

Abstract
Despite dose-limiting nephrotoxicity concerns, polymyxin B has resurged as the treatment of last resort for multidrug-resistant Gram-negative bacterial infections. However, the pharmacokinetic, pharmacodynamic, and nephrotoxic properties of polymyxin B still are not thoroughly understood. The objective of this study was to provide additional insights into the overall biodistribution and disposition of polymyxin B in an animal model. Sprague-Dawley rats were dosed with intravenous polymyxin B (3 mg/kg of body weight). Drug concentrations in the serum, urine, bile, and tissue (brain, heart, lungs, liver, spleen, kidneys, and skeletal muscle) samples over time were assayed by a validated methodology. Among all the organs evaluated, polymyxin B distribution was highest in the kidneys. The mean renal tissue/serum polymyxin B concentration ratios were 7.45 (95% confidence interval [CI], 4.63 to 10.27) at 3 h and 19.62 (95% CI, 5.02 to 34.22) at 6 h postdose. Intrarenal drug distribution was examined by immunostaining. Using a ratiometric analysis, proximal tubular cells showed the highest accumulation of polymyxin B (Mander's overlap coefficient, 0.998) among all cell types evaluated. Less than 5% of the administered dose was recovered in urine over 48 h, but all 4 major polymyxin B components were detected in the bile over 4 h. These findings corroborate previous results that polymyxin B is highly accumulated in the kidneys, but the elimination likely is via a nonrenal route. Biliary excretion could be one of the routes of polymyxin B elimination, and this should be further explored. The elucidation of mechanism(s) of drug uptake in proximal tubular cells is ongoing.
INTRODUCTION
The emergence of multidrug-resistant bacterial infections has become a medical crisis worldwide (1, 2). Infections caused by Gram-negative bacteria, such as Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter spp., are extremely challenging to treat (3–6). These infections also are associated with high rates of mortality and morbidity (7, 8). Moreover, there are few new antibacterial agents available in the clinical drug development pipeline for these life-threatening infections. Consequently, this has led to the revival of old antibiotics, such as the polymyxins, as the treatment of last resort for infections caused by multidrug-resistant Gram-negative pathogens (9–13).
Polymyxins (primarily polymyxin B and polymyxin E [colistin]) are cyclic polypeptide antibiotics isolated from Bacillus polymyxa (14). Commercially available polymyxin B is a mixture of several related analogs, primarily polymyxin B1, B2, and B3 and isoleucine B1 (15, 16). Polymyxin B first became available for clinical use in the 1950s, but its clinical use has been limited largely due to its nephrotoxic potential.
Despite being available for clinical use for more than 50 years, there is still a paucity of published reports correlating the pharmacokinetics of polymyxin B with its toxicity profile. Furthermore, we lack a thorough understanding of the biodistribution pattern, cellular disposition, elimination pathways, and transport characteristics of polymyxin B in vivo. Therefore, studies to delineate the pharmacokinetic, pharmacodynamic, and toxicodynamic profile of polymyxin B are warranted. Undoubtedly, such information will play a pivotal role in designing the optimal polymyxin B dosing strategies, which will maximize the clinical efficacy as well as safety of the drug.
The objective of this study was to provide additional insights into the overall biodistribution and disposition characteristics of polymyxin B using an animal model. It is anticipated that the outcomes from this investigation will unravel the intricacies in our current understanding of pharmacokinetic properties of polymyxin B. The results obtained from this research could guide future studies in designing the optimal dosing strategies for polymyxin B.
(This study was presented in part at the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, 5 to 9 September 2014 [17], as well as in part at the 55th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Diego, 17 to 21 September 2015 [18]).
MATERIALS AND METHODS
Chemicals and reagents.
Polymyxin B sulfate (USP) powder, trichloroacetic acid, phosphate-buffered saline with 0.01% Tween 80 (PBST), 4% paraformaldehyde (PFA), and 30% sucrose solution in phosphate-buffered saline (PBS) were purchased from Sigma-Aldrich (St. Louis, MO). Carbutamide was purchased from Aldrich Chemical Co. (Milwaukee, WI). Liquid chromatography-mass spectrometry (LC-MS)-grade acetonitrile and water were obtained from Mallinckrodt Baker (Philipsburg, NJ). LC-MS-grade formic acid was purchased from Fluka Analytical (St. Louis, MO). Murine anti-polymyxin B antibody was purchased from Thermo Scientific (Rockford, IL). Normal goat serum, streptavidin DyLight 488 (green) and 649 (red), and fluorogel-II with 4′,6-diamidino-2-phenylindole (DAPI) (blue) were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Anti-mouse IgG secondary antibody, avidin-biotin blocking kits, and biotinylated lectins were purchased from Vector Laboratories (Burlingame, CA). The natural sources, targeted nephron regions, and optimal dilutions of various lectins are listed in Table 1.
TABLE 1.
Lectins used for the staining of rat kidney sections
| Lectin | Source | Target | Final dilution of lectin in PBST (μg/ml) |
|---|---|---|---|
| PHA-E | Red kidney bean | Proximal convoluted tubules (PCT) | 10 |
| SBA | Soybean | Distal convoluted tubules (DCT) | 20 |
| WGA | Wheat germ | Glomerulus | 20 |
Animals.
Female Sprague-Dawley rats (225 to 250 g) (Harlan, Indianapolis, IN) were used. Jugular veins were cannulated to facilitate intravenous drug administration. The rats received food and water ad libitum. All animals were cared for in accordance with the highest humane and ethical standards, as approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Houston.
Polymyxin B assay.
A validated ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) method was modified to determine the concentrations of polymyxin B in rat serum, bile, brain, heart, lungs, liver, spleen, kidneys, and skeletal muscle tissues (19). Briefly, serum, bile, or tissue homogenate was spiked with 10 μl of an internal standard (5 μg/ml carbutamide) and extracted with 150 μl of 5% trichloroacetic acid. The samples were vortexed for 1 min followed by centrifugation as previously described (19). The supernatant was transferred to a new tube and evaporated to dryness under a stream of ambient air. The residue then was reconstituted with 100 μl of a mixture of acetonitrile and 0.1% formic acid (1:1, vol/vol). The samples again were centrifuged at 18,000 × g for 20 min at 4°C, and 10 μl of the supernatant was injected into the UPLC-MS/MS system for quantitative analysis. The bile samples were treated in a fashion similar to that for serum or tissue homogenate, except for the inclusion of blank rat serum (>2 times the volume of bile) with the bile as an attempt to minimize the matrix effect caused by bile components.
The calibration curves in serum/bile/tissue samples were constructed using at least eight concentration standards. The linearity of the calibration curves was determined by the best fit of peak area ratios (analyte/internal standard) versus concentrations and fitted using a linear regression (1/x2 weighting) method. The lower limit of quantification (LLOQ) was determined based on a signal-to-noise ratio of 10:1. The assay was validated based on accuracy, precision, and inter- and intraday variability, which were well within 15% of the coefficient of variation (CV) and did not exceed 20% of the CV for the LLOQ (19).
A validated microbiological assay method also was modified to determine the concentrations of polymyxin B in rat urine samples as previously described (20). Bordetella bronchiseptica ATCC 10580, obtained from the American Type Culture Collection (Manassas, VA), was used as the reference organism. The calibration curves were made by plotting the inhibition zone diameter (in millimeters) versus the logarithmic standard polymyxin B drug concentration in urine. The assay was validated previously based on accuracy, precision, and inter- and intraday variability; LLOQ was determined to be 2 μg/ml.
Biodistribution of polymyxin B.
Prior to each experiment, polymyxin B powder was dissolved in sterile water for injection and diluted to the desired concentration. The rats were given a single intravenous dose of polymyxin B (3 mg/kg of body weight) and sacrificed after 3 h (n = 3), 6 h (n = 6), and 24 h (n = 3). The harvested tissues were weighed and homogenized in deionized water (1:2). The mean tissue/serum drug concentration ratios were estimated for each tissue matrix. The groups were compared using one-way analysis of variance (ANOVA), followed by Tukey's post hoc test. P values of <0.05 were considered significant.
Intrarenal distribution of polymyxin B. (i) Harvesting and processing of rat kidney.
The rats (n = 3) were given a single intravenous dose of polymyxin B (4 mg/kg) and sacrificed at 6 h postdose. The kidneys were perfusion fixed with 4% paraformaldehyde and stored overnight. Subsequently, the kidneys were cryoprotected in 30% sucrose solution in PBS and finally cryosectioned at 20-μm intervals into thin tissue cross-sections.
(ii) Identification of renal cell type and immunostaining for polymyxin B.
Different anatomical regions of the nephron were identified by staining with specific lectin markers. The fresh frozen kidney tissue sections were placed in PBST maintained at pH 7.4 for 5 to 10 min before staining. Briefly, the sections were preincubated in avidin-biotin blocking solution, followed by incubation with biotinylated lectins diluted in PBST to the concentrations specified in Table 1, each for 30 min at room temperature. After washing 3 times in PBST, the sections were incubated for 30 min at room temperature with streptavidin DyLight 649 (10 μg/ml diluted in PBST). Subsequently, the sections were washed in PBST, followed by washing 3 times in distilled water. The sections then were mounted using the fluorogel-II mounting agent with DAPI. Controls were prepared by the same staining procedure, except that the lectins were replaced by PBST.
The double immunostaining of renal tissue sections was carried out using polymyxin B antibody and lectins. The sections were incubated with 5% normal goat serum followed by incubation with avidin-biotin blocking solution for 30 min at room temperature. Subsequently, the sections were treated with polymyxin B antibody (1:20 dilution in 5% normal goat serum in PBS) for 1 h at 4°C and later incubated with biotinylated mouse anti-polymyxin B antibody (1:150 dilution in 5% normal goat serum). After washing 3 times in PBST, the sections were incubated with streptavidin DyLight 488 (10 μg/ml, diluted in PBST) for 30 min each at room temperature. Subsequently, staining with a specific lectin marker was performed on the same tissue section as described above. Controls were prepared by the same staining procedure, except primary antibody and lectins were replaced by PBST.
(iii) Colocalization of polymyxin B antibody with lectin.
Images were captured using an Olympus IX61 DSU. Several regions of interest (ROI) were randomly selected from individual overlay/colocalized images for each cell type. Selected ROI(s) from each overlay image were further analyzed using a computer-assisted software Image J (version 1.47); the Mander's overlap coefficient (Rcolo), which signifies the percentage of overlap of two signal intensities, was calculated using the Just Another Colocalization Plug-in (JACoP). A coefficient value above 0.7 was deemed to be a good degree of colocalization.
Elimination of polymyxin B. (i) Pre- and postdose urine sampling.
The rats (n = 3) were housed individually in separate metabolic cages to facilitate urine collection. Blank urine was collected from each rat prior to the administration of a single intravenous dose of polymyxin B (3 mg/kg). Subsequently, the cumulative urine of each rat was collected in aliquots for up to 48 h following drug administration. The urine was concentrated 2× by evaporation to reduce the lower limit of detection.
(ii) Bile sampling.
The bile ducts of anesthetized rats were cannulated to facilitate bile sampling (n = 5). Prior to drug administration, the blank bile was collected (over 30 min) from each rat. The rats were given a single intravenous dose of polymyxin B (3 mg/kg), and the bile was further collected cumulatively for another 4 h.
RESULTS
Polymyxin B assay.
The linear concentration ranges for serum, bile, brain, heart, lungs, liver, spleen, kidneys, and muscle tissue homogenate were 0.1 to 12.8 μg/ml, 0.05 to 12.8 μg/ml, 0.05 to 12.8 μg/ml, 0.05 to 12.8 μg/ml, 0.05 to 12.8 μg/ml, 0.0125 to 12.8 μg/ml, 0.05 to 12.8 μg/ml, 0.05 to 12.8 μg/ml, and 0.1 to 6.4 μg/ml, respectively. The linear regression coefficients were ≥0.99 in all calibration curves. For the microbial assay of polymyxin B in urine, the linear concentration range was 2 to 64 μg/ml.
Biodistribution of polymyxin B.
Among all of the organs evaluated, polymyxin B distribution was highest in the kidneys. The mean renal tissue/serum polymyxin B concentration ratios were 7.45 (95% confidence interval [CI], 4.63 to 10.27) at 3 h postdose and 19.62 (95% CI, 5.02 to 34.22) at 6 h postdose, as shown in Fig. 1. As shown in Table 2, all tissue concentrations at 24 h were lower than those observed at 6 h, but the mean tissue/serum ratio at 24 h could not be calculated because the polymyxin B concentration in the serum was below the limit of detection. The kidney concentration at 24 h was roughly 1.4 to 5.8 times those observed in organs other than the brain. Polymyxin B levels in the brain remained consistently lower than that observed in serum. All other organs exhibited a distribution profile similar to that of serum (Fig. 1).
FIG 1.
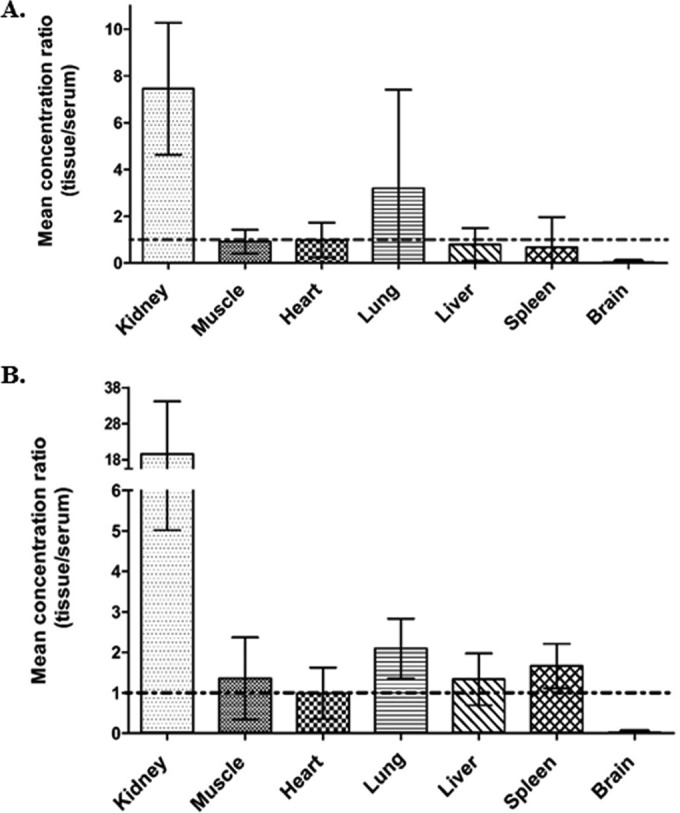
Tissue/serum concentration ratio of polymyxin B in various tissues 3 h (A) and 6 h (B) postdose. Vertical error bars represent the 95% confidence intervals.
TABLE 2.
Distribution of polymyxin B in rat serum and tissue homogenates
| Time (h) | Polymyxin B concn (means ± SD) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Serum (μg/ml) | Kidney (μg/g) | Muscle (μg/g) | Liver (μg/g) | Heart (μg/g) | Lung (μg/g) | Spleen (μg/g) | Brain (μg/g) | |
| 3 | 1.46 ± 0.38 | 10.57 ± 1.21 | 1.31 ± 0.41 | 1.10 ± 0.29 | 1.50 ± 0.78 | 4.93 ± 3.71 | 0.86 ± 0.43 | 0.04 ± 0.04 |
| 6 | 0.69 ± 0.29 | 11.32 ± 5.06 | 0.77 ± 0.34 | 0.82 ± 0.24 | 0.57 ± 0.18 | 1.33 ± 0.41 | 1.11 ± 0.50 | 0.02 ± 0.02 |
| 24 | <LLOQ | 0.76 ± 0.41 | 0.53 ± 0.18 | 0.13 ± 0.04 | 0.28 ± 0.06 | 0.25 ± 0.04 | 0.50 ± 0.07 | 0.01 ± 0.02 |
Intrarenal distribution of polymyxin B.
A gross cross-section of the kidney revealed that the intrarenal distribution of polymyxin B was heterogeneous, as shown in Fig. 2. Polymyxin B accumulated mainly in the renal cortex and outer stripe of the outer medulla.
FIG 2.

Confocal images of a kidney cross-section. Panel A represents a kidney cross-section from a control rat, whereas panel B represents a cross-section from a polymyxin B-treated rat. Blue represents counterstaining with nuclear stain (DAPI), whereas green color represents staining with anti-polymyxin B antibody.
The differential staining pattern was satisfactory with each lectin marker (data not shown). Strong, focal staining of the brush border epithelium of proximal tubular cells was seen with Phaseolus vulgaris erythroagglutinin (PHA-E) lectin. Soybean agglutinin (SBA) lectin predominantly stained the distal convoluted tubular cells. The staining pattern of SBA lectin was reasonably distinct; cytoplasmic staining of distal cells was observed. For glomeruli, a strong, global, and diffuse staining pattern was seen with wheat germ agglutinin (WGA) lectin.
The double staining with polymyxin B antibody and lectins is shown in Fig. 3. Polymyxin B was found to be accumulated primarily in the proximal tubular cells, with a Mander's overlap coefficient (Rcolo) of 0.998 for proximal tubular cells, 0.366 for distal tubular cells, and 0.097 for the glomerulus.
FIG 3.

Double staining with polymyxin B antibody and lectins. (A to C) Control section; (D to F) staining with anti-polymyxin B antibody; (G to I) staining with lectin; (J to L) multichannel images of double staining with polymyxin B antibody and lectin. Green color represents staining with anti-polymyxin B antibody, and red color represents staining with a specific lectin marker. The green-red colocalized signals produced a yellow signal in the overlay images.
Elimination of polymyxin B.
The concentration of polymyxin B in rat urine was below the limit of detection; less than 5% of the administered dose (or pharmacologically active metabolites, if any) could be recovered from the urine over 48 h. In contrast, all 4 major polymyxin B components (polymyxin B1, B2, and B3 and isoleucine-polymyxin B1) were detected in the bile collected over 4 h (data not shown).
DISCUSSION
As we are faced with a dearth of viable treatment options for infections caused by multidrug-resistant Gram-negative bacteria, polymyxins have undergone a resurgence as a treatment of last resort. However, their clinical utility is hindered by concerns of dose-limiting nephrotoxicity and a limited understanding of polymyxin pharmacokinetics.
Over the years, several attempts have been made to characterize the pharmacokinetics and nephrotoxicity of polymyxin B. Kunin et al. examined the biodistribution of polymyxin B in rabbits and observed that the distribution was nonuniform in various organs (21, 22). Our group also reported that polymyxin B was preferentially accumulated in rat kidneys with a prolonged residence time (23). In a mouse model, polymyxin B was found to be substantially accumulated in the renal proximal tubular cells (24), which was previously observed to be the primary renal injury site in a rat model (25). Furthermore, polymyxin B recovery from urine was reported to be very low in several studies. Abdelraouf et al. reported that less than 1% of polymyxin B was recovered in an unchanged form from the urine more than 48 h after a single intravenous dose (23, 25). Based on the urinary excretion data of polymyxin B available from 17 critically ill patients, Sandri et al. also reported the median urinary recovery to be 4.0% (range, 1.0 to 17.4%) of the administered dose (26). In line with previous findings, Zavascki et al. reported that less than 1% of the dose was recovered unchanged from urine samples following intravenous administration of polymyxin B in critically ill patients (27).
In this study, we provided additional insights. First, despite using different animal species, methods for sample preparation, and analysis, our biodistribution results are concordant with previous results, suggesting that polymyxin B distribution to different organs is not homogenous and that the drug is preferentially accumulated in the kidneys. However, in contrast to previous studies by Kunin et al. (21, 22), we observed consistently low drug concentrations in the brain. The earlier studies by Kunin et al. used a nonspecific microbiological assay to quantify the polymyxin B in various tissue homogenates rather than a sensitive, specific UPLC-MS/MS assay. Moreover, in the previous study a liquid-liquid extraction method was used for sample preparation; the amount of drug was estimated separately in the organic and aqueous extraction layers in the bound and free forms, respectively (21). This procedure might have overestimated the drug concentration, as the final level of drug recovery from serum and tissue homogenate was found to be greater than the dose administered. Our brain tissue biodistribution results were in accordance with a recent review of clinical findings (28). We were somewhat surprised to observe a higher concentration of polymyxin B in lung tissues than in serum at 6 h (but not at 3 h). This finding is in contrast to our previously reported low polymyxin B levels in the epithelial lining fluid (ELF) in mice (19). Since the lungs consisted of several subcompartments (lung parenchyma, alveolar epithelium, macrophages, ELF, blood-alveolar barrier, etc.), assaying drug concentrations in the whole organ could be subject to a high degree of variability.
Second, Yun et al. provided circumstantial evidence linking polymyxin B distribution to the site of renal injury (24). We have previously shown histological evidence that the polymyxin B-induced injury was confined mainly to the proximal tubular cells of the rat kidneys (25). Using the same model, we evaluated drug accumulation in multiple cell types in a semiquantitative fashion, establishing a more concrete association between the site of drug accumulation and the site of renal injury. These findings are expected to guide future investigations focusing on intervention(s) to reduce nephrotoxicity. In addition to demonstrating the low-level recovery of polymyxin B from urine, the use of a microbiological assay in this study provided additional evidence that renal excretion of active metabolite(s) is unlikely. Finally, to the best of our knowledge, the present study is the first to examine the biliary excretion of polymyxin B. We detected all 4 major polymyxin B components in the bile over 4 h. Biliary excretion could be one of the routes of polymyxin B elimination; this should be explored further.
There are several limitations to this study. As a preliminary investigation, we examined polymyxin B elimination in the bile for only 4 h. For future investigations, surgically modified rats could be used to evaluate biliary drug excretion for a longer duration. The sites of polymyxin B accumulation within the kidneys were identified at the cellular level. For better resolution, the investigations could be performed at the subcellular/organelle level. Finally, the biodistribution and disposition characterization of polymyxin B was investigated following a single intravenous dose. Further investigations to assess the steady-state pharmacokinetics of polymyxin B after multiple doses are warranted.
In conclusion, this study provides valuable insights into the overall in vivo distribution properties of polymyxin B. Drug accumulation within the kidneys is heterogeneous and is confined primarily to the proximal tubular cells. Among different renal cell types evaluated, proximal tubular cells show the highest accumulation of polymyxin B. In conjunction with previous findings, it is likely that there is a direct link between the site of drug accumulation and injury at the cellular level within the kidneys. Polymyxin B could be eliminated unchanged via the biliary route. Elucidation of the mechanism(s) of drug uptake in proximal tubular cells is ongoing.
Funding Statement
This study received no funding support.
REFERENCES
- 1.Vergidis PI, Falagas ME. 2008. Multidrug-resistant Gram-negative bacterial infections: the emerging threat and potential novel treatment options. Curr Opin Investig Drugs 9:176–183. [PubMed] [Google Scholar]
- 2.Curcio D. 2014. Multidrug-resistant Gram-negative bacterial infections: are you ready for the challenge? Curr Clin Pharmacol 9:27–38. doi: 10.2174/15748847113089990062. [DOI] [PubMed] [Google Scholar]
- 3.de Bentzmann S, Plesiat P. 2011. The Pseudomonas aeruginosa opportunistic pathogen and human infections. Environ Microbiol 13:1655–1665. doi: 10.1111/j.1462-2920.2011.02469.x. [DOI] [PubMed] [Google Scholar]
- 4.Kielhofner M, Atmar RL, Hamill RJ, Musher DM. 1992. Life-threatening Pseudomonas aeruginosa infections in patients with human immunodeficiency virus infection. Clin Infect Dis 14:403–411. doi: 10.1093/clinids/14.2.403. [DOI] [PubMed] [Google Scholar]
- 5.Decre D, Verdet C, Emirian A, Le Gourrierec T, Petit JC, Offenstadt G, Maury E, Brisse S, Arlet G. 2011. Emerging severe and fatal infections due to Klebsiella pneumoniae in two university hospitals in France. J Clin Microbiol 49:3012–3014. doi: 10.1128/JCM.00676-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montefour K, Frieden J, Hurst S, Helmich C, Headley D, Martin M, Boyle DA. 2008. Acinetobacter baumannii: an emerging multidrug-resistant pathogen in critical care. Crit Care Nurse 28:15–26. [PubMed] [Google Scholar]
- 7.Baijal R, Amarapurkar D, Praveen Kumar HR, Kulkarni S, Shah N, Doshi S, Gupta D, Jain M, Patel N, Kamani P, Issar SK, Dharod M, Shah A, Chandnani M, Gautam S. 2014. A multicenter prospective study of infections related morbidity and mortality in cirrhosis of liver. Indian J Gastroenterol 33:336–342. doi: 10.1007/s12664-014-0461-3. [DOI] [PubMed] [Google Scholar]
- 8.Tumbarello M, Trecarichi EM, De Rosa FG, Giannella M, Giacobbe DR, Bassetti M, Losito AR, Bartoletti M, Del Bono V, Corcione S, Maiuro G, Tedeschi S, Celani L, Cardellino CS, Spanu T, Marchese A, Ambretti S, Cauda R, Viscoli C, Viale P, Isgri S. 2015. Infections caused by KPC-producing Klebsiella pneumoniae: differences in therapy and mortality in a multicentre study. J Antimicrob Chemother 70:2133–2143. [DOI] [PubMed] [Google Scholar]
- 9.Kvitko CH, Rigatto MH, Moro AL, Zavascki AP. 2011. Polymyxin B versus other antimicrobials for the treatment of pseudomonas aeruginosa bacteraemia. J Antimicrob Chemother 66:175–179. doi: 10.1093/jac/dkq390. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez L, Gooderham WJ, Bains M, McPhee JB, Wiegand I, Hancock RE. 2010. Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob Agents Chemother 54:3372–3382. doi: 10.1128/AAC.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zavascki AP, Goldani LZ, Li J, Nation RL. 2007. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother 60:1206–1215. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 12.Zavascki AP, Li J, Nation RL, Superti SV, Barth AL, Lutz L, Ramos F, Boniatti MM, Goldani LZ. 2009. Stable polymyxin B susceptibility to Pseudomonas aeruginosa and Acinetobacter spp. despite persistent recovery of these organisms from respiratory secretions of patients with ventilator-associated pneumonia treated with this drug. J Clin Microbiol 47:3064–3065. doi: 10.1128/JCM.01035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan Z, Tam VH. 2008. Polymyxin B: a new strategy for multidrug-resistant Gram-negative organisms. Expert Opin Investig Drugs 17:661–668. doi: 10.1517/13543784.17.5.661. [DOI] [PubMed] [Google Scholar]
- 14.Shoji J, Hinoo H, Wakisaka Y, Koizumi K, Mayama M, Matsuura S. 1977. Isolation of two new polymyxin group antibiotics. (Studies on antibiotics from the genus Bacillus. XX.) J Antibiot 30:1029–1034. doi: 10.7164/antibiotics.30.1029. [DOI] [PubMed] [Google Scholar]
- 15.Orwa JA, Govaerts C, Busson R, Roets E, Van Schepdael A, Hoogmartens J. 2001. Isolation and structural characterization of polymyxin B components. J Chromatogr A 912:369–373. doi: 10.1016/S0021-9673(01)00585-4. [DOI] [PubMed] [Google Scholar]
- 16.He J, Ledesma KR, Lam WY, Figueroa DA, Lim TP, Chow DS, Tam VH. 2010. Variability of polymyxin B major components in commercial formulations. Int J Antimicrob Agents 35:308–310. doi: 10.1016/j.ijantimicag.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Manchandani P, Eriksen JL, Tam VH. 2014. Abstr 54th Intersci Conf Antimicrob Agents Chemother, abstr A-030. [Google Scholar]
- 18.Manchandani P, Ledesma KR, Zhou J, Tam VH. 2015. Abstr 55th Intersci Conf Antimicrob Agents Chemother, abstr A-930. [Google Scholar]
- 19.He J, Gao S, Hu M, Chow DS, Tam VH. 2013. A validated ultra-performance liquid chromatography-tandem mass spectrometry method for the quantification of polymyxin B in mouse serum and epithelial lining fluid: application to pharmacokinetic studies. J Antimicrob Chemother 68:1104–1110. doi: 10.1093/jac/dks536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He J, Figueroa DA, Lim TP, Chow DS, Tam VH. 2010. Stability of polymyxin B sulfate diluted in 0.9% sodium chloride injection and stored at 4 or 25 degrees C. Am J Health Syst Pharm 67:1191–1194. doi: 10.2146/ajhp090472. [DOI] [PubMed] [Google Scholar]
- 21.Kunin CM, Bugg A. 1971. Binding of polymyxin antibiotics to tissues: the major determinant of distribution and persistence in the body. J Infect Dis 124:394–400. doi: 10.1093/infdis/124.4.394. [DOI] [PubMed] [Google Scholar]
- 22.Kunin CM, Bugg A. 1971. Recovery of tissue bound polymyxin B and colistimethate. Proc Soc Exp Biol Med 137:786–790. doi: 10.3181/00379727-137-35667. [DOI] [PubMed] [Google Scholar]
- 23.Abdelraouf K, He J, Ledesma KR, Hu M, Tam VH. 2012. Pharmacokinetics and renal disposition of polymyxin B in an animal model. Antimicrob Agents Chemother 56:5724–5727. doi: 10.1128/AAC.01333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yun B, Azad MA, Wang J, Nation RL, Thompson PE, Roberts KD, Velkov T, Li J. 2015. Imaging the distribution of polymyxins in the kidney. J Antimicrob Chemother 70:827–829. doi: 10.1093/jac/dku441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdelraouf K, Braggs KH, Yin T, Truong LD, Hu M, Tam VH. 2012. Characterization of polymyxin B-induced nephrotoxicity: implications for dosing regimen design. Antimicrob Agents Chemother 56:4625–4629. doi: 10.1128/AAC.00280-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, Behle TF, Bordinhao RC, Wang J, Forrest A, Nation RL, Li J, Zavascki AP. 2013. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis 57:524–531. doi: 10.1093/cid/cit334. [DOI] [PubMed] [Google Scholar]
- 27.Zavascki AP, Goldani LZ, Cao G, Superti SV, Lutz L, Barth AL, Ramos F, Boniatti MM, Nation RL, Li J. 2008. Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin Infect Dis 47:1298–1304. doi: 10.1086/592577. [DOI] [PubMed] [Google Scholar]
- 28.Falagas ME, Bliziotis IA, Tam VH. 2007. Intraventricular or intrathecal use of polymyxins in patients with Gram-negative meningitis: a systematic review of the available evidence. Int J Antimicrob Agents 29:9–25. doi: 10.1016/j.ijantimicag.2006.08.024. [DOI] [PubMed] [Google Scholar]