

Abstract
Despite ongoing efforts, the available treatments for Chagas' disease are still unsatisfactory, especially in the chronic phase of the disease. Our previous study reported the strong trypanocidal activity of the dibenzylideneacetones A3K2A1 and A3K2A3 against Trypanosoma cruzi (Z. Ud Din, T. P. Fill, F. F. de Assis, D. Lazarin-Bidóia, V. Kaplum, F. P. Garcia, C. V. Nakamura, K. T. de Oliveira, and E. Rodrigues-Filho, Bioorg Med Chem 22:1121–1127, 2014, http://dx.doi.org/10.1016/j.bmc.2013.12.020). In the present study, we investigated the mechanisms of action of these compounds that are involved in parasite death. We showed that A3K2A1 and A3K2A3 induced oxidative stress in the three parasitic forms, especially trypomastigotes, reflected by an increase in oxidant species production and depletion of the endogenous antioxidant system. This oxidative imbalance culminated in damage in essential cell structures of T. cruzi, reflected by lipid peroxidation and DNA fragmentation. Consequently, A3K2A1 and A3K2A3 induced vital alterations in T. cruzi, leading to parasite death through the three pathways, apoptosis, autophagy, and necrosis.
INTRODUCTION
Chagas' disease was discovered more than 100 years ago (1), but the available treatments are still restricted to only two nitro-derivative compounds: benznidazole and nifurtimox. These compounds have limited efficacy, especially in the chronic phase of the disease, and serious side effects (2). Chagas' disease is caused by the protozoan parasite Trypanosoma cruzi, with an estimated 6 to 7 million people infected worldwide, mainly in 21 Latin America countries. However, cases of the disease have been increasingly detected in the United States, Canada, Europe, and Western Pacific countries (3).
The search for new active compounds that are more effective against Chagas' disease and devoid of serious side effects is increasing (4, 5). Few studies have reported the efficacy of trypanocidal compounds against the three forms of T. cruzi (6, 7), and little is known about their mechanisms of action. The scarcity of such studies and lack of effective and selective compounds against T. cruzi can be explained by the complex life cycle and distinct morphological and functional forms of this parasite (8, 9).
Dibenzylideneacetones (DBAs) are a class of compounds that have an acyclic dienone attached to aryl groups in both β-positions. Some studies have demonstrated the chemotherapeutic properties of DBAs, including antiplasmodial (10) and anticancer (11, 12, 13, 14) effects. Our research group recently tested a library of new DBAs against T. cruzi and Leishmania amazonensis and reported interesting results (15). (1E,4E)-2-Methyl-1-(4-nitrophenyl)-5-phenylpenta-1,4-dien-3-one (A3K2A1) and (1E,4E)-2-methyl-1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one (A3K2A3) were the most active compounds against T. cruzi.
Considering the trypanocidal activity of A3K2A1 and A3K2A3, the present study sought to characterize the biochemical and morphological alterations induced by these compounds in the three parasitic forms of T. cruzi and elucidate the mechanisms of action that are involved in the cell death of this parasite.
Our results provided insights into the mechanisms of action of A3K2A1 and A3K2A3 and strongly suggest that these compounds are effective against the three main parasitic forms of T. cruzi. Altogether, our results indicate that the effects of A3K2A1 and A3K2A3 are related to an increase in oxidant species production and depletion of the endogenous antioxidant system, leading to parasite death.
MATERIALS AND METHODS
Chemicals.
Actinomycin D, antimycin A (AA), bovine serum albumin, carbonyl cyanide m-chlorophenylhydrazone (CCCP), digitonin, dimethyl sulfoxide (DMSO), diphenyl-1-pyrenylphosphine (DPPP), 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), monodansylcadaverine (MDC), Nile red, penicillin, rhodamine 123 (Rh123), thiobarbituric acid, and wortmannin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Invitrogen (Grand Island, NY, USA). Annexin V-fluorescein isothiocyanate (FITC), 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM diacetate), the mitochondrion-targeted probe 3,8-phenanthridinediamine-5-(6-triphenylphosphoniumhexyl)-5,6-dihydro-6-phenyl (Mitosox), propidium iodide (PI), and RNase were obtained from Invitrogen (Eugene, OR, USA). Streptomycin was obtained from Gibco BRL-Life Technologies (Grand Island, NY, USA). The protein assay kit was obtained from Bio-Rad (Hercules, CA, USA). All other reagents were of analytical grade.
Synthesis of compounds.
The compounds (1E,4E)-2-methyl-1-(4-nitrophenyl)-5-phenylpenta-1,4-dien-3-one (A3K2A1) and (1E,4E)-2-methyl-1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one (A3K2A3) were synthesized as previously described by Ud Din et al. (15). The molecular structures of the compounds are shown in Fig. 1.
FIG 1.
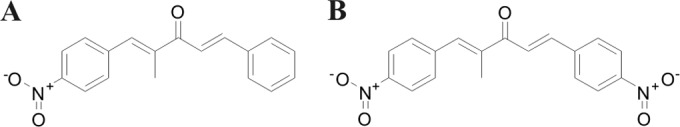
Structures of dibenzylideneacetones (1E,4E)-2-methyl-1-(4-nitrophenyl)-5-phenylpenta-1,4-dien-3-one (A) and (1E,4E)-2-methyl-1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one (B).
Stock solutions of A3K2A1 and A3K2A3 were prepared aseptically in DMSO and diluted in culture medium so that the DMSO concentration did not exceed 1% in the experiments.
The three forms of T. cruzi were treated with the compounds at concentrations that correspond to the IC50s (concentrations that inhibited 50% of the parasites) previously determined (15). Epimastigotes were treated at 28°C with 3.5 μM A3K2A1 and 1.8 μM A3K2A3. Amastigotes were treated at 37°C with 3.9 μM A3K2A1 and 1.2 μM A3K2A3. Trypomastigotes were treated at 37°C with 17.5 μM A3K2A1 and 15.4 μM A3K2A3.
Parasites and cell cultures.
All of the experiments were performed using the Y strain of T. cruzi. Epimastigotes were axenically maintained at 28°C with weekly transfers in liver infusion tryptose (LIT) medium supplemented with 10% heat-inactivated FBS, pH 7.4 (16). Trypomastigotes and amastigotes were obtained from the supernatants of previously infected monolayers of LLCMK2 cells (i.e., monkey [Macaca mulatta] kidney epithelial cells; CCL-7; American Type Culture Collection, Rockville, MD, USA) in DMEM supplemented with 2 mM l-glutamine and 10% FBS and buffered with sodium bicarbonate in a 5% CO2-air mixture at 37°C (17). Trypomastigotes and amastigotes were separated by differential centrifugation at 850 × g for 5 min. The trypomastigote forms were harvested from the supernatant, and the amastigote forms were collected from the pellet.
Cell membrane integrity assay.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then washed and incubated with 0.2 μg/ml PI for 10 min. Digitonin (40.0 μM) was used as a positive control. Data acquisition and analysis were performed using a FACSCalibur flow cytometer equipped with CellQuest software. A total of 10,000 events were acquired in the region that was previously established as the one that corresponded to the parasites. Alterations in the fluorescence of PI were quantified as the percentage increase in fluorescence compared with the control (untreated parasites).
Detection of mitochondrial-derived superoxide anions.
The parasites were loaded with 5 μM Mitosox for 10 min at 22°C and then washed with Krebs-Henseleit (KH) buffer (pH 7.3) that contained 15 mM NaHCO3, 5 mM KCl, 120 mM NaCl, 0.7 mM Na2HPO4, and 1.5 mM NaH2PO4. The parasites were then treated with A3K2A1 or A3K2A3 for up to 24 h. The fluorescence was measured in a Victor X3 spectrofluorometer (PerkinElmer) at an excitation wavelength (λex) of 510 nm and an emission wavelength (λem) of 580 nm (18). Oxidized Mitosox becomes highly fluorescent upon binding to nucleic acids. AA (10.0 μM) was used as a positive control.
Detection of reactive oxygen species production.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then loaded with 10 μM H2DCFDA in the dark for 45 min. The fluorescence was determined by oxidation of H2DCFDA to the fluorescent product 2′,7′dichlorofluorescein (DCF) in a Victor X3 spectrofluorometer at an λex of 488 nm and an λem of 530 nm (19).
Detection of nitric oxide.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then loaded with 1 μM DAF-FM diacetate in the dark for 30 min. Afterward, the parasites were washed, resuspended in phosphate-buffered saline (PBS), and incubated for an additional 15 min. Fluorescence was determined in a fluorescence microplate reader (Victor X3; PerkinElmer) at an λex of 495 nm and an λem of 515 nm. DAF-FM diacetate, which is deacetylated to DAF-FM by intracellular esterases, emits fluorescence when it reacts with nitric oxide (NO).
Evaluation of reduced thiol levels.
In the trypanothione system, trypanothione (T) is reduced to a dithiol, T(SH)2, by trypanothione reductase (TR). The inhibition of TR decreases total reduced-thiol levels (19). The parasites were treated with A3K2A1 or A3K2A3 for 3, 24, and 48 h. Afterward, the cells (1 × 107 cells/ml) were centrifuged, resuspended in 10 mM Tris-HCl buffer (pH 2.5), and sonicated. Acidic pH was used during sonication to prevent the oxidation of free thiol groups. Cellular debris was removed by centrifugation, and 100 μl of the supernatant and 100 μl of 500 mM phosphate buffer (pH 7.5) were taken from each microtiter well, followed by the addition of 20 μl of 1 mM DTNB to each well. Absorbance was measured at 412 nm.
Mitochondrial membrane potential assay.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then incubated with 5 μg/ml Rh123 for 15 min to verify the ΔΨm. CCCP (100.0 μM) was used as a positive control. The data acquisition and analysis were performed using a FACSCalibur flow cytometer equipped with CellQuest software. A total of 10,000 events were acquired in the region corresponding to the parasites. Alterations in Rh123 fluorescence were quantified using an index of variation (IV) that was obtained from the equation (MT − MC)/MC, where MT is the median fluorescence for the treated parasites and MC is the median fluorescence for the control parasites. Negative IV values correspond to depolarization of the mitochondrial membrane.
Lipid peroxidation assay.
The extent of lipid peroxidation was determined as the amount of thiobarbituric acid-reactive substances (TBARS) in terms of malondialdehyde (MDA) and using the probe DPPP. For the MDA assay, the parasites were treated with A3K2A1 or A3K2A3 for 24 h. After incubation, the samples were heated in a solution that contained 0.37% thiobarbituric acid, 15% trichloroacetic acid, and 0.25 N HCl for 45 min at 95°C. After cooling, absorbance was read at 532 nm, and the TBARS concentration was calculated based on an ε value of 153,000 M−1 cm−1 (20).
For the DPPP assay, the parasites were treated under the conditions described above. Afterward, parasites were loaded with 50 μM DPPP for 15 min at 22°C, and fluorescence was determined in a fluorescence microplate reader (Victor X3; PerkinElmer) at an λex of 355 nm and an λem of 460 nm. DPPP is essentially nonfluorescent until it is oxidized to a phosphine oxide (DPPP-O) by peroxides (21).
Cell cycle analysis.
Amastigotes and epimastigotes were treated with A3K2A1 or A3K2A3 for 24 h and then fixed in 70% cold methanol–PBS at 4°C for 1 h. Afterward, the parasites were washed in PBS, and 10 μl PI-RNase A was added, followed by incubation at 37°C for 45 min. The data acquisition and analysis were performed using a FACSCalibur flow cytometer equipped with CellQuest software. A total of 10,000 events were acquired in the region that corresponded to the parasites. The percentages of cells in each stage of the cell cycle were determined (22).
Evaluation of lipid bodies.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then incubated with 10 μg/ml Nile red for 30 min at 22°C. The fluorescence was observed under an Olympus BX51 fluorescence microscope (Olympus, Tokyo, Japan), and the images were recorded with an Olympus UC30 camera (Olympus, Tokyo, Japan). Additionally, fluorescence was measured in a fluorescence microplate reader (Victor X3; PerkinElmer) at an λex of 485 nm and an λem of 535 nm (23).
Phosphatidylserine exposure.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then washed and resuspended in 100 μl of binding buffer (140 mM NaCl, 5 mM CaCl2, and 10 mM HEPES-Na, pH 7.4), followed by the addition of 5 μl of annexin V-FITC, a calcium-dependent phospholipid-binding protein, for 15 min at room temperature. Binding buffer (400 μl) and 50 μl of PI were then added. AA (125.0 μM) was used as a positive control. Data acquisition and analysis were performed using a FACSCalibur flow cytometer equipped with CellQuest software. A total of 10,000 events were acquired in the region that was previously established as the one that corresponds to the parasites. Cells that were stained with annexin V (PI positive or negative) were considered apoptotic, and cells that were positive only for PI were considered necrotic (24).
Cell volume determination.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then washed twice in PBS, resuspended in PBS, and analyzed using fluorescence-activated cell sorting and a FACSCalibur flow cytometer. Actinomycin D (20.0 mM) was used as a positive control. A total of 10,000 events were acquired in the region that was previously established as the one that corresponded to the parasites. Histograms were generated, and the analysis was performed using CellQuest software. Forward light scatter (FSC-H) represents the cell volume.
Evaluation of autophagic vacuoles.
The parasites were treated with A3K2A1 or A3K2A3 for 24 h and then incubated with 0.05 mM MDC, a fluorescent probe that accumulates in autophagic vacuoles (25, 26), in PBS for 15 min. After incubation, the cells were washed twice in PBS. MDC staining was analyzed using an Olympus BX51 fluorescence microscope, images were captured using a UC30 camera, and fluorescence intensity was evaluated by ImageJ 1.44o. In parallel experiments, before the induction of autophagy, the cells were pretreated with 0.5 μM wortmannin, a potent inhibitor of PI3 kinase, an enzyme involved in autophagy regulation (27).
Statistical analysis.
The data are expressed as the means and standard deviations from at least three independent experiments. The data were analyzed using one- and two-way analysis of variance (ANOVA), with significant differences among means identified by Tukey and Bonferroni post hoc tests, respectively. Values of P of ≤0.05 were considered statistically significant. The statistical analyses were performed using Prism 5 software (GraphPad, San Diego, CA, USA).
RESULTS
A3K2A1 and A3K2A3 cause cell membrane disruption in T. cruzi.
Both compounds caused a significant alteration of cell membrane integrity in the three parasite forms compared with untreated cells (Fig. 2). The histograms show an increase in the intensity of PI fluorescence in trypomastigotes (48% for A3K2A1 and 70% for A3K2A3) (Fig. 2Ba), amastigotes (37% for A3K2A1 and 49% for A3K2A3) (Fig. 2Bb), and epimastigotes (approximately 20% for both compounds) (Fig. 2Bc). The positive control, digitonin, increased fluorescence by 92.4%, 44.6%, and 29.2% in trypomastigotes, amastigotes, and epimastigotes, respectively.
FIG 2.

Cell membrane integrity assay of Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s using PI staining. (A) Typical histograms for trypomastigotes (a, d, and g), amastigotes (b, e, and h), and epimastigotes (c, f, and i). (a, b, and c) Untreated parasites; (d, e, and f) A3K2A1-treated parasites; (g, h, and i) A3K2A3-treated parasites. The percentages of PI-positive cells are shown in the upper right and left quadrants. The percentages of PI-negative cells are shown in the lower right and left quadrants. (B) PI-positive cells in trypomastigotes (a), amastigotes (b), and epimastigotes (c). Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 increase mitochondrion-derived O2˙− production in T. cruzi.
The treatment with both compounds caused a similar and significant time-dependent increase in the production of mitochondrial O2˙− in the three forms of T. cruzi compared with the control group. This increase was more pronounced in trypomastigotes (approximately 120%) (Fig. 3A) than in amastigotes (approximately 90%) (Fig. 3B) and epimastigotes (approximately 55%) (Fig. 3C). The positive control, AA, also induced 97%, 84%, and 48% increases in mitochondrial O2˙− production in trypomastigotes, amastigotes, and epimastigotes, respectively.
FIG 3.
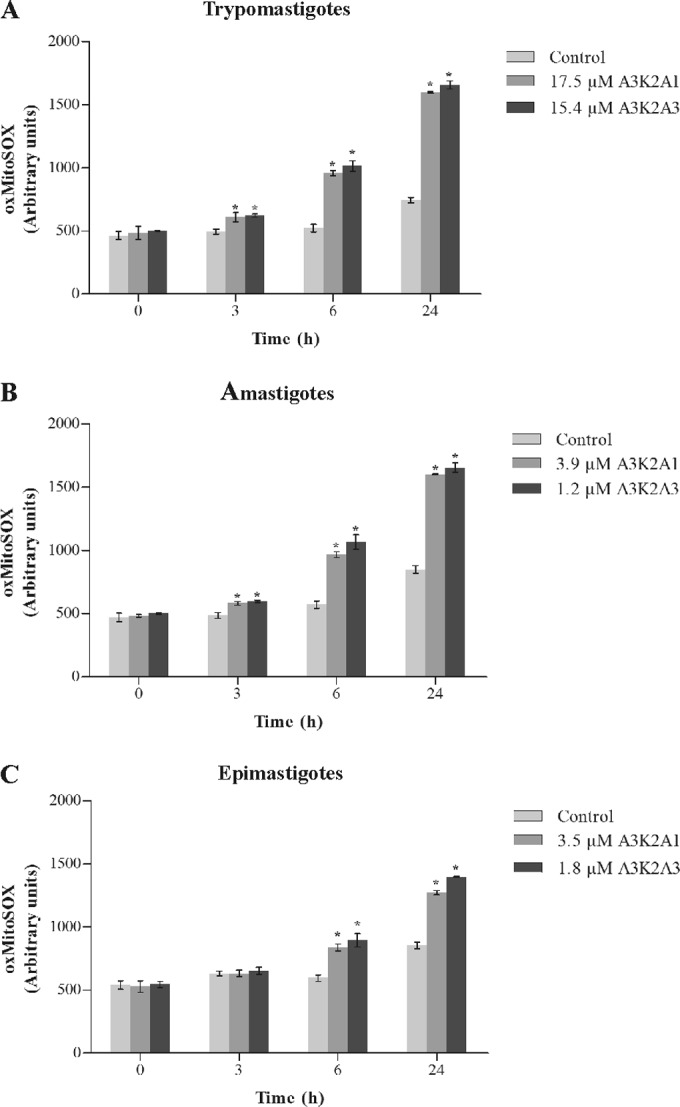
Mitochondrial O2˙− production in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for up to 24 h at concentrations corresponding to IC50s using the fluorescence probe Mitosox. Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 increase total reactive oxygen species in T. cruzi.
The treatment with both compounds led to similar and significant increase in total reactive oxygen species (ROS) after 24 h treatment compared with the control group (Fig. 4). A3K2A1 and A3K2A3 caused total ROS increases, of, respectively, 157% and 169% for trypomastigotes (Fig. 4A), 149% and 162% for amastigotes (Fig. 4B), and 91% and 99% for epimastigotes (Fig. 4C).
FIG 4.
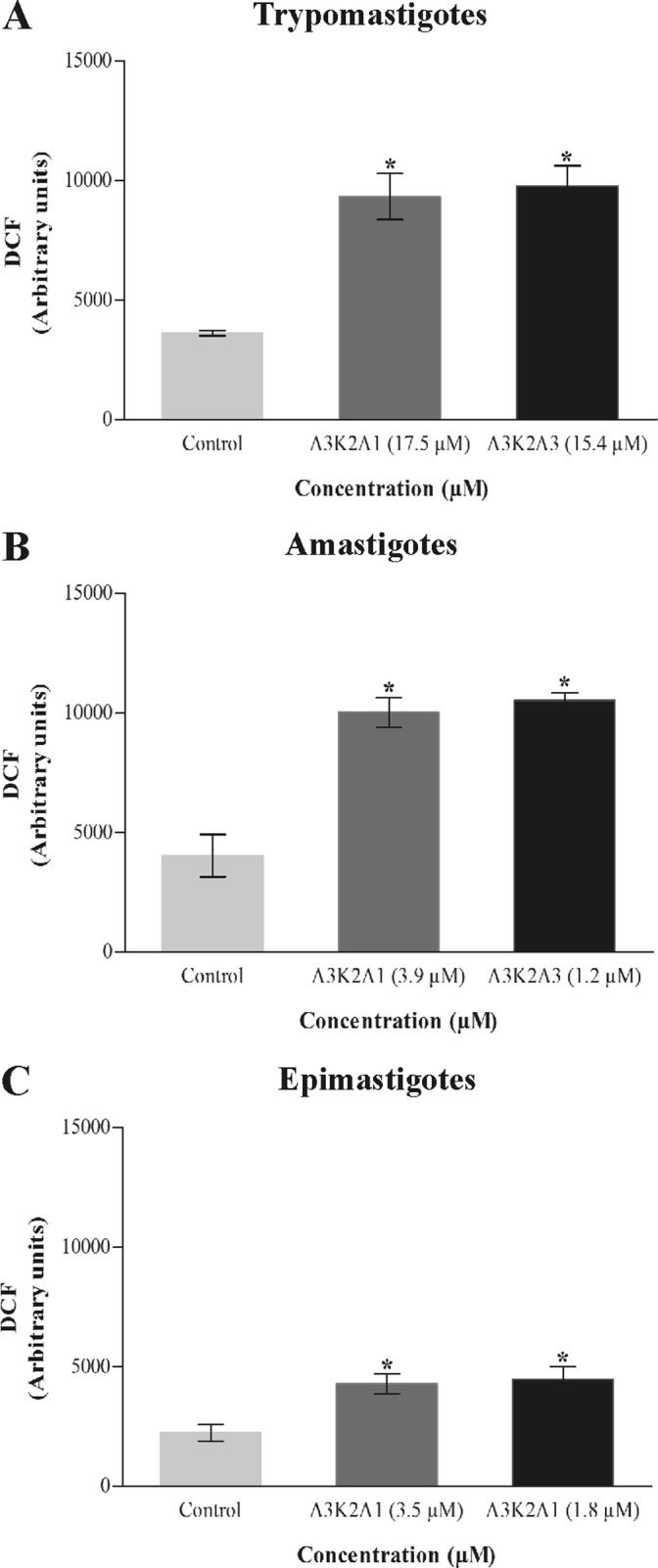
Total ROS in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s using the nonfluorescent probe H2DCFDA. (A) Trypomastigotes. (B) Amastigotes. (C) Epimastigotes. Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 increase nitric oxide in T. cruzi.
The compounds caused a significant increase in NO after 24 h treatment compared with control group (Fig. 5). This increase was more noticeable for trypomastigotes (105% and 312% for A3K2A1 and A3K2A3, respectively) (Fig. 5A), followed by amastigotes (86% and 112%) (Fig. 5B) and epimastigotes (65% and 45%) (Fig. 5C).
FIG 5.
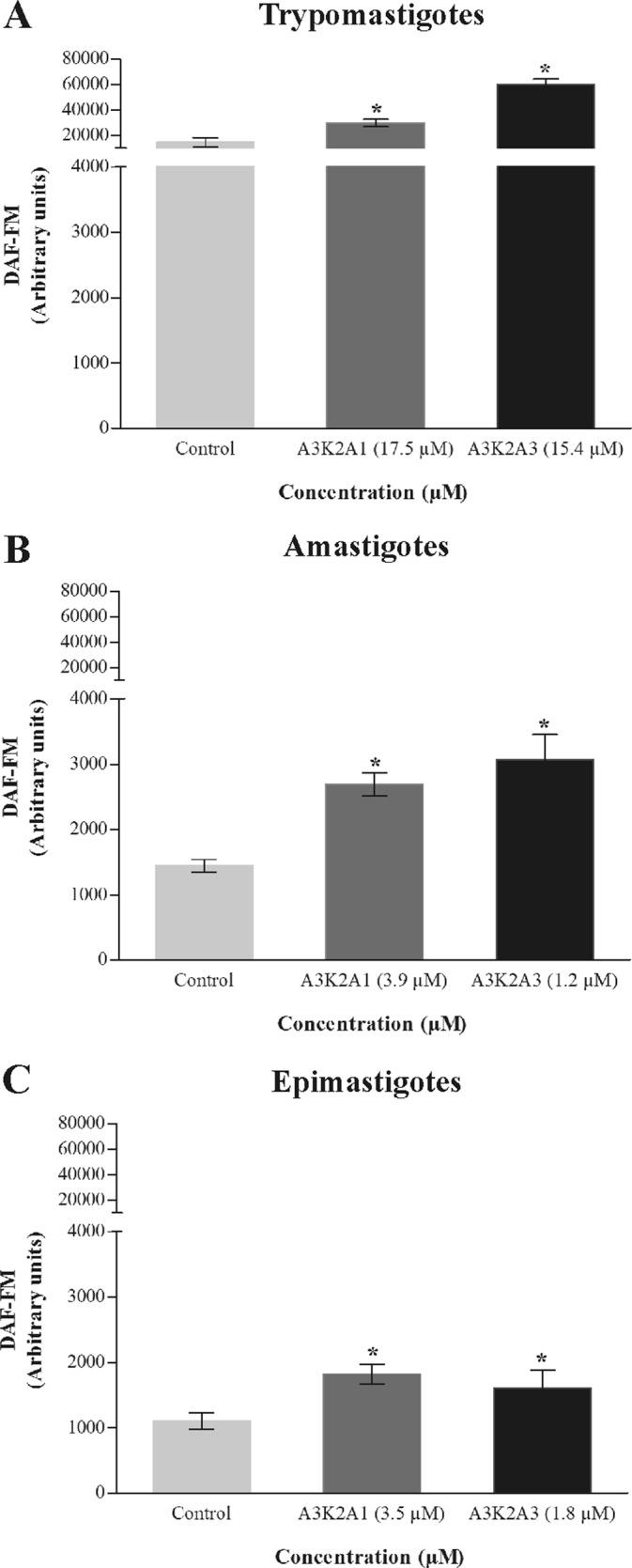
Nitric oxide in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s using the fluorescent probe DAF-FM diacetate. (A) Trypomastigotes. (B) Amastigotes. (C) Epimastigotes. Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 decrease reduced thiol levels of T. cruzi.
Both compounds caused a decrease in total reduced thiol levels that were significantly lower than the control group, only after 48 h treatment in the three parasite forms (Table 1). Trypomastigotes were the most sensitive parasite form with approximately 40% decrease while for amastigotes and epimastigotes, the decreases ranged from 10% to 26%.
TABLE 1.
Reduced thiol levels in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for up to 48 h using DTNB staining
| Parasitic form | Compound | Concn (μM) | Thiol level (%) after treatment timea |
||
|---|---|---|---|---|---|
| 3 h | 24 h | 48 h | |||
| Trypomastigotes | Control | 100.00 ± 0.003 | 100.00 ± 0.008 | 100.00 ± 0.018 | |
| A3K2A1 | 17.5 | 95.58 ± 0.006 | 89.67 ± 0.271 | 59.06* ± 0.005 | |
| A3K2A3 | 15.4 | 95.86 ± 0.004 | 89.88 ± 0.001 | 53.85* ± 0.005 | |
| Amastigotes | Control | 100.00 ± 0.005 | 100.00 ± 0.006 | 100.00 ± 0.008 | |
| A3K2A1 | 3.9 | 96.90 ± 0.004 | 96.78 ± 0.003 | 80.68* ± 0.008 | |
| A3K2A3 | 1.2 | 99.85 ± 0.001 | 94.94 ± 0.001 | 73.90* ± 0.004 | |
| Epimastigotes | Control | 100.00 ± 0.012 | 100.00 ± 0.003 | 100.00 ± 0.005 | |
| A3K2A1 | 3.5 | 99.93 ± 0.002 | 98.08 ± 0.009 | 88.62* ± 0.002 | |
| A3K2A3 | 1.8 | 97.48 ± 0.004 | 98.08 ± 0.002 | 88.49* ± 0.004 | |
Data are expressed as percentages. *, significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 induce mitochondrial depolarization of T. cruzi.
Both compounds caused a marked decrease in total Rh123 fluorescence intensity in the three parasite forms after treatment for 24 h, indicating mitochondrial depolarization (Fig. 6; Table 2). The loss of ΔΨm was more pronounced in trypomastigotes and amastigotes (about 70%) (Table 2). The positive control, CCCP, induced 89.7%, 74.4%, and 90.5% changes in the ΔΨm in trypomastigotes, amastigotes, and epimastigotes, respectively.
FIG 6.

Mitochondrial membrane potential assay in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s using Rh123 staining. (A and D) Trypomastigotes. (B and E) Amastigotes. (C and F) Epimastigotes. Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
TABLE 2.
Mitochondrial membrane potential assay in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h using Rh123 staining
| Parasite form | Compound | Concn (μM) | Median fluorescenceb | IVa |
|---|---|---|---|---|
| Trypomastigotes | Control | 774.05 | 0.00 | |
| A3K2A1 | 17.5 | 136.39* | −0.82 | |
| A3K2A3 | 15.4 | 129.44* | −0.83 | |
| Amastigotes | Control | 1,816.50 | 0.0 | |
| A3K2A1 | 3.9 | 359.70* | −0.80 | |
| A3K2A3 | 1.2 | 272.02* | −0.85 | |
| Epimastigotes | Control | 2,722.82 | 0.0 | |
| A3K2A1 | 3.5 | 894.25* | −0.67 | |
| A3K2A3 | 1.8 | 762.66* | −0.72 |
IV = (MT − MC)/MC, where MT is the median of the fluorescence for treated parasites and MC is that for control parasites.
*, significant difference relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 induce lipid peroxidation in T. cruzi.
Figure 7 shows that treated parasites exhibited a significant increase in lipid peroxidation compared with the control group. In both, MDA and DPPP assays, the increase was more pronounced for trypomastigotes (Fig. 7A and B), followed by amastigotes (Fig. 7C and D) and epimastigotes (Fig. 7E and F).
FIG 7.

Lipid peroxidation in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s, based on MDA (A, C, and E) and DPPP (B, D, and F). (A and B) Trypomastigotes. (C and D) Amastigotes. (E and F) Epimastigotes. Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 induce cell cycle arrest in the sub-G0/G1 phase in T. cruzi.
The compounds caused a significant increase in the sub-G0/G1 cell population (i.e., cells that undergo the fragmentation of nuclear chromatin by DNases, a typical feature of apoptosis [28]) and a decrease in the G2/M phase (DNA duplication) compared with the control group (Fig. 8). The increase in the sub-G0/G1 cell population was observed in amastigotes (42% and 53% for A3K2A1 and A3K2A3, respectively) (Fig. 8A) and in epimastigotes (36% and 46%) (Fig. 8B).
FIG 8.
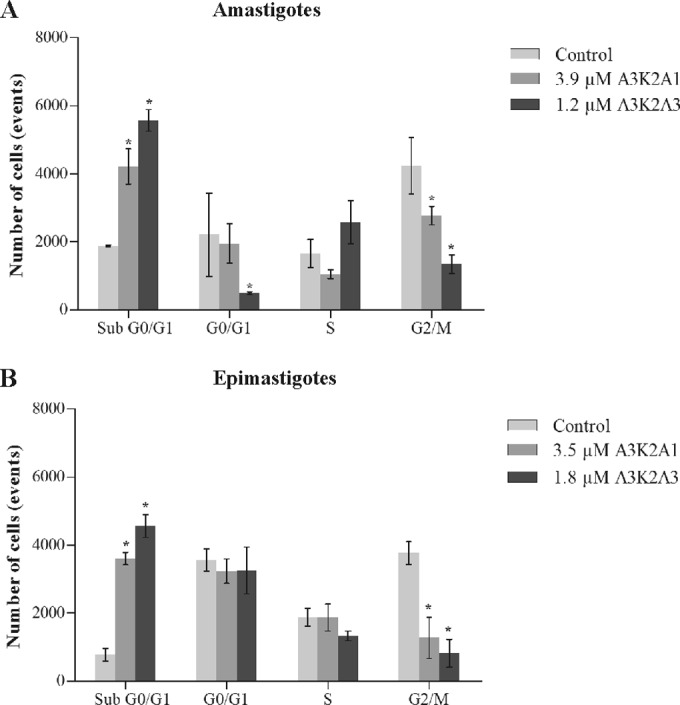
Cell cycle of Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s. (A) Amastigotes. (B) Epimastigotes. Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 increase lipid bodies in T. cruzi.
Figure 9 shows increases in lipid bodies in treated parasites. This increase was significant and more pronounced in amastigotes (215% and 264% for A3K2A1 and A3K2A3, respectively) (Fig. 9B), followed by trypomastigotes (62% and 107%) (Fig. 9A) and epimastigotes (24% and 31%) (Fig. 9C).
FIG 9.

Lipid storage bodies in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s using Nile red labeling. (A) Fluorescence microscopy images for trypomastigotes (a, b, and c), amastigotes (d, e, and f), and epimastigotes (g, h, and i). (a, b, and c) Untreated parasites; (b, e, and h) A3K2A1-treated parasites; (c, f, and i) A3K2A3-treated parasites. Bars = 20 μm. Fluorescence intensity obtained by fluorimetry in trypomastigotes (B), amastigotes (C), and epimastigotes (D). Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 induce phosphatidylserine exposure in T. cruzi.
Figure 10 shows that both compounds caused in the parasites a significant increase in annexin V fluorescence intensity compared with untreated parasites, with the exception of epimastigotes treated with 3.5 μM A3K2A1. Phosphatidylserine exposure was more pronounced in trypomastigotes (approximately 50%) (Fig. 10Ba) than in amastigotes (approximately 40%) (Fig. 10Bb) and in epimastigotes (approximately 12%) (Fig. 10Bc). The positive control, actinomycin D, increased phosphatidylserine exposure by 32%, 28%, and 8% in trypomastigotes, amastigotes, and epimastigotes, respectively.
FIG 10.

Phosphatidylserine exposure in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s using annexin V/FITC and PI. (A) Typical histograms for trypomastigotes (a, d, and g), amastigotes (b, e, and h), and epimastigotes (c, f, and i). (a, b, and c) Untreated parasites; (d, e, and f) A3K2A1-treated parasites; (g, h, and i) A3K2A3-treated parasites. Percentages of annexin V-positive cells are shown in the upper and lower right quadrants. Percentages of annexin V-negative cells are shown in the upper and lower left quadrants. (B) Annexin V-positive cells in trypomastigotes (a), amastigotes (b), and epimastigotes (c). Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 decrease the cell volume of T. cruzi.
As shown in Fig. 11, a significant decrease in cell volume was observed after treatment of the three parasite forms with each compound compared with the control group. The decrease in cell volume was more pronounced in trypomastigotes (75% and 83% for A3K2A1 and A3K2A3, respectively) (Fig. 11A and D) than in amastigotes (70% and 37%) (Fig. 11B and E) and epimastigotes (35% and 39%) (Fig. 11C and F). The positive control, actinomycin D, also decreased the cell volume by 39%, 69%, and 28% in trypomastigotes, amastigotes, and epimastigotes, respectively.
FIG 11.

Cell volume in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s. (A and D) Trypomastigotes. (B and E) Amastigotes. (C and F) Epimastigotes. Asterisks indicate significant differences relative to the control group (P ≤ 0.05).
A3K2A1 and A3K2A3 induce the formation of autophagic vacuoles in T. cruzi.
Figure 12 shows the presence of fluorescence in rounded structures in treated parasites, indicating the formation of autophagic vacuoles compared with untreated parasites. This effect could be partially prevented by wortmannin pretreatment. This increase in the formation of autophagic vacuoles was significant and more pronounced in amastigotes (90% and 105% for A3K2A1 and A3K2A3, respectively) (Fig. 12C), and trypomastigotes (84% and 102%) (Fig. 12B) than epimastigotes (67% and 71%) (Fig. 12D).
FIG 12.

Autophagic vacuoles in Trypanosoma cruzi treated with A3K2A1 or A3K2A3 for 24 h at concentrations corresponding to IC50s using MDC labeling. Fluorescence microscopy images are as follows: for trypomastigotes, (a) untreated parasites, (b) A3K2A1, (c) A3K2A3, (a′) untreated parasites plus wortmannin, (b′) A3K2A1 plus wortmannin, (c′) A3K2A3 plus wortmannin; for amastigotes, (d) untreated parasites, (e) A3K2A1, (f) A3K2A3, (d′) untreated parasites plus wortmannin, (e′) A3K2A1 plus wortmannin, (f′) A3K2A3 plus wortmannin; and for epimastigotes, (g) untreated parasites, (h) A3K2A1, (i) A3K2A3, (g′) untreated parasites plus wortmannin, (h′) A3K2A1 plus wortmannin, (i′) A3K2A3 plus wortmannin. Bars = 20 μm. (B through D) Fluorescence intensity was obtained with ImageJ. (B) Trypomastigotes. (C) Amastigotes. (D) Epimastigotes. Asterisks indicate significant differences relative to the control group (untreated cells) (P ≤ 0.05).
DISCUSSION
Several studies have been conducted over the past 100 years to discover new trypanocidal agents (4, 29). However, no substitute has yet been developed for the nitro compounds benznidazole and nifurtimox. The development of new synthetic molecules containing a nitroaromatic moiety is an interesting approach (30); however, their potential toxicity should be very carefully addressed (31, 32). In a previous study, the DBAs A3K2A1 and A3K2A3, characterized by the presence of nitro groups attached directly to an aromatic ring, showed selective activity against T. cruzi (15). The present study sought to investigate their mechanism of trypanocidal action.
We initially focused our studies on investigating alterations in the plasma membrane by staining the parasites with PI. The three parasite forms, especially trypomastigotes (i.e., the infective bloodstream form in the mammalian host), exhibited marked alterations in plasma membrane integrity.
The literature reports that the bioactivity of nitro compounds is related to reduction of the nitro group (30). As A3K2A1 and A3K2A3 are nitro compounds, we also investigated their effects on the production of reactive species in T. cruzi. We observed an increase in the production of ROS in treated parasites. Reactive oxygen species are molecules that are derived from the incomplete one-electron reduction of molecular oxygen, and high concentrations might induce oxidative damage (33). These compounds also increase reactive nitrogen species (RNS) as evidenced by the production of NO, a short-lived free radical than can react with proteins and nucleic acids (34). Thus, A3K2A1 and A3K2A3 may induce oxidative imbalance in T. cruzi, attributable to an increase in ROS/RNS. Oxidative stress is a result of enhanced free-radical formation and/or a defect in antioxidant defenses (35). Here, we observed a decrease in reduced thiol levels in the three parasite forms. Thiols are the main functional group of trypanothione, an antioxidant molecule that is central to the unique thiol-redox system that acts in trypanosomatids. In these parasites, the enzymatic regeneration of the thiol pool appears to depend on TR. Similar to the glutathione system that is found in the mammalian host, the trypanothione system protects the parasite from oxidative damage (36). Interestingly, we found that trypomastigotes were more susceptible to the action of A3K2A1 and A3K2A3. Trypomastigotes are the T. cruzi form with higher trypanothione levels in the thiol-redox system (37), which prompted us to speculate that both compounds are especially active against the trypanothione-dependent antioxidant systems. Similar results were obtained previously for T. cruzi by using another trypanocidal compound (38). Increases in oxidant species induce mitochondrial dysfunction, such as alterations in the ΔΨm (39). The ΔΨm is essential for maintaining the physiological function of the respiratory chain, and a significant loss of ΔΨm depletes cells of energy, with subsequent death (40). In the present study, Rh123 staining revealed marked reductions of the ΔΨm, especially in trypomastigotes. Trypomastigotes exhibit reductions of ΔΨm as a result of remodeling during the life cycle of T. cruzi (41). Several studies of trypanocidal compounds that target T. cruzi mitochondria have been published (7, 38, 42). The mitochondrion in T. cruzi is deficient in the detoxification of ROS/RNS and thus especially susceptible to oxidative stress (43).
Increases in reactive species can damage cell structures, including lipids, nucleic acids, and proteins (34, 35). We found that the three forms of T. cruzi treated with A3K2A1 and A3K2A3 induced lipid peroxidation (MDA and DPPP assays), which interferes with the functional and structural integrity of cell membranes, inducing membrane leakage (44). We also found that A3K2A1 and A3K2A3 induced DNA fragmentation in both proliferative forms of T. cruzi, reflected by an increase in the population of cells in the sub-G0/G1 phase. DNA fragmentation might impair cell duplication, leading to cell cycle arrest, followed by apoptosis (45). A3K2A1 and A3K2A3 also led to lipid body formation, a hallmark of cellular stress that results from mitochondrial deregulation, revealed here by Nile red. The formation and fast accumulation of lipid bodies constitute a characteristic of apoptotic cell death (46, 47). Other studies have reported an increase in lipid body formation in trypanosomatids under stress conditions (23, 48).
All of the biochemical and morphological alterations induced by A3K2A1 and A3K2A3 led to cumulative damage through a cascade of events that are incompatible with cell survival. We found that both compounds might induce parasite death through different pathways. We believe that apoptosis reflected here by mitochondrial depolarization, DNA fragmentation, lipid body accumulation, phosphatidylserine exposure (a distinct set of biochemical changes in cells that undergo apoptosis [49]), and reduced parasite volume might be the preferential mechanism of cell death, even considering all the controversial published data concerning apoptosis in parasitic protozoa (50, 51, 52). Based on the presence of autophagic vacuoles, revealed by MDC labeling, autophagy may also be a pathway of T. cruzi cell death, mainly induced by the increases in cellular ROS levels (53). In addition to apoptosis and autophagy, the plasma membrane disruption, revealed by PI labeling, suggests cell death by necrosis. Evidence indicates that a key event in the transition from apoptosis to autophagy or necrosis is excessive mitochondrial oxidant species production (54, 55).
One issue is whether the increase in oxidant species formation induced by A3K2A1 and A3K2A3 is a cause or consequence of mitochondrial dysfunction. Mitochondrial dysfunction may lead to an increase in ROS/RNS; conversely, an increase in ROS/RNS may induce mitochondrial dysfunction (56). Based on our results, our hypothesis is that the mechanism of action of A3K2A1 and A3K2A3 involves an increase in ROS/RNS production, followed by mitochondrial depolarization. This hypothesis is based on the early increase in O2˙− (after 3 h treatment) compared with late ΔΨm depolarization (after 24 h treatment). An excess of oxidant species might act on any membrane of T. cruzi, including the mitochondrial membrane, which can further impair mitochondrial function and induce more ROS/RNS production (57, 58). Moreover, an excess of reactive species might induce depletion of the endogenous antioxidant system (demonstrated here by a decrease in reduced thiol levels after 48 h treatment), which further exacerbates the condition of oxidative stress (59) (Fig. 13). This possibility is also supported by the chemical structures of the nitro compounds A3K2A1 and A3K2A3 (30). We believe that the increase in ROS/RNS is a cause of mitochondrial dysfunction.
FIG 13.

Proposed mechanism of trypanocidal action of dibenzylideneacetones. A3K2A1 and A3K2A3 increase ROS/RNS production, which triggers mitochondrial depolarization (loss of ΔΨm), thus inducing more ROS/RNS production followed by a decrease in reduced thiol levels. This condition of oxidative stress affects all cell structures and functions, leading to parasite death.
All of the effects induced by A3K2A1 and A3K2A3 are closely related to their structure-activity relationship. Several studies have reported the efficacy of compounds that possess one or more nitro groups against many pathogens (60, 61, 62). Thus, as expected, A3K2A3, which contains two nitro groups in its structure, had better trypanocidal activity than A3K2A1, which contains only one nitro group.
In conclusion, the present findings demonstrated that A3K2A1 and A3K2A3 act as strong activators of T. cruzi death by disrupting redox system homeostasis. A3K2A3 was more effective, inducing parasite death with expressive alterations in T. cruzi machinery components and their function. Both trypomastigotes and amastigotes are infective forms of T. cruzi that are present in the mammalian host, and they were more susceptible to the actions of these nitro compounds. Our results support further studies and may open the way for the development of new chemotherapeutic agents against T. cruzi.
ACKNOWLEDGMENTS
This work was supported through grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq, Capacitação de Aperfeiçoamento de Pessoal de Nível Superior—CAPES, Financiadora de Estudos e Projetos—FINEP, PRONEX/Fundação Araucária, and Complexo de Centrais de Apoio a Pesquisa COMCAP—Universidade Estadual de Maringá.
We have no conflicts of interest.
REFERENCES
- 1.Chagas C. 1909. Nova tripanossomíase humana: estudos sobre a morfologia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., agente etiológico de nova entidade mórbida do homem. Mem Inst Oswaldo Cruz 1:159–218. doi: 10.1590/S0074-02761909000200008. [DOI] [Google Scholar]
- 2.Coura JR. 2009. Present situation and new strategies for Chagas disease chemotherapy—a proposal. Mem Inst Oswaldo Cruz 104:549–554. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2015. Chagas disease (American trypanosomiasis). http://www.who.int/mediacentre/factsheets/fs340/en/. Acessed 20 May 2015. [PMC free article] [PubMed]
- 4.Izumi E, Ueda-Nakamura T, Dias Filho BP, Veiga Júnior VF, Nakamura CV. 2011. Natural products and Chagas' disease: a review of plants compounds studied for activity against Trypanosoma cruzi. Nat Prod Rep 28:809–823. doi: 10.1039/c0np00069h. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigues JH, Ueda-Nakamura T, Corrêa AG, Sangi DP, Nakamura CV. 2014. A quinoxaline derivative as a potent chemotherapeutic agent, alone or in combination with benznidazole, against Trypanosoma cruzi. PLoS One 9:e85706. doi: 10.1371/journal.pone.0085706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menna-Barreto RFS, Laranja GAT, Silva MCC, Coelho MGP, Paes MC, Oliveira MM, Castro SL. 2008. Anti-Trypanosoma cruzi activity of Pterodon pubescens seed oil: geranylgeraniol as the major bioactive component. Parasitol Res 103:111–117. doi: 10.1007/s00436-008-0937-0. [DOI] [PubMed] [Google Scholar]
- 7.Caleare AO, Lazarin-Bidóia D, Cortez DAG, Ueda-Nakamura T, Dias Filho BP, Silva SO, Nakamura CV. 2013. Trypanocidal activity of 1,3,7-trihydroxy-2-(3-methylbut-2-enyl)-xanthone isolated from Kielmeyera coriacea. Parasitol Int 62:405–411. doi: 10.1016/j.parint.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Kollien A, Schaub G. 2000. The development of Trypanosoma cruzi in triatominae. Parasitol Today 16:381–387. doi: 10.1016/S0169-4758(00)01724-5. [DOI] [PubMed] [Google Scholar]
- 9.Maya JD, Cassels BK, Iturriaga-Vásquez P, Ferreira J, Faúndez M, Galanti N, Ferreira A, Morello A. 2007. Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp Biochem Physiol 146:601–620. doi: 10.1016/j.cbpa.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Aher RB, Wanare G, Kawathekar N, Kumar RR, Kaushik N, Sahal KD, Chauhan VS. 2011. Dibenzylideneacetone analogues as novel Plasmodium falciparum inhibitors. Bioorganic Med Chem Lett 21:3034–3036. doi: 10.1016/j.bmcl.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 11.Bhandarkar SS, Bromberg J, Carrillo C, Selvakumar P, Sharma RK, Perry BN, Govindarajan B, Fried L, Sohn A, Reddy K, Arbiser JL. 2008. Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma grown in vitro and in vivo. Clin Cancer Res 14:5743–5748. doi: 10.1158/1078-0432.CCR-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prasad S, Yadav VR, Ravindran J, Aggarwal BB. 2011. ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res 71:538–549. doi: 10.1158/0008-5472.CAN-10-3121. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Yu HJ, Shin J, Nam JS, Kang BS, Cho SD. 2013. Apoptotic effect of dibenzylideneacetone on oral cancer cells via modulation of specificity protein 1 and Bax. Oral Dis 19:767–774. doi: 10.1111/odi.12062. [DOI] [PubMed] [Google Scholar]
- 14.Lee HE, Choi ES, Jung JY, You MJ, Kim LH, Cho SD. 2014. Inhibition of specificity protein 1 by dibenzylideneacetone, a curcumin analogue, induces apoptosis in mucoepidermoid carcinomas and tumor xenografts through Bim and truncated Bid. Oral Oncol 50:189–195. doi: 10.1016/j.oraloncology.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Ud Din Z, Fill TP, de Assis FF, Lazarin-Bidóia D, Kaplum V, Garcia FP, Nakamura CV, de Oliveira KT, Rodrigues-Filho E. 2014. Unsymmetrical 1,5-diaryl-3-oxo-1,4-pentadienyls and their evaluation as antiparasitic agents. Bioorg Med Chem 22:1121–1127. doi: 10.1016/j.bmc.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 16.Camargo EP. 1964. Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev Inst Med Trop São Paulo 6:93–100. [PubMed] [Google Scholar]
- 17.Andrews NW, Colli W. 1982. Adhesion and interiorization of Trypanosoma cruzi in mammalian cells. J Protozool 29:264–269. doi: 10.1111/j.1550-7408.1982.tb04024.x. [DOI] [PubMed] [Google Scholar]
- 18.Piacenza L, Irigoin F, Alvarez MN, Peluffo G, Taylor MC, Kelly JM, Wilkinson SR, Radi R. 2007. Mitochondrial superoxide radicals mediate programmed cell death in Trypanosoma cruzi: cytoprotective action of mitochondrial iron superoxide dismutase overexpression. Biochem J 403:323–334. doi: 10.1042/BJ20061281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shukla AK, Patra S, Dubey VK. 2012. Iridoid glucosides from Nyctanthes arbortristis result in increased reactive oxygen species and cellular redox homeostasis imbalance in Leishmania parasite. Eur J Med Chem 54:49–58. doi: 10.1016/j.ejmech.2012.04.034. [DOI] [PubMed] [Google Scholar]
- 20.Pompella A, Maellaro E, Casini AF, Ferrali M, Ciccoli L, Comporti M. 1987. Measurement of lipid peroxidation in vivo: a comparison of different procedures. Lipids 22:206–211. doi: 10.1007/BF02537304. [DOI] [PubMed] [Google Scholar]
- 21.Okimoto Y, Watanabe A, Niki E, Yamashita T, Noguchi N. 2000. A novel fluorescent probe diphenyl-1-pyrenylphosphine to follow lipid peroxidation in cell membranes. FEBS Lett 474:137–140. doi: 10.1016/S0014-5793(00)01587-8. [DOI] [PubMed] [Google Scholar]
- 22.Ferreira C, Soares DC, Barreto-Junior CB, Nascimento MT, Freire-de-Lima L, Delorenzi JC, Lima ME, Atella GC, Folly E, Carvalho TM, Saraiva EM, Pinto-da-Silva LH. 2011. Leishmanicidal effects of piperine, its derivatives, and analogues on Leishmania amazonensis. Phytochemistry 72:2155–2164. doi: 10.1016/j.phytochem.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Stefanello TF, Panice MR, Ueda-Nakamura T, Sarragiotto MH, Auzély-Velty R, Nakamura CV. 2014. N-Butyl-[1-(4-methoxy)phenyl-9H-β-carboline]-3-carboxamide prevents cytokinesis in Leishmania amazonensis. Antimicrob Agents Chemother 58:7112–7120. doi: 10.1128/AAC.03340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jimenez V, Paredes R, Sosa MA, Galanti N. 2008. Natural programmed cell death in T. cruzi epimastigotes maintained in axenic cultures. J Cell Biochem 105:688–698. doi: 10.1002/jcb.21864. [DOI] [PubMed] [Google Scholar]
- 25.Munafo DB, Colombo MI. 2001. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 114:3619–3629. [DOI] [PubMed] [Google Scholar]
- 26.Biederbick A, Kern HF, Elsasser HP. 1995. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 66:3–14. [PubMed] [Google Scholar]
- 27.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. 1997. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 28.Kaczanowski S, Mohammed S, Reece SE. 2011. Evolution of apoptosis-like programmed cell death in unicellular protozoan parasites. Parasit Vectors 4:44. doi: 10.1186/1756-3305-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valdez RH, Tonin LTD, Ueda-Nakamura T, Dias Filho BP, Díaz JAM, Sarragiotto MH, Nakamura CV. 2009. Biological activity of 1,2,3,4-tetrahydro-β-carboline-3-carboxamides against Trypanosoma cruzi. Acta Trop 110:7–14. doi: 10.1016/j.actatropica.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 30.Patterson S, Wyllie S. 2014. Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends Parasitol 30:289–298. doi: 10.1016/j.pt.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boiani M, Piacenza L, Hernández P, Boiani L, Cerecetto H, González M, Denicola A. 2010. Mode of action of nifurtimox and N-oxide-containing heterocycles against Trypanosoma cruzi: is oxidative stress involved? Biochem Pharmacol 79:1736–1745. doi: 10.1016/j.bcp.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Rodríguez J, Arán VJ, Boiani L, Olea-Azar C, Lavaggi ML, González M, Cerecetto H, Maya JD, Carrasco-Pozo C, Cosoy HS. 2009. New potent 5-nitroindazole derivatives as inhibitors of Trypanosoma cruzi growth: synthesis, biological evaluation, and mechanism of action studies. Bioorg Med Chem 17:8186–8196. doi: 10.1016/j.bmc.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 33.Cadenas E, Davies KJA. 2000. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29:222–230. doi: 10.1016/S0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 34.Ali EMM, Hamdy SM, Mohamed TM. 2012. Nitric oxide synthase and oxidative stress: regulation of nitric oxide synthase, p 61–71. In Lushchak V. (ed), Oxidative stress—molecular mechanisms and biological effects. Intech, Rijeka, Croatia. [Google Scholar]
- 35.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. 2012. Oxidative stress and antioxidant defense. World Allergy Organ J 5:9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irigoín F, Cibils L, Comini MA, Wilkinson SR, Flohe L, Radi R. 2008. Insights into the redox biology of Trypanosoma cruzi: trypanothione metabolism and oxidant detoxification. Free Radic Biol Med 45:733–742. doi: 10.1016/j.freeradbiomed.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 37.Ariyanayagam MR, Oza SL, Mehlert A, Fairlamb AH. 2003. Bis(glutathionyl)spermine and other novel trypanothione analogues in Trypanosoma cruzi. J Biol Chem 278:27612–27619. doi: 10.1074/jbc.M302750200. [DOI] [PubMed] [Google Scholar]
- 38.Lazarin-Bidóia D, Desoti VC, Ueda-Nakamura T, Dias-Filho BP, Nakamura CV, Silva SO. 2013. Further evidence of the trypanocidal action of eupomatenoid-5: confirmation of involvement of reactive oxygen species and mitochondria owing to a reduction in trypanothione reductase activity. Free Radic Biol Med 60:17–28. doi: 10.1016/j.freeradbiomed.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 39.Kang J, Pervaiz S. 2012. Mitochondria: redox metabolism and dysfunction. Biochem Res Int 2012:896751. doi: 10.1155/2012/896751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joshi DC, Bakowska JC. 2011. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J Vis Exp 51:1–4. doi: 10.3791/2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonçalves RLS, Menna-Barreto RFS, Polycarpo CR, Gadelha FR, Castro SL, Oliveira MF. 2011. A comparative assessment of mitochondrial function in epimastigotes and bloodstream trypomastigotes of Trypanosoma cruzi. J Bioenerg Biomembr 43:651–661. doi: 10.1007/s10863-011-9398-8. [DOI] [PubMed] [Google Scholar]
- 42.Menna-Barreto RFS, Gonçalves RLS, Costa EM, Silva RSF, Pinto AV, Oliveira MF, Castro SL. 2009. The effects on Trypanosoma cruzi of novel synthetic naphthoquinones are mediated by mitochondrial dysfunction. Free Radic Biol Med 47:644–653. [DOI] [PubMed] [Google Scholar]
- 43.Urbina JA. 1999. Parasitological cure of Chagas disease: is it possible? Is it relevant? Mem Inst Oswaldo Cruz 94:349–355. doi: 10.1590/S0074-02761999000700068. [DOI] [PubMed] [Google Scholar]
- 44.Pamplona R. 2008. Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim Biophys Acta 1777:1249–1262. doi: 10.1016/j.bbabio.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Fulda S, Gorman AM, Hori O, Samali A. 2010. Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SJ, Zhang J, Choi AMK, Kim HP. 2013. Mitochondrial dysfunction induces formation lipid droplets as a generalized response to stress. Oxid Med Cell Longev 2013:327167. doi: 10.1155/2013/327167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boren J, Brindle KM. 2012. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ 19:1561–1570. doi: 10.1038/cdd.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dantas AP, Barbosa HS, Castro SL. 2003. Biological and ultrastructural effects of the anti-microtubule agent taxol against Trypanosoma cruzi. J Submicrosc Cytol Pathol 35:287–294. [PubMed] [Google Scholar]
- 49.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nuñez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G. 2009. Classification of cell death recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smirlis D, Duszenko M, Ruiz AJ, Scoulica E, Bastien P, Fasel N, Soteriadou K. 2010. Targeting essential pathways in trypanosomatids gives insights into protozoan mechanisms of cell death. Parasit Vectors 3:107. doi: 10.1186/1756-3305-3-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiménez-Ruiz A, Alzate JF, MacLeod ET, Lüder CGK, Fasel N, Hurd H. 2010. Apoptotic markers in protozoan parasites. Parasit Vectors 3:104. doi: 10.1186/1756-3305-3-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Proto WR, Coombs GH, Mottram JC. 2013. Cell death in parasitic protozoa: regulated or incidental? Nat Rev Microbiol 11:58–66. doi: 10.1038/nrmicro2929. [DOI] [PubMed] [Google Scholar]
- 53.Filomeni G, Zio D, Cecconi F. 2014. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 2014:1–12. doi: 10.1038/cdd.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaminskyy VO, Zhivotovsky B. 2014. Free radicals in cross talk between autophagy and apoptosis. Antioxid Redox Signal 21:86–102. doi: 10.1089/ars.2013.5746. [DOI] [PubMed] [Google Scholar]
- 55.Addabbo F, Montagnani M, Goligorsky MS. 2009. Mitochondria and reactive oxygen species. Hypertension 53:885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vamecq J, Dessein AF, Fontaine M, Briand G, Porchet N, Latruffe N, Andreolotti P, Cherkaoui-Malki M. 2012. Mitochondrial dysfunction and lipid homeostasis. Curr Drug Metab 13:1388–1400. doi: 10.2174/138920012803762792. [DOI] [PubMed] [Google Scholar]
- 57.Rhoads DM, Umbach AL, Subbaiah CC, Siedow JN. 2006. Mitochondrial reactive oxygen species. Contribution to oxidative stress and interorganellar signaling. Plant Physiol 141:357–366. doi: 10.1104/pp.106.079129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupta S, Bhatia V, Wen JJ, Wu Y, Huang MH, Garg NJ. 2009. Trypanosoma cruzi infection disturbs mitochondrial disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic Biol Med 47:1414–1421. doi: 10.1016/j.freeradbiomed.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mahmood KAS, Ahmed JH, Jawad AM. 2009. Non-steroidal anti-inflammatory drugs (NSAIDS), free radicals and reactive oxygen species (ROS): a review of literature. Med J Basrah Univ 27:46–53. [Google Scholar]
- 60.Oliveira RB, Passos APF, Alves RO, Romanha AJ, Prado MAF, Souza Filho JD, Alves RJ. 2003. In vitro evaluation of the activity of aromatic nitrocompounds against Trypanosoma cruzi. Mem Inst Oswaldo Cruz 98:141–144. doi: 10.1590/S0074-02762003000100018. [DOI] [PubMed] [Google Scholar]
- 61.Mukherjee A, Dutta S, Shanmugavel M, Mondhe DL, Sharma PR, Singh SK, Saxena AK, Sanyal U. 2010. 6-Nitro-2-(3-hydroxypropyl)-1H-benz[de]isoquinoline-1,3-dione, a potente antitumor agent, induces cell cycle arrest and apoptosis. J Exp Clin Cancer Res 29:175. doi: 10.1186/1756-9966-29-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bastos TM, Barbosa MIF, Silva MM, Júnior JWC, Meira CS, Guimarães ET, Ellena J, Moreira DRM, Batista AA, Soares MBP. 2014. Nitro/nitrosyl ruthenium complexes are potent and selective anti-Trypanosoma cruzi agents causing autophagy and necrotic parasite death. Antimicrob Agents Chemother 58:6044–6055. doi: 10.1128/AAC.02765-14. [DOI] [PMC free article] [PubMed] [Google Scholar]