

Abstract
Around the world, Burkholderia spp. are emerging as pathogens highly resistant to β-lactam antibiotics, especially ceftazidime. Clinical variants of Burkholderia pseudomallei possessing the class A β-lactamase PenI with substitutions at positions C69 and P167 are known to demonstrate ceftazidime resistance. However, the biochemical basis for ceftazidime resistance in class A β-lactamases in B. pseudomallei is largely undefined. Here, we performed site saturation mutagenesis of the C69 position and investigated the kinetic properties of the C69F variant of PenI from B. pseudomallei that results in a high level of ceftazidime resistance (2 to 64 mg/liter) when expressed in Escherichia coli. Surprisingly, quantitative immunoblotting showed that the steady-state protein levels of the C69F variant β-lactamase were ∼4-fold lower than those of wild-type PenI (0.76 fg of protein/cell versus 4.1 fg of protein/cell, respectively). However, growth in the presence of ceftazidime increases the relative amount of the C69F variant to greater than wild-type PenI levels. The C69F variant exhibits a branched kinetic mechanism for ceftazidime hydrolysis, suggesting there are two different conformations of the enzyme. When incubated with an anti-PenI antibody, one conformation of the C69F variant rapidly hydrolyzes ceftazidime and most likely contributes to the higher levels of ceftazidime resistance observed in cell-based assays. Molecular dynamics simulations suggest that the electrostatic characteristics of the oxyanion hole are altered in the C69F variant. When ceftazidime was positioned in the active site, the C69F variant is predicted to form a greater number of hydrogen-bonding interactions than PenI with ceftazidime. In conclusion, we propose “a new twist” for enhanced ceftazidime resistance mediated by the C69F variant of the PenI β-lactamase based on conformational changes in the C69F variant. Our findings explain the biochemical basis of ceftazidime resistance in B. pseudomallei, a pathogen of considerable importance, and suggest that the full repertoire of conformational states of a β-lactamase profoundly affects β-lactam resistance.
INTRODUCTION
The expanded-spectrum (ES) cephalosporin ceftazidime is a first-line agent used to treat patients infected with Burkholderia pseudomallei (1). B. pseudomallei is the causative agent of the multisystem disease melioidosis, is a potential biothreat agent, and possesses many innate antimicrobial resistance genes (2). In some parts of the world, melioidosis is characterized by a 20 to 50% mortality rate with treatment, and the death rate increases to >80% for patients with sepsis (2, 3). Resistance to ceftazidime is common in clinical isolates of B. pseudomallei and is linked to increased expression of serine β-lactamases (classes A and D) and expression of different isoforms of the class A β-lactamase, as well as to deletion of penicillin binding protein 3 (ΔPBP3) (4–12).
The class A β-lactamase PenI, also referred to as PenA and BPS, produced by B. pseudomallei is part of the PenA family of class A β-lactamases that are widely distributed in the genus Burkholderia (5, 7, 13–15). Single-amino-acid substitutions at positions C69 and P167 are associated with ceftazidime resistance in PenI-expressing B. pseudomallei (5–7, 11). In class A β-lactamases, the evolution of ceftazidime resistance is mediated by amino acid substitutions in the Ω loop (residues 164 to 179), the B3 β-strand (residues 234 to 240), and, in rare instances, the oxyanion hole (i.e., residue 69) (Fig. 1A) (16–18). The mechanistic contributions of amino acid substitutions at these sites that enhance catalytic efficiency toward ceftazidime are poorly understood and merit further exploration. Ascertaining the details and mechanism of ceftazidime resistance is critical to identifying future successful therapies.
FIG 1.

(A) Structure of the PenI β-lactamase and PenI's active site displaying residues C69, S70, and T237 in green. The backbone amides of S70 and T237 form the electrophilic center of the β-lactamase that draws in the carbonyl of the β-lactam (blue) for nucleophilic attack by the S70 hydroxyl side chain. C69 resides on the back wall of the active site and does not participate in the catalytic mechanism. (B) Alignment of amino acid positions 66 to 73 from a variety of class A β-lactamases showing the diversity at position 69.
The focus of this work is Ambler residue 69 in PenI, which forms the “back wall” of the oxyanion hole in class A β-lactamases and is located on helix 2 (H2) proximal to the nucleophilic S70 between the B3 β-strand and the Ω loop (Fig. 1A). The oxyanion hole is a “pocket” formed by the backbone amides of S70 and T237 that creates an electrophilic center attracting the β-lactam's carbonyl for binding and subsequent hydrolysis. Conservation of amino acids at position 69 is minimal, as class A enzymes possess different amino acids (e.g., M, C, A, T, and Q) at this position (Fig. 1B). Most often, substitutions at position 69 in other class A β-lactamases (e.g., SHV, TEM, OHIO-1, and BlaC) are associated with β-lactam–β-lactamase inhibitor resistance (e.g., ampicillin-clavulanic acid) (16, 19–27). For example, M69I, -V, and –L variants of SHV-1 are resistant to inhibition by clavulanic acid. β-Lactamase inhibitor resistance in the SHV family of β-lactamases is hypothesized to occur due to the “deformation” of the oxyanion hole (25, 26).
On the other hand, studies using an entire panel of M69 variants of SHV-1 revealed that residue 69 played a larger role in determining substrate specificity, since M69K, -Y, and -F variants demonstrated increased resistance to ceftazidime (16). This phenotype was attributed to movement of the B3 β-strand and repositioning of amino acids in the active site that mimicked the case with extended-spectrum β-lactamases (ESBLs). Clearly, the mechanism that allows ceftazidime resistance due to amino acid substitutions at position 69 of class A β-lactamases is complex.
In this study, we explore the role of Ambler position C69 and its contribution to ceftazidime resistance in PenI using microbiological, biochemical, analytical, and proteomic methods. Interestingly, our data reveal that 9 of 19 substitutions at this position increase ceftazidime resistance by >2 doubling dilutions. We chose to study the C69F variant in detail as (i) it conferred an MIC of 64 mg/liter despite lower protein expression levels; (ii) the change to Phe is found in other enzymes, such as SHV; and (iii) the C69Y variant of PenI was previously studied (6, 7). Our data suggest that ceftazidime resistance in the C69F variant is mediated by unique conformational species of the C69F variant, one of which hydrolyzes ceftazidime rapidly when stabilized. Our findings provide new insight in the quest to understand β-lactamase-mediated resistance in this clinically important pathogen.
MATERIALS AND METHODS
Strains and mutagenesis.
Construction of the wild-type β-lactamase vectors has been previously described (14). Site saturation and site-directed mutagenesis were performed using the Quikchange XL site-directed-mutagenesis kit (Agilent) according to the manufacturer's protocol. Briefly, primers that included all 19 amino acid substitutions at Ambler position 69 for blaPenI were designed. PCR was conducted using the phagemid pBC SK(+) blaPenI as the template for the C69 site saturation primer sets. To digest the template, 1.0 μl of DpnI was added to all of the reaction mixtures, and the reaction mixtures were incubated at 37°C for 1 h. Escherichia coli DH10B cells were electroporated with 1.0 μl of each reaction mixture and plated on lysogeny broth (LB) agar plates containing 20 mg/liter chloramphenicol as the selecting agent. Single colonies were chosen for plasmid purification, and DNA sequencing by MCLAB (Molecular Cloning Laboratories) was conducted to verify the mutations. Site-directed mutagenesis was also performed as described above, but using the pET24a(+) blaPenI vector with site-directed primers containing the nucleotide sequence for the corresponding C69F substitution. After the mutations were verified, pET24a(+) plasmids were transformed into E. coli Origami 2(DE3) cells. All protein expression constructs carrying pET24a(+) blaPenI are missing the signal peptide corresponding to the first 90 nucleotides.
Cell-based assays.
E. coli DH10B expressing blaPenI and position 69 from pBC SK(+) were phenotypically characterized using agar dilution MICs. Mueller-Hinton (MH) agar was used for the cell-based assays, according to the Clinical and Laboratory Standards Institute guidelines, using a Steers replicator (28). Ampicillin, piperacillin, cephalothin, cefotaxime, and clavulanic acid were purchased from Sigma-Aldrich. Ceftazidime was acquired from Research Products International Corp. Imipenem-cilastatin was obtained from its commercial source. Sulbactam and aztreonam were bought from Astratech Inc., while tazobactam was purchased from Chem-Impex International Inc.
Immunoblotting.
Cells expressing PenI and all of the variants were grown in LB to log phase (with an optical density at 600 nm [OD600] between 0.6 and 0.7). The cells were pelleted and lysed using stringent periplasmic fractionation to prepare crude extracts, as previously described (29). The extracts were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes. The membranes were blocked in 5% nonfat dry milk in 20 mM Tris-Cl with 150 mM NaCl, pH 7.4 (Tris-buffered saline [TBS]), for 1 h and probed in 5% nonfat dry milk in TBS with 1 μg/ml of polyclonal anti-PenI antibody (7) and a loading control, a 1:10,000 dilution of monoclonal anti-DNAK antibody (Stressgen), for 1 h. The membranes were washed five times for 10 min each time with TBS with 0.05% Tween 20 (TBST), and for protein detection, blots were incubated for 1 h in 1:10,000 dilutions of horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse secondary antibodies in 5% nonfat dry milk in TBS. The blots were washed five times for 10 min each time with TBST and developed using the ECL-Plus developing kit (GE Healthcare Life Sciences) or the SuperSignal West Femto Chemiluminescent Substrate (Thermoscientific) according to the manufacturers' instructions. A Fotodyne Luminary/FX was used to capture images.
Quantitative immunoblotting of β-lactamases.
Previously described methods were used to determine the amount of β-lactamase protein per cell (30). Briefly, cells were grown and periplasmic extracts were prepared as described above for immunoblotting. Cells pretreated with ceftazidime at a concentration of 1 µg/ml were grown overnight in LB and then subcultured and grown to an OD600 of 0.6 to 0.7 in LB with ceftazidime at 1 μg/ml. An aliquot of cells was used to determine the total cell number by serial dilution, plating, and colony counts. A standard curve with known amounts of purified PenI and crude extracts was used for immunoblotting as described above. Band densities on the blot images were assigned using EZQuant gel analysis software. The standard curve and total cell number were used to calculate the amount of protein per cell.
β-Lactamase purification.
The purification of PenI was described previously (14). Briefly, E. coli Origami 2(DE3) cells expressing PenI and the C69F variant β-lactamases expressed from the pET24a(+) plasmid were grown in superoptimal broth to an OD600 of 0.6, 200 μM isopropyl-β-d-1-thiogalactopyranoside was added, and the cells were grown for an additional 3 h at 37°C. The cells were pelleted and lysed. The crude extracts prepared from PenI and the variants were used for preparative isoelectric focusing (pIEF) with a pH gradient from 3.5 to 10, followed by a nitrocefin overlay for detection (31). The samples from pIEF were dialyzed against 10 mM phosphate-buffered saline (PBS), pH 7.4, for up to 48 h to remove residual nitrocefin. The purity of the fractions from all of the preparations was determined by SDS-PAGE. The gels were stained with Coomassie brilliant blue R250. The protein concentrations were determined by measuring the absorbance at λ280 and Beer's law using the proteins' extinction coefficients (Δε) (21,555 M−1 cm−1 for PenI and the variants at 280 nm) (14). The theoretical masses of the β-lactamases, as well as the extinction coefficients, were obtained using the ProtParam tool on the ExPASy Bioinformatics Resource Portal. To maintain full enzymatic activity, PenI and the variant β-lactamases were stored in 10 mM PBS, pH 7.4, with 25% glycerol at −20°C.
Steady-state kinetics.
The steady-state kinetic parameters were determined using an Agilent 8453 Diode Array spectrophotometer. All of the assays were completed in 10 mM PBS at pH 7.4 at 25°C. The interactions between the PenI β-lactamase (E) and the β-lactam (S) substrate studies were initially interpreted according to the scheme presented in Fig. 2.
FIG 2.
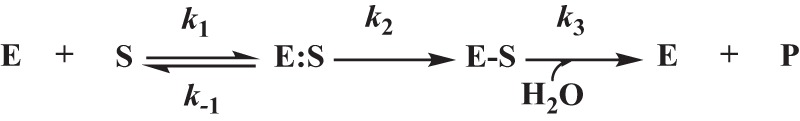
Interactions between the β-lactamase (E) and the β-lactam (S) were initially interpreted according to the scheme shown. Here, the formation of the noncovalent complex, E:S, is represented by the dissociation constant, Ks, which is equivalent to k−1/k1. k2 is the first-order rate constant for the acylation step, or the formation of the E-S complex. k3 is the rate constant for the hydrolysis of the E-S acyl-enzyme and product (P) release. The Michaelis constant, Km, is equivalent to Ks × (k3/k2 + k3).
The spectroscopic properties of nitrocefin and ceftazidime, as well as the poor catalytic efficiency of the C69F variant, limited the ability to determine Km and Vmax values. To study ceftazidime hydrolysis, 10 μM PenI or the C69F variant was mixed with 25 μM ceftazidime. Ceftazidime hydrolysis was measured during a 2,000 s time course at an absorbance of λ260.
Additionally, progress curves were obtained by inhibiting nitrocefin hydrolysis with increasing concentrations of ceftazidime for a selected time course. For these assays, ceftazidime and nitrocefin were initially mixed, and then the enzyme was diluted in the reaction mixture. PenI was maintained at 350 nM with ceftazidime concentrations ranging from 20 to 320 mM to observe inhibition of 50 μM nitrocefin hydrolysis for 300 s. The C69F variant was held at 10 μM (to obtain measurable nitrocefin hydrolysis) with a ceftazidime concentration range of 10 μM up to 1 mM with 50 μM nitrocefin for 2,200 s. Progress curves were fitted to obtain kobs values using the following equation: y = Vf × x + (V0 − Vf) × [1 − exp(−kobs × x)]/kobs + A0, where, Vf is the final velocity, V0 is the initial velocity, and A0 is the initial absorbance at 482 nm. The kobs values versus the ceftazidime concentration were plotted. If a saturation curve was obtained from the kobs versus the ceftazidime concentration graph, then a modified Michaelis-Menten equation was used to determine KI and kinact (32). Conversely, if a line was obtained, then the slope of the line was determined to be k2/K (33). The KI and k2/K values were not corrected for the use of nitrocefin, as the Km and Vmax values could not be determined for the C69F variant.
Pre-steady-state kinetics.
The pre-steady-state kinetic parameters were determined using an Applied Photophysics SX20 stopped-flow apparatus. All of the assays were completed in 10 mM PBS at pH 7.4 at 25°C. To assess for a “burst” in ceftazidime hydrolysis, 10 μM the C69F variant was mixed with 25 μM ceftazidime, and ceftazidime hydrolysis at an absorbance of λ260 was measured. In order to evaluate the changes in protein conformation, 10 μM the C69F variant was preincubated for 10 min with a polyclonal anti-PenI antibody (which was also used for immunoblotting [see above]) according to the method developed by Citri et al. (34). The preincubated C69 variant-antibody complex was again mixed with 25 μM ceftazidime, and ceftazidime hydrolysis at an absorbance of λ260 was measured.
ESI-MS of ceftazidime.
Electrospray ionization-mass spectrometry (ESI-MS) of ceftazidime and the hydrolyzed products of ceftazidime was performed on an Agilent Technologies 6460 Triple Quad mass spectrometer equipped with an electrospray ion source. A check tune was conducted in positive mode and passed on the Agilent 6460 Triple Quad using ESI-L low-concentration tuning mix (Agilent; G1969-85000). Samples consisted of 2 mM ceftazidime, 2 mM ceftazidime with 100 mM sodium hydroxide (NaOH), 2 mM ceftazidime with 100 μM PenI, and 2 mM ceftazidime with 100 μM the C69F variant. These samples were incubated at 37°C for 4 h, and then the protein was removed using centrifugation on Nanosep centrifugation columns with a 10,000 Da molecular mass cutoff (Pall Corp.) for 10 min at 10,000 rpm. One microliter of flowthrough from each centrifugation was run on an Agilent Infinity 1260 high-performance liquid chromatograph (HPLC) with an Agilent Poroshell 120 EC-C18 2.7-μm, 3.0- by 50-mm column with the column temperature maintained at 25°C. The mobile phase consisted of 0.1% formic acid in water. The small molecules were eluted using a gradient with final conditions of 10% mobile phase and 90% organic phase (100% acetonitrile with 0.1% formic acid). The samples were run in MS2 positive scan mode with a starting mass of 400 atomic mass units (amu) and an ending mass of 600 amu, a scan time of 500, the fragmenter set at 70, and the cell accelerator voltage at 4. The settings for each of the data runs were as follows: capillary voltage at 4.0 kV, gas temperature at 350°C, gas flow at 10 liters/min, nebulizer at 40 lb/in2, sheath gas temperature at 350°C, and sheath gas flow at 11 liters/min. The spectra were analyzed using the Agilent Mass Hunter Qualitative Analysis Program B.06.00.
ESI-MS of β-lactamases.
ESI-MS of the intact PenI and the C69F variant with and without ceftazidime was performed on a Waters Synapt G2-Si quadrupole-time of flight mass spectrometer equipped with a LockSpray dual-electrospray ion source, using glu-1-fibrinopeptide B as the lock mass. The Synapt G2-Si was calibrated with a sodium iodide solution using a 50 to 2,000 m/z mass range. Each β-lactamase (10 μM) was incubated with the β-lactam (10 μM in most cases for a 1:1 β-lactam-to-β-lactamase ratio, but up to 1 mM with PenI for a 100:1 ceftazidime-to-PenI ratio) for various incubation times. The reactions were terminated by the addition of 0.1% formic acid and 1% acetonitrile. The samples were run using a Waters Acquity H class ultraperformance liquid chromatograph (UPLC) on an Acquity UPLC BEH C18 1.7-μm, 2.1- by 100-mm column. The mobile phase consisted of 0.1% formic acid in water. The β-lactamase and β-lactamase–β-lactam complexes were eluted using a gradient with final conditions of 15% mobile phase and 85% organic phase (100% acetonitrile with 0.1% formic acid). Lock mass spectra were collected during the sample injections. The tune settings for each of the data runs were as follows: capillary voltage at 3.5 kV, sampling cone at 35, source offset at 35, source temperature at 100°C, desolvation temperature at 500°C, cone gas at 100 liters/h, desolvation gas at 800 liters/h, and nebulizer bar at 6.0. The spectra were analyzed using MassLynx v4.1, modified for the lock mass deviations by applying a gain factor, and deconvoluted using the MaxEnt1 program.
Molecular modeling.
The crystal coordinates of PenI (Protein Data Bank [PDB] 3W4P) were used to construct the acyl-enzyme representations of PenI and the C69F variant with ceftazidime, as previously described, using the Discovery Studio 4.1 (DS 4.1) (Accelrys, Inc., San Diego, CA) molecular-modeling software (17, 35). The variant was built by substituting phenylalanine for the cysteine at position 69.
Ceftazidime was constructed for molecular modeling using the Fragment Builder tools and was minimized using a Standard Dynamics Cascade protocol of DS 4.1. Ceftazidime (missing its R2 side chain [see below]) was automatically positioned in the active site of PenI β-lactamase using the Flexible Docking module of DS 4.1. The protocol allowed for the flexible placement of a β-lactamase inhibitor in the active site of PenI. After docking, the most favorable pose of ceftazidime was chosen (i.e., demonstrating a short distance [2 to 3 Å] between Ser70: O (hydroxyl side chain of Ser70) and C7 of ceftazidime, as well as anticipated active-site contacts), and the acyl complex was created. The complex of the C69F variant with ceftazidime was created in a similar fashion. To check the stability and to look for possible conformational changes of the complex, Molecular dynamics simulation (MDS) was conducted for 6 ps on PenI-ceftazidime and the C69F-ceftazidime complexes, as previously described and validated (35).
RESULTS AND DISCUSSION
Cell-based assays of E. coli expressing blaPenI and the C69 variants.
An isogenic E. coli library was created to assess the impact of single-amino-acid substitutions. We observed that, in general, changes at residue 69 in PenI increased resistance to ceftazidime, but not to other β-lactams or β-lactam–β-lactamase inhibitor combinations. As Table 1 shows, 13 of 19 variants demonstrated an increase in the ceftazidime MIC compared to the wild-type blaPenI counterpart when expressed in an isogenic background, E. coli DH10B carrying the pBC SK(+) phagemid. The C69F and -Y variants displayed the highest levels of ceftazidime MICs at 64 mg/liter, and the former (C69F) was chosen for further study. This increase in the MIC was much more dramatic than that observed with another class A β-lactamase, SHV-1. For SHV-1, only a two-doubling-dilution increase in the ceftazidime MIC was observed for the M69F variant (8 mg/liter) compared to wild-type SHV-1 (2 mg/liter) expressed in E. coli DH10B (16).
TABLE 1.
Cell-based assays to determine MIC values of E. coli DH10B expressing blaPenI and the C69X variants of PenI for selected β-lactams and β-lactam–β-lactamase inhibitor combinations
| Strain | MICa (mg/liter) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| AMP | THIN | TAX | CAZ | AZT | IMI | AMP-CLAVb | AMP-SULb | PIP-TAZOc | |
| E. coli DH10B pBC SK(+) | 4 | 2 | 0.06 | 0.25 | 1 | 0.5 | 50/0.06 | 50/1 | 2/0.25 |
| E. coli DH10B pBC SK(+) blaPenId | 512 | 256 | 4 | 2 | 8 | 0.25 | 50/0.06 | 50/4 | 4/0.5 |
| C69A | 512 | 512 | 2 | 4 | 1 | 0.25 | 50/0.125 | 50/4 | 4/0.5 |
| C69D | 8 | 4 | 0.125 | 0.5 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
| C69E | 8 | 16 | 1 | 4 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
| C69F | 8 | 16 | 4 | 64 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
| C69G | 256 | 64 | 0.25 | 2 | 1 | 0.25 | 50/0.125 | 50/2 | 8/1 |
| C69H | 64 | 64 | 2 | 16 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
| C69I | 256 | 32 | 0.5 | 16 | 1 | 0.25 | 50/0.06 | 50/1 | 8/1 |
| C69K | 8 | 16 | 1 | 16 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
| C69L | 512 | 128 | 0.5 | 4 | 1 | 0.5 | 50/0.06 | 50/4 | 8/1 |
| C69 M | 256 | 256 | 2 | 16 | 1 | 0.5 | 50/0.06 | 50/1 | 4/0.5 |
| C69N | 256 | 128 | 0.5 | 8 | 1 | 0.5 | 50/0.06 | 50/2 | 8/1 |
| C69P | 4 | 2 | 0.06 | 0.25 | 1 | 0.25 | 50/0.06 | 50/1 | 2/0.25 |
| C69Q | 64 | 16 | 1 | 16 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
| C69R | 8 | 8 | 1 | 16 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
| C69S | 256 | 32 | 0.125 | 0.5 | 1 | 0.5 | 50/0.06 | 50/4 | 8/1 |
| C69T | 512 | 256 | 0.5 | 1 | 1 | 0.5 | 50/0.06 | 50/4 | 16/2 |
| C69V | 512 | 64 | 0.5 | 8 | 1 | 0.5 | 50/0.25 | 50/4 | 8/1 |
| C69W | 4 | 8 | 0.125 | 0.5 | 1 | 0.25 | 50/0.06 | 50/1 | 2/0.25 |
| C69Y | 4 | 16 | 8 | 64 | 1 | 0.25 | 50/0.06 | 50/1 | 4/0.5 |
β-Lactam and β-lactamase inhibitor abbreviations: AMP, ampicillin; THIN, cephalothin; TAX, cefotaxime; CAZ, ceftazidime; AZT, aztreonam; IMI, imipenem; CLAV, clavulanic acid; SUL, sulbactam; and TAZO, tazobactam. Experiments were completed in triplicate. Elevated CAZ MICs are in boldface.
AMP was maintained at a constant concentration of 50 mg/liter, and the clavulanic acid and sulbactam concentrations were varied.
Piperacillin and tazobactam were varied at a ratio of 8:1.
All of the C69 variants were expressed from pBC SK(+) blaPenI in E. coli DH10B.
Interestingly, three variants, C69D, -P, and -W, were more susceptible to both ampicillin and ceftazidime, unlike the other 16 variants. We hypothesize that the amino acid substitutions of aspartic acid, proline, and tryptophan must alter the oxyanion hole pocket due to the introduction of a negative charge, a kink in the strand, and a steric clash caused by a large hydrophobic side chain, respectively. These changes would directly impact the binding and hydrolysis of ampicillin and ceftazidime. The understanding of these mechanisms will be the focus of future study.
Steady-state β-lactamase expression of PenI and C69 variants in E. coli.
The pattern of susceptibility of the C69 variants prompted us to measure the β-lactamase expression levels of PenI and the C69 variants of these cells grown to log phase in LB. Here, we asked whether the differences in the ceftazidime resistance profiles of the C69 variants when expressed in E. coli could be the result of changes in β-lactamase expression (Fig. 3A). Interestingly, expression of the C69F variant of PenI was less than steady-state wild-type PenI expression. Growing the C69F variant with subinhibitory concentrations of ceftazidime returned the expression of C69F to steady-state levels (Fig. 3B). A number of factors (e.g., altered transcription, altered translation, protein half-life, proteolytic degradation, protein stability, and/or transport into the periplasmic space) have the potential to influence the increased protein content. Deeper analysis of these factors will be the focus of future work.
FIG 3.
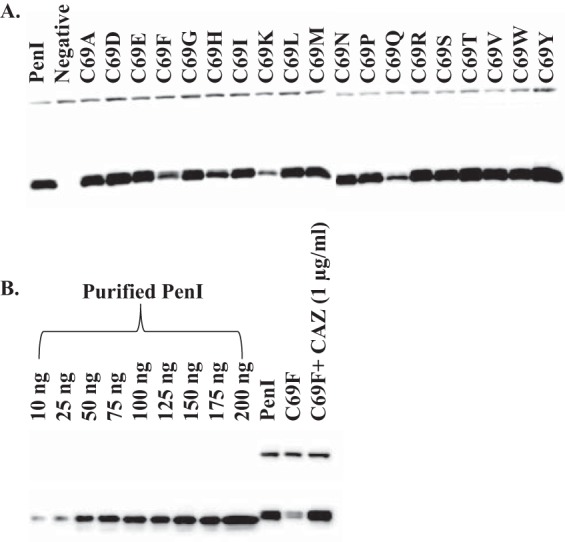
Immunoblots of PenI and variants. (A) PenI β-lactamase (bottom band) expression levels of E. coli DH10B cells carrying pBC SK(+) blaPenI (PenI) and the C69 variants grown in LB to log phase; DNAK was used as a loading control (top band). Lane “Negative” is E. coli DH10B with an empty pBC SK(+) phagemid. (B) PenI β-lactamase expression levels of E. coli DH10B cells carrying pBC SK(+) blaPenI (PenI) and the C69F variant grown in LB to log phase with or without ceftazidime (CAZ) at 1 μg/ml. A standard curve was derived from the densities of the bands for known concentrations of purified PenI (10 to 200 ng).
Ceftazidime turnover by PenI and the C69F variant.
To assess the β-lactam resistance profile of the C69F variant compared to PenI, the β-lactamases were purified for steady-state and pre-steady-state kinetics. We also investigated the β-lactam products formed following hydrolysis by the β-lactamases.
Measuring hydrolysis of 25 μM ceftazidime by 10 μM PenI and the C69F variant revealed that the C69F variant hydrolyzed an amount of ceftazidime similar to that hydrolyzed by PenI within 1,500 s, but at different overall rates (Fig. 4A and B). PenI's hydrolysis of ceftazidime follows an exponential decrease that is faster than that of the C69F variant. This hydrolysis could be due to poor affinity of ceftazidime for PenI, resulting in an instantaneous velocity for the reaction that decreases as the ceftazidime is consumed. Notably, the progress curve of the C69F variant with ceftazidime was biphasic with an initial burst in hydrolysis, suggesting a branched pathway followed by a linear velocity similar to a zero-order process, which may be attributable to better affinity of ceftazidime for the C69F variant than PenI.
FIG 4.

Kinetics of PenI and the C69F variant. (A) Hydrolysis of 25 μM ceftazidime by 10 μM PenI. (B) Biphasic hydrolysis of 25 μM ceftazidime by 10 μM the C69F variant measured on a stopped-flow apparatus. (C) Hydrolysis of 50 μM nitrocefin by 350 nM PenI inhibited by increasing concentrations of ceftazidime. (D) Hydrolysis of 50 μM nitrocefin by 10 μM the C69F variant inhibited by increasing concentrations of ceftazidime. (E) The kobs values obtained by fitting the progress curves in panel C to the equation in Materials and Methods plotted versus the ceftazidime concentration. (F) The kobs values obtained by fitting the progress curves in panel D to the equation in Materials and Methods plotted versus the ceftazidime concentration. a.u., arbitrary units.
Two primary branched pathways for select substrates have been described for certain β-lactamases. As initially reported by Faraci and Pratt in 1985 (36), one branched pathway can occur as a result of the loss of the R2 side chain of cephalosporins (Fig. 5A). Another branched pathway can be observed due to a reversible conformational change in the enzyme during hydrolysis of a cephalosporin (Fig. 5B) (34). To aid in distinguishing between these two possible branched mechanisms in C69F turnover of ceftazidime, we performed small-molecule mass spectrometry and pre-steady-state kinetics.
FIG 5.

The biphasic kinetics observed for the C69F variant can be explained by two different branched kinetic schemes. (A) As previously described by Faraci and Pratt (36), loss of the R2 of cephalosporins is a common mechanism during cephalosporin hydrolysis that can lead to a branched mechanism, depicted by the rate constant k3. (B) Alternatively, earlier, Citri et al. (34) found that β-lactamases can undergo a potentially reversible conformation change to a less active form, E*, here depicted by the rate constant k5, resulting in an alternative branched mechanism. (C) Chemical structures of hydrolysis of ceftazidime (S), acyl-enzyme complexes, loss of the R2 side chain (S−R2), and potential final products (P and P−R2).
If the branched mechanism is due to loss of the R2 side chain, then we would observe two ceftazidime products (P and P−R2) (Fig. 5A and C). To test this possible mechanism, we incubated ceftazidime alone, ceftazidime with NaOH (as a control), ceftazidime with PenI, and ceftazidime with the C69F variant at 37°C for 4 h. In each case, the β-lactamase was removed using size exclusion column chromatography, and the products were evaluated using HPLC and triple quad mass spectrometry.
Unreacted ceftazidime produced the expected mass of unhydrolyzed substrate at 547.1 amu (Fig. 6A). The reaction that served as a control (NaOH) revealed a peak of 486.1 amu, which corresponds to hydrolyzed exomethylene ceftazidime product (P−R2); the hydrolyzed product (P) of ceftazidime of 565 amu was not observed (Fig. 6B). The exomethylene product of ceftazidime is predicted to occur as a result of catalysis by the β-lactamase and was previously observed by other groups (17, 37). Both PenI and the C69F variant incubated with ceftazidime produced the same two products of 474.0 amu and 486.1 amu, corresponding to the complete removal of the R2 side chain and hydrolyzed exomethylene ceftazidime product (P−R2), respectively (Fig. 6C and D). The products observed from hydrolysis of ceftazidime were missing the R2 side chain, which leads us to hypothesize that a conformation change in the β-lactamase may be major driving force behind the kinetics observed. However, we note that the hydrolyzed ceftazidime with an intact R2 side chain may be too unstable under these conditions to be detected by this method. Thus, the contribution of the loss of the R2 side chain of ceftazidime to a branched mechanism cannot be entirely ruled out.
FIG 6.

Small-molecule mass spectrometry revealed the observed products after incubation at 37°C for 4 h, restricting the mass range window to 400 to 600 amu. (A) Ceftazidime (2 mM) alone showed a major peak of the full-size ceftazidime at 547 amu and a very minor peak at 468 amu representative of ceftazidime with the breakdown of the R2 side chain to the exomethylene; increasing the fragmenter and cell accelerator voltage while conducting mass spectrometry can result in artifactual breakdown to the 468-amu ceftazidime. (B) Ceftazidime (2 mM) hydrolyzed by 100 mM NaOH revealed one major peak of 486 amu, corresponding to the hydrolyzed ceftazidime with breakdown of the R2 side chain to the exomethylene. (C) Ceftazidime (2 mM) hydrolyzed by 100 μM PenI identified two peaks of 474 and 486 amu, which correspond to hydrolyzed ceftazidime with complete loss of the R2 side chain and hydrolyzed ceftazidime with breakdown of the R2 side chain to the exomethylene, respectively. (D) Ceftazidime (2 mM) hydrolyzed by 100 μM the C69F variant identified two peaks of 474 and 486 amu, which correspond to hydrolyzed ceftazidime with complete loss of the R2 side chain and hydrolyzed ceftazidime with breakdown of the R2 side chain to the exomethylene, respectively. The full-size hydrolyzed ceftazidime of 565 amu was not observed under any tested condition.
Pre-steady-state kinetics was next used to further explore this pathway, following the method developed by Citri and colleagues (34). The burst amplitude for 10 μM the C69F variant with 25 μM ceftazidime was approximately 1.5 μM, nearly 7-fold lower than the enzyme concentration used in the assay, and represents a nonstoichiometric burst (Fig. 7A) (38). Partitioning between the parallel reactions of a branched mechanism may be the cause of this nonstoichiometric burst. Preincubating 10 μM the C69F variant with the anti-PenI polyclonal antibody for 10 min increased the burst amplitude to 17 μM when reacted with 25 μM ceftazidime (Fig. 7B). These results are consistent with the work conducted by Citri and colleagues with the Bacillus cereus class A β-lactamase, showing that adding an antibody stabilizes a particular conformation of a specific β-lactamase, resulting in increased hydrolysis. These data also support our contention that there is a conformational change in the β-lactamase (34).
FIG 7.

(A) Burst in ceftazidime (25 μM) hydrolysis observed with 10 μM the C69F variant. Extrapolation (green line) of the steady-state velocity to the y axis revealed a burst amplitude of ∼1.5 μM. (B) Burst in ceftazidime (25 μM) hydrolysis observed with 10 μM the C69F variant that was preincubated with anti-PenI polyclonal antibody for 10 min. Extrapolation (green line) of the steady-state velocity to the y axis revealed a burst amplitude of ∼17 μM.
Ceftazidime steady-state kinetic parameters were unable to be accurately measured due to the spectroscopic properties of ceftazidime and poor hydrolytic activity of the enzymes; thus, the ability of ceftazidime to inhibit PenI and the C69F variant was assessed using a competition experiment. Progress curves of nitrocefin inhibition by ceftazidime revealed that 320 mM ceftazidime was required to almost fully inhibit PenI versus only 1 mM ceftazidime for the C69F variant (Fig. 4C and D). Subsequently, the kobs values were determined for each progress curve, and when the kobs values were plotted versus the concentration of ceftazidime, two different graphs were obtained. A saturation curve for PenI was observed and fitted to obtain a KI observed value of 23 ± 3 mM and a kinact value of 0.026 s−1 (Fig. 4E). A linear plot was detected with the C69F variant corresponding to a slope or k2/K observed value of 102 ± 10 M−1 s−1 (Fig. 4F). The linearity of the C69F variant suggests rapid acylation, as well as better affinity for ceftazidime than PenI. With SHV-1, the M69F variant possessed only a 3-fold-lower Km value than wild-type SHV-1 (16). The different kinetic properties of the C69F variant of PenI and the M69F variant of SHV-1 were correlated with the higher ceftazidime MIC for the C69F variant.
Protein mass spectrometry.
To further investigate our kinetic observations, mass spectrometry of purified PenI missing its signal peptide sequence (theoretical mass = 28,350 amu) and the C69F variant missing its signal peptide sequence (theoretical mass = 28,394 amu) with ceftazidime (theoretical mass = 546 amu), using a 1:1 ratio of ceftazidime to β-lactamase, was conducted. Observable adducts were not detected between PenI and ceftazidime over a 15-s to 15-min time course. After longer times, up to 1 h, an acyl-enzyme adduct of PenI and ceftazidime was also not detected under these conditions (Fig. 8A). However, at higher ratios of ceftazidime to PenI—a ratio of 100:1—a small population of acyl-enzyme (28,815 ± 3 amu) could be observed after 1 min incubation (Fig. 8A). The mass of the adduct corresponded to the exomethylene form of ceftazidime (measured mass = 467 ± 3 amu) (Fig. 5C). Conversely, the C69F variant was found to rapidly acylate ceftazidime (28,857 ± 3 amu). Deacylation (28,390 ± 3 amu) occurred within 15 min; only the 467 ± 3 amu exomethylene form of ceftazidime adduct was observed with the variant (Fig. 8B). These data are in agreement with the small-molecule mass spectrometry.
FIG 8.
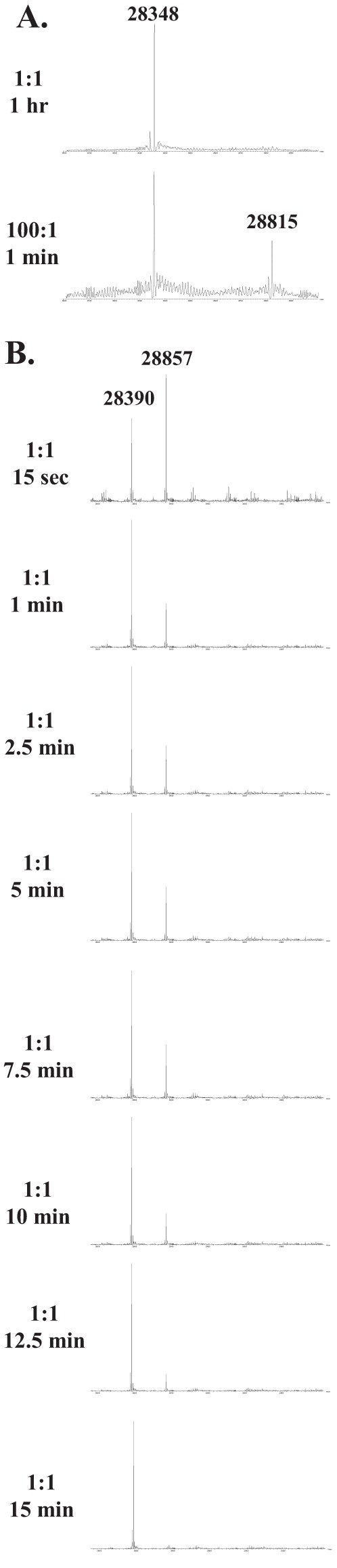
Mass spectrometry of PenI and the C69F variant with ceftazidime (error, ±3 amu). (A) Ceftazidime (10 μM) and PenI (10 μM) at a 1:1 ratio revealed only apo-enzyme (28,348 amu) after 1 h. However, at a 100:1 ratio of ceftazidime to PenI, the acyl-enzyme (28,348 amu + 467 amu = 28,815 amu) was observed within 1 min. (B) Ceftazidime (10 μM) and the C69F variant (10 μM) at a 1:1 ratio over a time course of 15 s to 15 min revealed acyl-enzyme formation (28,857 amu) within 15 s and returned to complete apo-enzyme (28,390 amu) by 15 min.
Molecular modeling.
X-ray crystallography of class A β-lactamases with substitutions at position 69 was previously unrevealing in fully elucidating how amino acid changes affected clavulanic acid binding (39). Earlier studies showed that structural changes were not readily observed between the TEM-1 β-lactamase and its M69L variant despite the presence of a resistance phenotype when expressed in E. coli. Only upon molecular modeling and MDS were any changes between the two β-lactamases detected. Thus, to assess the contributions of the substitutions at position C69 to the structure of the β-lactamase, molecular representations of PenI and the C69F variant were generated and MDS was conducted.
The apo forms of the β-lactamases revealed that the geometry of the oxyanion hole was different in the C69F variant, as the oxyanion hole water molecule was deeper in the pocket by ∼1 Å than in PenI (Fig. 9A). The deeper oxyanion hole in the C69F variant may serve to draw the carbonyl of ceftazidime into the active site more readily than PenI, explaining the lower values seen with competition experiments. Next, the exomethylene form of ceftazidime was docked into the active sites of PenI and the C69F variant. An acyl bond was formed with the deprotonated oxygen of the hydroxyl side chain of S70 and the ceftazidime carbonyl. Two important differences were evident between the two acyl-enzyme models. First, the R1 side chain of ceftazidime possessed two different orientations, similar to the structure of cefotaxime in TOHO-1 (PDB 2ZQA); thus, different orientations of the R1 side chain are conceivable. Second, in the C69F variant model, ceftazidime formed 13 possible hydrogen-bonding interactions with active-site residues, mainly (8 interactions) with the B3 β-strand; a unique interaction with D240 not seen with PenI was also observed (Fig. 9B). The C-4 carboxylate formed typically observed interactions with the B3 β-strand (e.g., hydrogen bonding with amino acids at positions 234, 235, and 237). Conversely, ceftazidime formed fewer potential hydrogen bonds with active-site residues of PenI, and the β-lactam C-4 carboxylate made uncommon interactions with the left side of the active site (with residues S130 and Y105) (Fig. 9C). Given the difference between PenI and the C69F variant, the modeled form of the C69F variant may be the conformation that hydrolyzes ceftazidime rapidly.
FIG 9.

(A) Overlay of the PenI (purple) and C69F (orange) apo-enzyme models showing that the oxyanion hole water position was ∼1 Å deeper in the electrophilic center in the C69F variant than with PenI. (B) Molecular modeling of the C69F variant acyl-enzyme with ceftazidime showing the possibility of up to 13 hydrogen-bonding partners, including residue D240. (C) Molecular modeling of the PenI acyl-enzyme with ceftazidime in the active site revealed a limited number of hydrogen-bonding interactions.
Mechanism for ceftazidime resistance.
Our data led us to propose a new pathway explaining enhanced ceftazidime kinetics in the C69F variant compared to PenI (Fig. 10). Using previously published approaches as a guide (34, 36), we showed that the C69F variant hydrolyzes ceftazidime along a branched kinetic pathway compared to wild-type PenI. We found that the products formed upon ceftazidime hydrolysis by PenI and the C69F variant lacked the R2 side chain. In addition, pre-steady-state kinetics suggested that the C69F variant may be present in at least two conformations; β-lactamases possessing different conformations have previously been observed (38, 40). We propose that the isoform of the C69F variant that hydrolyzes ceftazidime rapidly was stabilized by preincubation with an anti-PenI antibody. Almost-complete hydrolysis of 25 μM ceftazidime was observed within 100 s compared to 1,500 s without the antibody preincubation. In the cell, the C69F variant may be stabilized in the “rapid form” in the periplasmic space, resulting in the high MIC of 64 mg/liter. Conversely, based upon (i) the observation that PenI possesses a KI observed value of 23 mM and (ii) our inability to capture the PenI-ceftazidime acyl-enzyme complex on mass spectrometry unless the concentration of ceftazidime was raised, we propose that PenI possesses poor affinity for ceftazidime compared to the C69F variant.
FIG 10.

Proposed kinetic mechanism for different ceftazidime kinetics of the PenI (purple) and C69F variant (orange) β-lactamases. The major difference is that the C69F variant possesses two enzyme conformations, resulting in a branched pathway toward ceftazidime hydrolysis compared to PenI.
What are the clinical and mechanistic implications of these findings? Our investigations reveal that the C69F variant of PenI is a unique β-lactamase. The wild-type enzyme, PenI, interacts with ceftazidime so poorly that ceftazidime is able to reach the PBP target, thus killing the bacterial cell. In contrast, when expressed in E. coli, the C69F variant confers a high ceftazidime MIC (64 mg/liter). To our surprise, we next discovered that a branched pathway exists and that this variant β-lactamase can be present in two conformations. Interestingly, one conformation of the C69F variant rapidly hydrolyzes ceftazidime and may be the dominant form expressed in the cell, resulting in high MICs; the other conformation is catalytically impaired. The presence of ceftazidime in the medium during growth also increases (e.g., stabilizes) the C69F β-lactamase present in the periplasm (41). Underexpressed β-lactamases and those with low hydrolytic activity may be overlooked in clinical isolates using nonphenotypic diagnostic procedures (e.g., mass spectrometry) for detection of antibiotic resistance mechanisms (42); this could result in poor therapeutic choices and treatment failure.
From a mechanistic standpoint, we posit that amino acid changes that affect the tertiary structure by permitting conformational changes that enhance hydrolytic activity can have a significant impact on substrate turnover. In the case of the C69F variant of PenI, changes in the oxyanion hole result in alterations in hydrogen bond formation that permit catalysis of a substrate not normally regarded as “favorable.” Considerations such as this have significant impact on new β-lactam and β-lactamase inhibitor design. Showing that conformational changes result in higher MICs also adds a new level of complexity to understanding β-lactamase evolution. Conversely, finding ways to inhibit conformational changes in enzymes with enhanced kinetic properties can lead to new insights into allosteric inhibitor design (43). Such unanticipated findings can open “new doors” in the design of β-lactamase inhibitors.
ACKNOWLEDGMENTS
We thank Michael E. Harris for helpful discussions of kinetics and the reviewers of the manuscript for their helpful suggestions.
Research reported in this publication was supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs to K.M.P.-W. and R.A.B., the Veterans Affairs Career Development Program to K.M.P.-W., Veterans Affairs Merit Review Program Award 1I01BX002872 to K.M.P.-W., Veterans Affairs Merit Review Program Award 1I01BX001974 to R.A.B., Geriatric Research Education and Clinical Center grant VISN 10 to R.A.B., and National Institute of Allergy and Infectious Diseases of the National Institutes of Health award numbers R01 AI100560 and R01 AI063517 to R.A.B. and U54 AI03657 to H.P.S. M.L.W. was supported by Medical Scientist Training Program Training Grant Case Western Reserve University-T32 GM07250.
Funding Statement
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Currie BJ. 2015. Melioidosis: evolving concepts in epidemiology, pathogenesis, and treatment. Semin Respir Crit Care Med 36:111–125. doi: 10.1055/s-0034-1398389. [DOI] [PubMed] [Google Scholar]
- 2.Samuel M, Ti TY. 2002. Interventions for treating melioidosis. Cochrane Database Syst Rev 4:CD001263. [DOI] [PubMed] [Google Scholar]
- 3.Cheng AC, Limmathurotsakul D, Chierakul W, Getchalarat N, Wuthiekanun V, Stephens DP, Day NP, White NJ, Chaowagul W, Currie BJ, Peacock SJ. 2007. A randomized controlled trial of granulocyte colony-stimulating factor for the treatment of severe sepsis due to melioidosis in Thailand. Clin Infect Dis 45:308–314. doi: 10.1086/519261. [DOI] [PubMed] [Google Scholar]
- 4.Godfrey AJ, Wong S, Dance DA, Chaowagul W, Bryan LE. 1991. Pseudomonas pseudomallei resistance to β-lactam antibiotics due to alterations in the chromosomally encoded β-lactamase. Antimicrob Agents Chemother 35:1635–1640. doi: 10.1128/AAC.35.8.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tribuddharat C, Moore RA, Baker P, Woods DE. 2003. Burkholderia pseudomallei class A β-lactamase mutations that confer selective resistance against ceftazidime or clavulanic acid inhibition. Antimicrob Agents Chemother 47:2082–2087. doi: 10.1128/AAC.47.7.2082-2087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sam IC, See KH, Puthucheary SD. 2009. Variations in ceftazidime and amoxicillin-clavulanate susceptibilities within a clonal infection of Burkholderia pseudomallei. J Clin Microbiol 47:1556–1558. doi: 10.1128/JCM.01657-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rholl DA, Papp-Wallace KM, Tomaras AP, Vasil ML, Bonomo RA, Schweizer HP. 2011. Molecular investigations of PenA-mediated β-lactam resistance in Burkholderia pseudomallei. Front Microbiol 2:139. doi: 10.3389/fmicb.2011.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chantratita N, Rholl DA, Sim B, Wuthiekanun V, Limmathurotsakul D, Amornchai P, Thanwisai A, Chua HH, Ooi WF, Holden MT, Day NP, Tan P, Schweizer HP, Peacock SJ. 2011. Antimicrobial resistance to ceftazidime involving loss of penicillin-binding protein 3 in Burkholderia pseudomallei. Proc Natl Acad Sci U S A 108:17165–17170. doi: 10.1073/pnas.1111020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niumsup P, Wuthiekanun V. 2002. Cloning of the class D β-lactamase gene from Burkholderia pseudomallei and studies on its expression in ceftazidime-susceptible and -resistant strains. J Antimicrob Chemother 50:445–455. doi: 10.1093/jac/dkf165. [DOI] [PubMed] [Google Scholar]
- 10.Sarovich DS, Price EP, Von Schulze AT, Cook JM, Mayo M, Watson LM, Richardson L, Seymour ML, Tuanyok A, Engelthaler DM, Pearson T, Peacock SJ, Currie BJ, Keim P, Wagner DM. 2012. Characterization of ceftazidime resistance mechanisms in clinical isolates of Burkholderia pseudomallei from Australia. PLoS One 7:e30789. doi: 10.1371/journal.pone.0030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarovich DS, Price EP, Limmathurotsakul D, Cook JM, Von Schulze AT, Wolken SR, Keim P, Peacock SJ, Pearson T. 2012. Development of ceftazidime resistance in an acute Burkholderia pseudomallei infection. Infect Drug Resist 5:129–132. doi: 10.2147/IDR.S35529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schweizer HP. 2012. Mechanisms of antibiotic resistance in Burkholderia pseudomallei: implications for treatment of melioidosis. Future Microbiol 7:1389–1399. doi: 10.2217/fmb.12.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poirel L, Rodriguez-Martinez JM, Plesiat P, Nordmann P. 2009. Naturally occurring class A β-lactamases from the Burkholderia cepacia complex. Antimicrob Agents Chemother 53:876–882. doi: 10.1128/AAC.00946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papp-Wallace KM, Taracila MA, Gatta JA, Ohuchi N, Bonomo RA, Nukaga M. 2013. Insights into β-lactamases from Burkholderia species, two phylogenetically related yet distinct resistance determinants. J Biol Chem 288:19090–19102. doi: 10.1074/jbc.M113.458315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung TK, Ho PL, Woo PC, Yuen KY, Chau PY. 2002. Cloning and expression of class A β-lactamase gene blaA(BPS) in Burkholderia pseudomallei. Antimicrob Agents Chemother 46:1132–1135. doi: 10.1128/AAC.46.4.1132-1135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helfand MS, Hujer AM, Sonnichsen FD, Bonomo RA. 2002. Unexpected advanced generation cephalosporinase activity of the M69F variant of SHV β-lactamase. J Biol Chem 277:47719–47723. doi: 10.1074/jbc.M207271200. [DOI] [PubMed] [Google Scholar]
- 17.Levitt PS, Papp-Wallace KM, Taracila MA, Hujer AM, Winkler ML, Smith KM, Xu Y, Harris ME, Bonomo RA. 2012. Exploring the role of a conserved class A residue in the Ω-Loop of KPC-2 β-lactamase: a mechanism for ceftazidime hydrolysis. J Biol Chem 287:31783–31793. doi: 10.1074/jbc.M112.348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celenza G, Luzi C, Aschi M, Segatore B, Setacci D, Pellegrini C, Forcella C, Amicosante G, Perilli M. 2008. Natural D240G Toho-1 mutant conferring resistance to ceftazidime: biochemical characterization of CTX-M-43. J Antimicrob Chemother 62:991–997. doi: 10.1093/jac/dkn339. [DOI] [PubMed] [Google Scholar]
- 19.Egesborg P, Carlettini H, Volpato JP, Doucet N. 2015. Combinatorial active-site variants confer sustained clavulanate resistance in BlaC β-lactamase from Mycobacterium tuberculosis. Protein Sci 24:534–544. doi: 10.1002/pro.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Minasov G, Shoichet BK. 2002. The structural bases of antibiotic resistance in the clinically derived mutant β-lactamases TEM-30, TEM-32, and TEM-34. J Biol Chem 277:32149–32156. doi: 10.1074/jbc.M204212200. [DOI] [PubMed] [Google Scholar]
- 21.Madec S, Blin C, Krishnamoorthy R, Picard B, Chaibi el B, Fouchereau-Peron M, Labia R. 2002. Substitution of Met-69 by Ala or Gly in TEM-1 β-lactamase confer an increased susceptibility to clavulanic acid and other inhibitors. FEMS Microbiol Lett 211:13–16. doi: 10.1111/j.1574-6968.2002.tb11196.x. [DOI] [PubMed] [Google Scholar]
- 22.Mammeri H, Gilly L, Laurans G, Vedel G, Eb F, Paul G. 2001. Catalytic and structural properties of IRT-21 β-lactamase (TEM-77) from a co-amoxiclav-resistant Proteus mirabilis isolate. FEMS Microbiol Lett 205:185–189. doi: 10.1111/j.1574-6968.2001.tb10945.x. [DOI] [PubMed] [Google Scholar]
- 23.Lin S, Thomas M, Shlaes DM, Rudin SD, Knox JR, Anderson V, Bonomo RA. 1998. Kinetic analysis of an inhibitor-resistant variant of the OHIO-1 β-lactamase, an SHV-family class A enzyme. Biochem J 333:395–400. doi: 10.1042/bj3330395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaibi EB, Peduzzi J, Farzaneh S, Barthelemy M, Sirot D, Labia R. 1998. Clinical inhibitor-resistant mutants of the β-lactamase TEM-1 at amino-acid position 69. Kinetic analysis and molecular modelling. Biochim Biophys Acta 1382:38–46. [DOI] [PubMed] [Google Scholar]
- 25.Bonomo RA, Dawes CG, Knox JR, Shlaes DM. 1995. Complementary roles of mutations at positions 69 and 242 in a class A β-lactamase. Biochim Biophys Acta 1247:113–120. doi: 10.1016/0167-4838(94)00187-L. [DOI] [PubMed] [Google Scholar]
- 26.Delaire M, Labia R, Samama JP, Masson JM. 1992. Site-directed mutagenesis at the active site of Escherichia coli TEM-1 β-lactamase. Suicide inhibitor-resistant mutants reveal the role of arginine 244 and methionine 69 in catalysis. J Biol Chem 267:20600–20606. [PubMed] [Google Scholar]
- 27.Totir MA, Padayatti PS, Helfand MS, Carey MP, Bonomo RA, Carey PR, van den Akker F. 2006. Effect of the inhibitor-resistant M69V substitution on the structures and populations of trans-enamine β-lactamase intermediates. Biochemistry 45:11895–11904. doi: 10.1021/bi060990m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clinical and Laboratory Standards Institute. 2015. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 10th ed, 2015 Approved standard M7-A10. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 29.Papp-Wallace KM, Taracila MA, Smith KM, Xu Y, Bonomo RA. 2012. Understanding the molecular determinants of substrate and inhibitor specificities in the carbapenemase KPC-2: exploring the roles of Arg220 and Glu276. Antimicrob Agents Chemother 56:4428–4438. doi: 10.1128/AAC.05769-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heidari Torkabadi H, Bethel CR, Papp-Wallace KM, de Boer PA, Bonomo RA, Carey PR. 2014. Following drug uptake and reactions inside Escherichia coli cells by Raman microspectroscopy. Biochemistry 53:4113–4121. doi: 10.1021/bi500529c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkler ML, Rodkey EA, Taracila MA, Drawz SM, Bethel CR, Papp-Wallace KM, Smith KM, Xu Y, Dwulit-Smith JR, Romagnoli C, Caselli E, Prati F, van den Akker F, Bonomo RA. 2013. Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J Med Chem 56:1084–1097. doi: 10.1021/jm301490d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, Bonomo RA. 2010. Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-lactamase. Antimicrob Agents Chemother 54:890–897. doi: 10.1128/AAC.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-beta-lactam beta-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Citri N, Samuni A, Zyk N. 1976. Acquisition of substrate-specific parameters during the catalytic reaction of penicillinase. Proc Natl Acad Sci U S A 73:1048–1052. doi: 10.1073/pnas.73.4.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papp-Wallace KM, Taracila M, Wallace CJ, Hujer KM, Bethel CR, Hornick JM, Bonomo RA. 2010. Elucidating the role of Trp105 in the KPC-2 β-lactamase. Protein Sci 19:1714–1727. doi: 10.1002/pro.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faraci WS, Pratt RF. 1985. Mechanism of inhibition of the PC1 β-lactamase of Staphylococcus aureus by cephalosporins: importance of the 3′-leaving group. Biochemistry 24:903–910. doi: 10.1021/bi00325a014. [DOI] [PubMed] [Google Scholar]
- 37.Vakulenko SB, Taibi-Tronche P, Toth M, Massova I, Lerner SA, Mobashery S. 1999. Effects on substrate profile by mutational substitutions at positions 164 and 179 of the class A TEM(pUC19) β-lactamase from Escherichia coli. J Biol Chem 274:23052–23060. doi: 10.1074/jbc.274.33.23052. [DOI] [PubMed] [Google Scholar]
- 38.Page MG. 1993. The kinetics of non-stoichiometric bursts of β-lactam hydrolysis catalysed by class C β-lactamases. Biochem J 295:295–304. doi: 10.1042/bj2950295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meroueh SO, Roblin P, Golemi D, Maveyraud L, Vakulenko SB, Zhang Y, Samama JP, Mobashery S. 2002. Molecular dynamics at the root of expansion of function in the M69L inhibitor-resistant TEM β-lactamase from Escherichia coli. J Am Chem Soc 124:9422–9430. doi: 10.1021/ja026547q. [DOI] [PubMed] [Google Scholar]
- 40.Waley SG. 1991. The kinetics of substrate-induced inactivation. Biochem J 279:87–94. doi: 10.1042/bj2790087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson JW, Fisher JF, Mobashery S. 2013. Bacterial cell-wall recycling. Ann N Y Acad Sci 1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lupo A, Papp-Wallace KM, Sendi P, Bonomo RA, Endimiani A. 2013. Non-phenotypic tests to detect and characterize antibiotic resistance mechanisms in Enterobacteriaceae. Diagn Microbiol Infect Dis 77:179–194. doi: 10.1016/j.diagmicrobio.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowman GR, Bolin ER, Hart KM, Maguire BC, Marqusee S. 2015. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc Natl Acad Sci U S A 112:2734–2739. doi: 10.1073/pnas.1417811112. [DOI] [PMC free article] [PubMed] [Google Scholar]