this carboxylate group contacted Zn1, while the other lay close (approximately 3.0 Å) to the aromatic ring of Y233. In the Y233N mutant, a hydrogen bond was formed between the side chain amide of the N233 residue and the carboxylate of the hydrolyzed ampicillin ligand. We then calculated binding affinities from these models. The estimated relative affinities of binding of hydrolyzed ampicillin to wild-type GIM-1 and GIM-1 Y233N were −8.7 kcal/mol and −5.0 kcal/mol, respectively, indicating that the binding of hydrolyzed ampicillin to wild-type GIM-1 is stronger than that to GIM-1 Y233N. The difference in the estimated affinities of binding of hydrolyzed ampicillin to wild-type GIM-1 and GIM-1 Y233N was 3.7 kcal/mol, which is on the order of one hydrogen bond. In the in silico models, the major difference was the anion pi interaction from the C-7 carboxyl on ampicillin to the aromatic ring of Y233 for wild-type GIM-1. This interaction which is a hydrogen bond in the GIM-1 Y233N mutant. The calculated relative binding affinities correlate with the kinetic measurements of ampicillin hydrolysis by wild-type GIM-1 and GIM-1 Y233N, where Km was lower for wild-type GIM-1, consistent with the higher affinity of the wild-type protein for ampicillin. The additional hydrogen bond in the GIM-1 Y233N complex to ampicillin then seems to be less favorable than the anion pi interaction of wild-type GIM-1 with ampicillin in our calculations and enzyme kinetics experiments. However, a direct comparison of the calculated and the measured binding affinities is difficult. The binding affinities calculated from the modeling were based on a hydrolyzed ampicillin with two negative charges, whereas the measured binding affinity, Km, was measured with unhydrolyzed ampicillin with one negative charge. Although our GIM-1 Y233N mutation resulted in the most profound changes in activity, previous reports have shown that N233 is not critical for enzyme activity (37–39).
FIG 6.
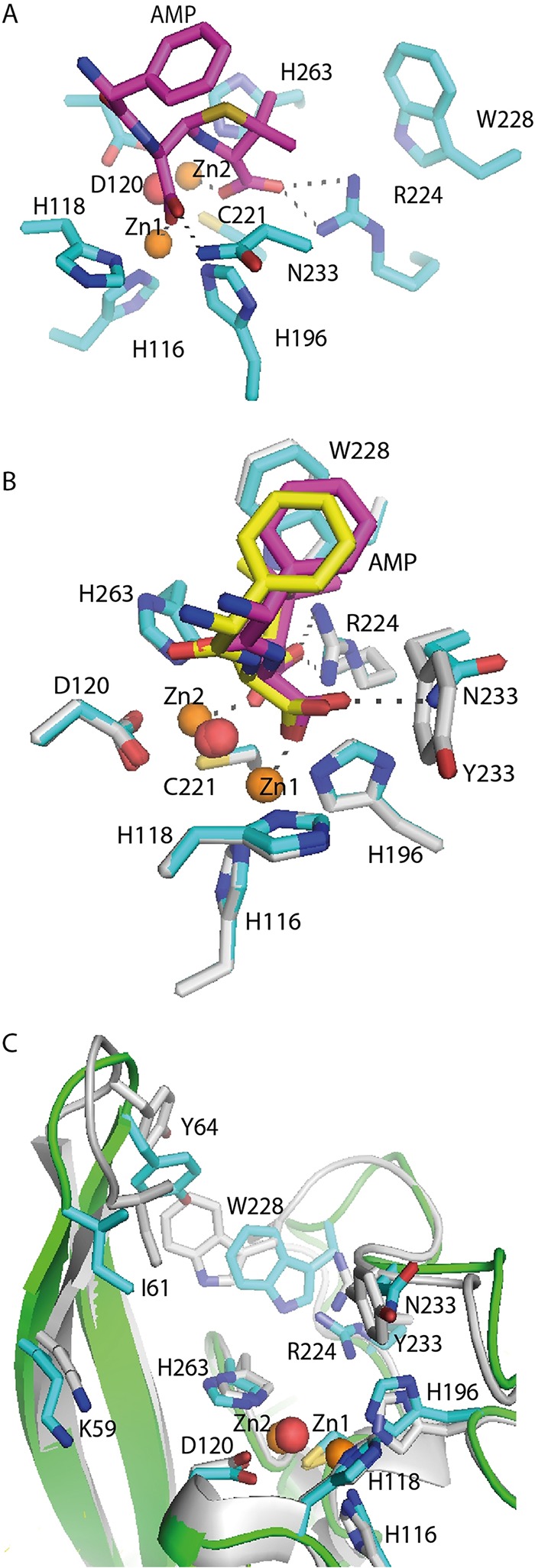
In silico model of GIM-1 Y233N with hydrolyzed ampicillin in the active site and the GIM-1 Y233N model superimposed on the model of native GIM-1. (A and B) A closeup of the active site in the in silico model of the GIM-1 Y233N mutant (cyan/magenta) (A) and of the model of the GIM-1 Y233N mutant superimposed on wild-type GIM-1 (gray/yellow) with hydrolyzed ampicillin (AMP) (B). (C) Superimposition of the model of GIM-1 (gray; PDB accession number 2YNW.B) and that of GIM-1 Y233N (green backbone and cyan side chains) showing the movement of the L1 and L3 loops. Red spheres, water molecules; orange spheres, zinc ions.
Thermostability measured by thermofluor-based assays.
In order to explain the differences in activity between wild-type GIM-1 and the mutants, thermostability measurements were performed. The fluorescence-based thermostability measurements revealed no significant differences in melting temperature (Tm) between GIM-1 and the mutants since all had Tms ranging from 56.8 ± 0.2 to 57.1 ± 0.2°C (Fig. 7) in 50 mM HEPES, pH 7.2. To investigate the effect of salt, the thermostability measurements were also performed in the same buffer with 150 mM NaCl, where the presence of NaCl decreased the melting points by approximately 3°C but did not reveal differences between wild-type GIM-1 and the mutants (data not shown). Thus, the changes in hydrogen bonding around the active site described above did not change the thermostability of GIM-1, in contrast to previous observations on VIM-7, where mutants making additional hydrogen bonds (F218Y and H224Y) also had increased Tms (14). Thus, differences in stability are unlikely to explain the observed differences in the catalytic properties of the different GIM-1 variants.
FIG 7.

Thermostability of GIM-1 and mutants. Fluorescence signals are for wild-type GIM-1 and the W228R, W228A, W228Y, W228S, Y233N, Y233A, Y233I, and Y233S mutants. Mean ± standard deviation melting temperatures are given in parentheses. The assay was performed in 50 mM HEPES (pH 7.2).
Conclusion.
This study shows that residues W228 and Y233 are important to the activity of GIM-1, as previously demonstrated for their equivalents in other MBLs (15–17). Our steady-state data reveal that a mutation at either position affects activity and is, in general, deleterious, but it can also influence specificity toward different β-lactam classes. Notably, introduction of a less bulky side chain of Ala or Ser at position 228 reduces the activity against most β-lactams but also increases the efficiency of hydrolysis of the type 1 substrates cefoxitin and meropenem above that of the wild type. A mutation at position 233 is generally more harmful to activity, but these effects were more marked for type 2 substrates than type 1 substrates. However, the retention of significant catalytic activity with a kcat/Km of ≥1 s−1/mM for all enzyme-substrate combinations tested, coupled with the lack of profound effects upon the MIC or no effects on the MICs, indicates that neither position is essential to the structural integrity or activity of GIM-1. This finding is in broad agreement with the conclusions of a comprehensive study of the effects of substitutions of N233 in the IMP-1 enzyme (17), which demonstrated that a wide range of amino acids could be tolerated at this position and that the effects of mutations varied markedly with different β-lactam substrates. These authors also noted that substitutions were generally associated with increases in Km values, again in accordance with what is reported here (17).
Consistent with the results of solution experiments, X-ray crystal structures of the four W228 mutants and the Y233A mutant did not reveal that these mutations introduce profound structural changes, as indicated by the low RMSD values for CA atoms upon superposition of the mutant structures with the structure of the wild-type enzyme. Interpretation of the structural data is also complicated by the involvement of the L1 loop in crystal contacts in our new structures. However, our data do reveal that mutations at both positions 228 and 233 can exert subtle effects upon the GIM-1 active-site architecture, in particular, by altering interactions involving H263 in the Zn2 site. In all five mutant structures, we observed a repositioning, in comparison to the positioning in the wild type, of residue D68 at the base of the L1 loop that in turn affected the location of the Zn2 ligand H263. The consequences of this rearrangement may include increases in the Zn-Zn distance, as seen by the partial or complete loss of the Zn2 ion in our crystallized samples. As demonstrated by several previous studies (see, e.g., references 36 and 40), disruption by mutation of hydrogen-bonding networks involving interactions with zinc-binding residues in so-called second-shell ligands or repositioning of Zn2 in the active site of class B1 MBLs can exert significant effects upon activity. It is thus reasonable to propose that the observed effects of mutations upon GIM-1 activity may arise from subtle modifications to the Zn2 site. While in the majority of instances these are likely to be deleterious, in some cases they may favor the hydrolysis of particular substrates, as evidenced by small increases in activity of, e.g., some W228 mutants against type 1 substrates.
Modeling of the mode of binding of hydrolyzed ampicillin to the GIM-1 active site also provides new insights into the contribution of residue 233 to substrate binding by class B1 MBLs. Multiple lines of evidence, including crystal structures (19), implicate N233 in the making of hydrogen-bonding interactions with the C-7 carboxylate group of hydrolyzed β-lactams via its side chain amide. In enzymes such as GIM-1, such interactions would not be possible due to the presence of Y233, and the deleterious effects of the Y233N mutation upon β-lactam hydrolysis might also be considered surprising. Our modeling suggests, however, that Y233 makes a positive contribution to β-lactam binding, as evidenced by close contact between the C-7 carboxylate of hydrolyzed ampicillin and the aromatic ring of the Y233 side chain and by the substantial difference in the calculated binding affinity between wild-type GIM-1 and the Y233N mutant. However, the relatively modest effect upon the activity of the mutant with the Y233A substitution, in which, on the basis of the crystal structure, interactions between the β-lactam C-3/C-4 carboxylate and the protein would not be expected to occur without a substantial conformational rearrangement, also indicates that this contribution is positive but limited. This is consistent with the generally accepted conclusion that interactions involving the metal ions, rather than specific residues within the active site, are more important with respect to β-lactam binding and hydrolysis by MBLs.
In summary, our data provide evidence for an important but nonessential role of two residues, W228 and Y233, in the activity and specificity of GIM-1 that distinguish this and similar MBLs, such as DIM-1, from other class B1 enzymes. In addition to the contribution of these bulky side chains to defining the active-site groove, it is clear that both W228 and Y233 help to define the active-site architecture, in particular about the Zn2 site, and that Y233 is also able to interact with bound β-lactam substrates.
Establishing how specific residues such as W228 and Y233 contribute to substrate binding by GIM-1 may assist with the development of inhibitors able to combat the β-lactam resistance conferred by the presence of MBLs, including GIM-1, by identifying interactions that may be exploited in designed ligands.
ACKNOWLEDGMENTS
We acknowledge Marit Sjo Lorentzen for help with the design of primers for the GIM-1 site-directed mutants, Osman A. B. S. M. Ghani for valuable help with the model-building process, and Ronny Helland for help during data collection. Provision of beam time at ID29 and BM30 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France, is highly valued.
Author contributions were as follows: H.-K.S.L., S.S., G.E.K.B., and Ø.S. designed the experiments; T.J.C. and S.S. performed the cloning, expression and purification; S.S., Ø.S., T.J.C., and J.S. performed the biochemical characterization; T.J.C. performed the crystallization; S.S. and H.-K.S.L. solved, evaluated, and docked the three-dimensional structures; and S.S., H.-K.S.L., Ø.S., G.E.K.B., and J.S. analyzed the data and wrote the paper. All authors have given approval to the final version of the manuscript.
Funding Statement
Funding from the Northern Norway Regional Health Authority, Tromsø, Research Foundation, and Research Council of Norway (FRIMEDBIO project number 213808) are all acknowledged. Research on metallo-β-lactamases in J.S.'s laboratory is supported by the United Kingdom Medical Research Council (United Kingdom-Canada team grant G1100135) and by the National Institute of Allergy and Infectious Diseases of the U.S. National Institutes of Health via grant R01AI100560.
REFERENCES
- 1.Bush K. 2013. Carbapenemases: partners in crime. J Global Antimicrob Resist 1:7–16. doi: 10.1016/j.jgar.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Palzkill T. 2013. Metallo-β-lactamase structure and function. Ann N Y Acad Sci 1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bush K. 2013. The ABCD's of β-lactamase nomenclature. J Infect Chemother 19:549–559. doi: 10.1007/s10156-013-0640-7. [DOI] [PubMed] [Google Scholar]
- 4.Toussaint KA, Gallagher JC. 2015. β-Lactam/β-lactamase inhibitor combinations: from then to now. Ann Pharmacother 49:86–98. doi: 10.1177/1060028014556652. [DOI] [PubMed] [Google Scholar]
- 5.Liscio JL, Mahoney MV, Hirsch EB. 2015. Ceftolozane/tazobactam and ceftazidime/avibactam: two novel β-lactam/β-lactamase inhibitor combination agents for the treatment of resistant Gram-negative bacterial infections. Int J Antimicrob Agents 46:266–271. doi: 10.1016/j.ijantimicag.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Castanheira M, Toleman MA, Jones RN, Schmidt FJ, Walsh TR. 2004. Molecular characterization of a β-lactamase gene, blaGIM-1, encoding a new subclass of metallo-β-lactamase. Antimicrob Agents Chemother 48:4654–4661. doi: 10.1128/AAC.48.12.4654-4661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rieber H, Frontzek A, Pfeifer Y. 2012. Emergence of metallo-β-lactamase GIM-1 in a clinical isolate of Serratia marcescens. Antimicrob Agents Chemother 56:4945–4947. doi: 10.1128/AAC.00405-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamprecht A, Poirel L, Gottig S, Seifert H, Kaase M, Nordmann P. 2013. Detection of the carbapenemase GIM-1 in Enterobacter cloacae in Germany. J Antimicrob Chemother 68:558–561. doi: 10.1093/jac/dks447. [DOI] [PubMed] [Google Scholar]
- 9.Kaase M, Szabados F, Pfennigwerth N, Anders A, Geis G, Pranada AB, Rossler S, Lang U, Gatermann SG. 2014. Description of the metallo-β-lactamase GIM-1 in Acinetobacter pittii. J Antimicrob Chemother 69:81–84. doi: 10.1093/jac/dkt325. [DOI] [PubMed] [Google Scholar]
- 10.Wendel AF, MacKenzie CR. 2015. Characterization of a novel metallo-β-lactamase variant, GIM-2, from a clinical isolate of Enterobacter cloacae in Germany. Antimicrob Agents Chemother 59:1824–1825. doi: 10.1128/AAC.05062-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galleni M, Lamotte-Brasseur J, Rossolini GM, Spencer J, Dideberg O, Frere JM, The Metallo-β-Lactamase Working Group . 2001. Standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother 45:660–663. doi: 10.1128/AAC.45.3.660-663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borra PS, Samuelsen Ø Spencer J, Walsh TR, Lorentzen MS, Leiros H-KS. 2013. Crystal structures of Pseudomonas aeruginosa GIM-1: active-site plasticity in metallo-β-lactamases. Antimicrob Agents Chemother 57:848–854. doi: 10.1128/AAC.02227-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oelschlaeger P, Schmid RD, Pleiss J. 2003. Modeling domino effects in enzymes: molecular basis of the substrate specificity of the bacterial metallo-β-lactamases IMP-1 and IMP-6. Biochemistry 42:8945–8956. doi: 10.1021/bi0300332. [DOI] [PubMed] [Google Scholar]
- 14.Leiros H-KS, Skagseth S, Edvardsen KS, Lorentzen MS, Bjerga GE, Leiros I, Samuelsen Ø. 2014. His224 alters the R2 drug binding site and Phe218 influences the catalytic efficiency of the metallo-β-lactamase VIM-7. Antimicrob Agents Chemother 58:4826–4836. doi: 10.1128/AAC.02735-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Materon IC, Beharry Z, Huang W, Perez C, Palzkill T. 2004. Analysis of the context dependent sequence requirements of active site residues in the metallo-β-lactamase IMP-1. J Mol Biol 344:653–663. doi: 10.1016/j.jmb.2004.09.074. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi Y, Jin W, Matsunaga K, Ikemizu S, Yamagata Y, Wachino J, Shibata N, Arakawa Y, Kurosaki H. 2007. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-β-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J Med Chem 50:6647–6653. doi: 10.1021/jm701031n. [DOI] [PubMed] [Google Scholar]
- 17.Brown NG, Horton LB, Huang W, Vongpunsawad S, Palzkill T. 2011. Analysis of the functional contributions of Asn233 in metallo-β-lactamase IMP-1. Antimicrob Agents Chemother 55:5696–5702. doi: 10.1128/AAC.00340-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garau G, Bebrone C, Anne C, Galleni M, Frere JM, Dideberg O. 2005. A metallo-β-lactamase enzyme in action: crystal structures of the monozinc carbapenemase CphA and its complex with biapenem. J Mol Biol 345:785–795. doi: 10.1016/j.jmb.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 19.Feng H, Ding J, Zhu D, Liu X, Xu X, Zhang Y, Zang S, Wang DC, Liu W. 2014. Structural and mechanistic insights into NDM-1 catalyzed hydrolysis of cephalosporins. J Am Chem Soc 136:14694–14697. doi: 10.1021/ja508388e. [DOI] [PubMed] [Google Scholar]
- 20.Garau G, Garcia-Saez I, Bebrone C, Anne C, Mercuri P, Galleni M, Frere JM, Dideberg O. 2004. Update of the standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother 48:2347–2349. doi: 10.1128/AAC.48.7.2347-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogulin EA, Perevyazova TA, Zheleznaya LA, Matvienko NI. 2004. Plasmid pRARE as a vector for cloning to construct a superproducer of the site-specific nickase N.BspD6I. Biochemistry (Moscow) 69:1123–1127. doi: 10.1023/B:BIRY.0000046886.19428.d5. [DOI] [PubMed] [Google Scholar]
- 22.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Laraki N, Franceschini N, Rossolini GM, Santucci P, Meunier C, de Pauw E, Amicosante G, Frere JM, Galleni M. 1999. Biochemical characterization of the Pseudomonas aeruginosa 101/1477 metallo-β-lactamase IMP-1 produced by Escherichia coli. Antimicrob Agents Chemother 43:902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siemann S, Evanoff DP, Marrone L, Clarke AJ, Viswanatha T, Dmitrienko GI. 2002. N-Arylsulfonyl hydrazones as inhibitors of IMP-1 metallo-β-lactamase. Antimicrob Agents Chemother 46:2450–2457. doi: 10.1128/AAC.46.8.2450-2457.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Saez I, Docquier JD, Rossolini GM, Dideberg O. 2008. The three-dimensional structure of VIM-2, a Zn-β-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form. J Mol Biol 375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 26.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Hao Q. 2011. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J 25:2574–2582. doi: 10.1096/fj.11-184036. [DOI] [PubMed] [Google Scholar]
- 30.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 31.Layton CJ, Hellinga HW. 2010. Thermodynamic analysis of ligand-induced changes in protein thermal unfolding applied to high-throughput determination of ligand affinities with extrinsic fluorescent dyes. Biochemistry 49:10831–10841. doi: 10.1021/bi101414z. [DOI] [PubMed] [Google Scholar]
- 32.Moali C, Anne C, Lamotte-Brasseur J, Groslambert S, Devreese B, Van Beeumen J, Galleni M, Frere JM. 2003. Analysis of the importance of the metallo-β-lactamase active site loop in substrate binding and catalysis. Chem Biol 10:319–329. doi: 10.1016/S1074-5521(03)00070-X. [DOI] [PubMed] [Google Scholar]
- 33.Reference deleted.
- 34.Oelschlaeger P, Mayo SL, Pleiss J. 2005. Impact of remote mutations on metallo-β-lactamase substrate specificity: implications for the evolution of antibiotic resistance. Protein Sci 14:765–774. doi: 10.1110/ps.041093405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez JM, Meini MR, Tomatis PE, Medrano Martin FJ, Cricco JA, Vila AJ. 2012. Metallo-β-lactamases withstand low Zn(II) conditions by tuning metal-ligand interactions. Nat Chem Biol 8:698–700. doi: 10.1038/nchembio.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez LJ, Moreno DM, Bonomo RA, Vila AJ. 2014. Host-specific enzyme-substrate interactions in SPM-1 metallo-β-lactamase are modulated by second sphere residues. PLoS Pathog 10:e1003817. doi: 10.1371/journal.ppat.1003817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haruta S, Yamamoto ET, Eriguchi Y, Sawai T. 2001. Characterization of the active-site residues asparagine 167 and lysine 161 of the IMP-1 metallo beta-lactamase. FEMS Microbiol Lett 197:85–89. doi: 10.1111/j.1574-6968.2001.tb10587.x. [DOI] [PubMed] [Google Scholar]
- 38.Materon IC, Palzkill T. 2001. Identification of residues critical for metallo-β-lactamase function by codon randomization and selection. Protein Sci 10:2556–2565. doi: 10.1110/ps.ps.40884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oelschlaeger P, Schmid RD, Pleiss J. 2003. Insight into the mechanism of the IMP-1 metallo-β-lactamase by molecular dynamics simulations. Protein Eng 16:341–350. doi: 10.1093/protein/gzg049. [DOI] [PubMed] [Google Scholar]
- 40.Tomatis PE, Rasia RM, Segovia L, Vila AJ. 2005. Mimicking natural evolution in metallo-β-lactamases through second-shell ligand mutations. Proc Natl Acad Sci U S A 102:13761–13766. doi: 10.1073/pnas.0503495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]