

GAN patient cells have abnormal aggregates of vimentin intermediate filaments, to which mitochondria appear to be tethered. Motility of mitochondria, but not lysosomes, is inhibited in these cells. Transfection with wild-type gigaxonin (the protein mutated in this disease) disperses these aggregates and bundles, and mitochondrial motility returns to normal.
Abstract
Giant axonal neuropathy (GAN) is a rare disease caused by mutations in the GAN gene, which encodes gigaxonin, an E3 ligase adapter that targets intermediate filament (IF) proteins for degradation in numerous cell types, including neurons and fibroblasts. The cellular hallmark of GAN pathology is the formation of large aggregates and bundles of IFs. In this study, we show that both the distribution and motility of mitochondria are altered in GAN fibroblasts and this is attributable to their association with vimentin IF aggregates and bundles. Transient expression of wild-type gigaxonin in GAN fibroblasts reduces the number of IF aggregates and bundles, restoring mitochondrial motility. Conversely, silencing the expression of gigaxonin in control fibroblasts leads to changes in IF organization similar to that of GAN patient fibroblasts and a coincident loss of mitochondrial motility. The inhibition of mitochondrial motility in GAN fibroblasts is not due to a global inhibition of organelle translocation, as lysosome motility is normal. Our findings demonstrate that it is the pathological changes in IF organization that cause the loss of mitochondrial motility.
INTRODUCTION
Giant axonal neuropathy (GAN) is typically diagnosed as a neurodegenerative disorder because its main phenotype involves progressive deterioration of the peripheral and central nervous systems (Asbury et al., 1972; Berg et al., 1972; Peiffer et al., 1977; Johnson-Kerner et al., 2014). The pathological hallmark of GAN in the nervous system is the formation of large aggregates of neuronal intermediate filaments (IFs) such as the type IV neurofilament triplet proteins, which cause the swelling and “giant axons” for which the disease is named (Asbury et al., 1972). These aggregates are not restricted to neurons, as similar IF aggregates have been reported for type III IFs such as vimentin in fibroblasts, Schwann cells, and endothelial cells and glial fibrillary acid protein in astrocytes (Peiffer et al., 1977; Pena, 1981; Bomont et al., 2000). In addition, GAN patients frequently have kinky hair, suggesting that keratins, type I and II IF proteins, are also affected (Bomont et al., 2000). GAN is caused by mutations in the GAN gene encoding gigaxonin, a member of the BTB-Kelch family of E3 ligase adapters known to target proteins for ubiquitination and proteasomal degradation (Bomont et al., 2000). Gigaxonin binds to the neurofilament triplet proteins vimentin, peripherin, and α-internexin via its Kelch domain (Mahammad et al., 2013; Johnson-Kerner et al., 2015b) and targets vimentin, peripherin, and neurofilament light chain (NFL) for degradation by the proteasome (Mahammad et al., 2013). Mutations in gigaxonin therefore cause defects in IF protein turnover, frequently leading to their accumulation into large bundles and aggregates (Mahammad et al., 2013; Johnson-Kerner et al., 2015a).
The dramatic changes in vimentin IF network organization in the fibroblasts obtained from skin biopsies of GAN patients are likely to cause significant alterations in cell physiology, as it is well established that the vimentin cytoskeletal system plays important roles in regulating the shape, motility, and mechanical properties of mesenchymal cells such as fibroblasts (Helmke et al., 2000; Ofek et al., 2009; Guo et al., 2013, 2014; Mendez et al., 2014; Murray et al., 2014; Lowery et al., 2015). With respect to cellular mechanics, recent studies show that vimentin IF networks are major contributors to noncortical cytoplasmic stiffness and represent important factors involved in stabilizing and tethering organelles exposed to random fluctuating forces derived from the activities of molecular motors (Guo et al., 2013, 2014). Furthermore, there is evidence supporting a role for vimentin IF in modulating the distribution of mitochondria (Goldman, 1971; Summerhayes et al., 1983; Tang et al., 2008) and regulating their motile properties by intermittently anchoring them within the cytoplasm (Nekrasova et al., 2011). In light of these findings and the abnormal organization and turnover of vimentin IF in GAN patient fibroblasts, we carried out experiments designed to determine whether there are alterations in the motility and distribution of mitochondria within these cells.
RESULTS
Mitochondria in GAN patient fibroblasts are associated with bundles and aggregates of vimentin IFs
In control human fibroblasts, the vimentin IF network is dispersed throughout much of the cytoplasm of spread cells (Figure 1A). In contrast, GAN patient fibroblasts contain abnormally large aggregates and bundles of vimentin IF (Figure 1, B–D; Pena, 1981; Mahammad et al., 2013). Typically ∼40% of the GAN patient cells used in this study (GAN-NHS-NS-08; see Materials and Methods) have ovoid aggregates frequently located near the nucleus, and >90% of cells contain numerous thick bundles (n = 200; Figure 1, B–D). Note that the formation of vimentin IF bundles and aggregates is not restricted to the mutations found in the GAN cells reported in this study (EXON1c.130C>T and EXON9c.1420G>C; see Materials and Methods) but also occurs in three other GAN mutant fibroblasts, as we previously showed (p.Glu486Lys in Exon 9 and a 57- to 131-kb microdeletion encompassing exons 3–11; IVS6_1g_a and L510X; S52G and C393X; Mahammad et al., 2013).
FIGURE 1:
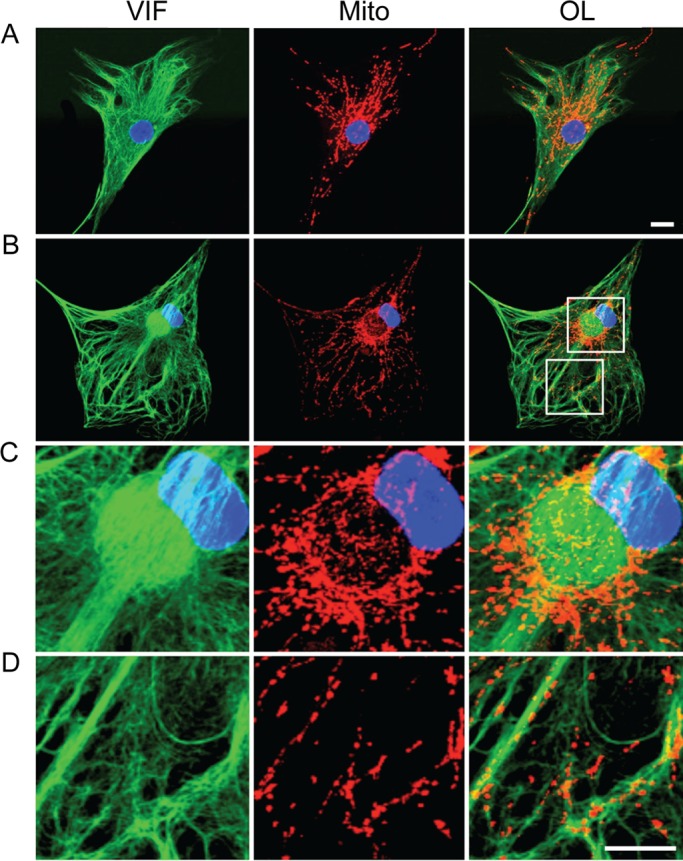
Mitochondria are associated with vimentin IFs. Indirect immunofluorescence with anti-vimentin (VIF), mitochondria stained with MitoTracker Red (Mito), and nucleus stained with Hoechst 33258. (A) Control fibroblasts with typical arrays of vimentin IFs (VIF; green) and containing a normal distribution of mitochondria (Mito; red in the same cell). OL, overlay; blue, nucleus. (B) A GAN fibroblast containing bundles and an ovoid aggregate of vimentin IFs (VIF, green) next to the nucleus (blue); mitochondria in the same cell (Mito, red). Boxed regions in B (OL) are shown at higher magnification to reveal details of mitochondrial association with the aggregate (C) and bundles (D) of vimentin IFs. There are also some mitochondria associated with IFs located between the thicker bundles (D). Confocal images. Scale bars, 10 μm.
Because vimentin IFs interact with mitochondria in normal fibroblasts (Summerhayes et al., 1983; Tang et al., 2008; Nekrasova et al., 2011), we determined whether these organelles are also associated with the vimentin IF aggregates and bundles in the GAN cells. In normal fibroblasts, mitochondria are dispersed throughout the cytoplasm, and their distribution frequently appears similar to that of the vimentin IF network (Summerhayes et al., 1983; Figure 1A). In GAN fibroblasts, mitochondria retain their association with the vimentin IFs comprising the aggregates and bundles (Figure 1, B–D). Confocal images of aggregates show that most mitochondria are associated with the surface of these structures, with some distributed more internally (Supplemental Figure S1). The higher resolution afforded by three-dimensional structured illumination microscopy (3D-SIM) further revealed that IFs are organized into cage-like structures at the surfaces of aggregates with closely associated mitochondria (Figure 2A and Supplemental Figures S1 and S2). Many of the mitochondria not associated with the ovoid aggregates are closely associated with the bundles of vimentin IFs present in the GAN cells (Figures 1D and 2B). The association between IFs and mitochondria has been confirmed in GAN cells by electron microscopic observations of bundles (Figure 2C). Previously, we showed electron microscopy that mitochondria were also closely associated with the surface and in some cases the interior of aggregates of vimentin IFs (Mahammad et al., 2013). Note that in control fibroblasts, mitochondria are also frequently seen in close proximity to vimentin IFs (Supplemental Figure S2).
FIGURE 2:
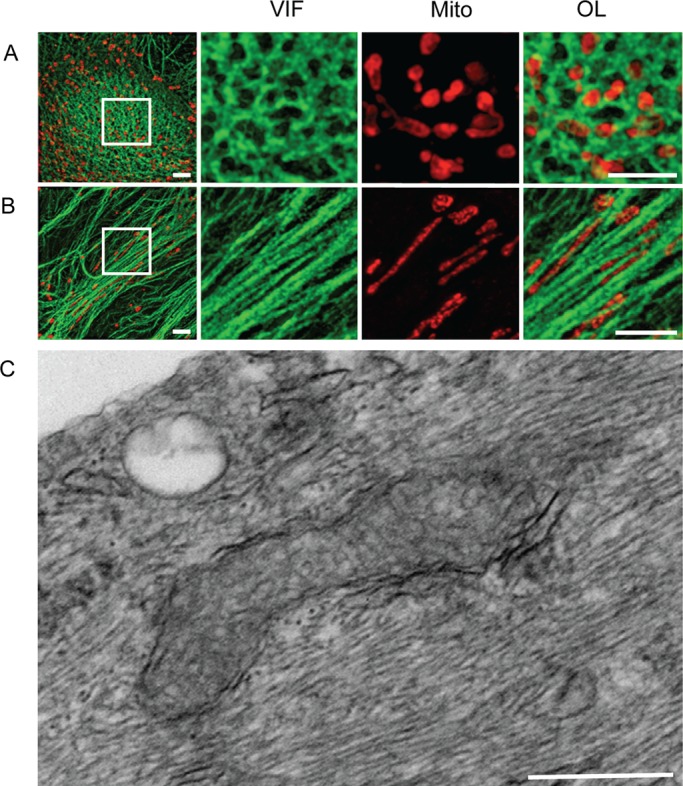
Superresolution and electron microscopy showing details of the association between mitochondria and vimentin IF aggregates and bundles in GAN fibroblasts. (A) The surface of a vimentin IF aggregate. The boxed area shown at higher magnification reveals a cage-like structure of vimentin IFs (VIF; green) forming meshwork mitochondria (Mito; red). OL, overlay. (B) Vimentin IF bundles; the boxed area shown at higher magnification on the right reveals bundles (VIF; green) and closely associated mitochondria (Mito; red). Images in A and B acquired by 3D-SIM. Scale bar, 500 nm. (C) Electron micrograph showing that many 10-nm-diameter vimentin IFs tightly surround mitochondria. Scale bar, 500 nm.
The motility of mitochondria is altered in GAN fibroblasts
The altered distribution of mitochondria and their close association with vimentin IF aggregates and bundles suggest that their motility might be affected. Therefore we compared the motile properties of mitochondria in GAN and control fibroblasts by time-lapse imaging after staining with MitoTracker Red CMXROS (MitoTracker Red). As expected, numerous mitochondria display anterograde and retrograde translocations as they move from one cytoplasmic region to another in control fibroblasts (Supplemental Movie S1). In contrast, individual mitochondria in GAN fibroblasts exhibit only Brownian-like motions, with no detectable translocations (Supplemental Movie S2). We carried out comparative quantitative analyses of the trajectories of the centroid positions of individual mitochondria. Specifically, we determined the motile properties of 62 mitochondria in time-lapse sequences of five different control fibroblasts and 90 mitochondria in five different GAN fibroblasts (Figure 3; see Materials and Methods). The data derived from tracking the trajectories of individual mitochondria show that in control cells, they display typical saltatory motions, frequently moving long distances in the same, a reverse, or a new direction (Figure 3A). In contrast, the mitochondria in GAN fibroblasts exhibit only short excursions (Figure 3B). These differences are further emphasized by measurements of the net displacements of mitochondria in both cell types, which demonstrate >80% reduction in GAN compared with control fibroblasts (Figure 3C). The results in Figure 3D show that mitochondria are anchored in a manner that restricts their movements such that they cannot engage in long translocations but instead appear to move as if extending from an anchorage site. The results show that the mean total distance traveled by mitochondria in 15 min is significantly less in GAN fibroblasts compared with control fibroblasts (22.9 ± 1 vs. 45.1 ± 6.1 μm, respectively; Figure 3D). Further analyses of the live-cell data in control cells show that mitochondria exhibit high fluctuations in instantaneous velocity, reaching a maximum of 2 μm/s (Figure 3E), whereas the instantaneous velocities in GAN cells are very low and relatively constant (Figure 3F). Similar associations between immotile mitochondria and vimentin IF bundles and aggregates have been detected in two other GAN fibroblasts (one containing mutations EXON4(724C>T) and R242X and the second containing the mutations IVS6_1g_a and L510X; unpublished data).
FIGURE 3:

The motility of mitochondria is inhibited in GAN patient fibroblasts. Trajectories of individual mitochondria in control (A) and GAN patient (B) fibroblasts. (C) Box plot of net displacement of mitochondria in control and GAN patient fibroblasts. The box represents the 25th and 75th percentiles, the median is denoted by the horizontal line, the mean is indicated by a small dot, and whiskers indicate SD. (D) Bar graph shows the differences in the total distance traveled by mitochondria in control and GAN patient cells (p < 0.001). (E, F) Instantaneous velocities of individual mitochondria (different colors) were determined in control and GAN patient cells. A total of 90 mitochondria in GAN fibroblasts and 62 mitochondria in control cells were analyzed as described in Materials and Methods.
Another use of MitoTracker Red is to assess the membrane potential of mitochondria, a parameter that has been shown to be affected by vimentin IFs (Chernoivanenko et al., 2015). Therefore we determined the mitochondrial membrane potential in control and GAN fibroblasts stained with MitoTracker Red for 30 min, followed by fixation and quantitative fluorescence intensity measurements (see Materials and Methods). The results demonstrate that there is no significant difference in membrane potential between normal and GAN fibroblasts bearing several different gigaxonin mutations (Supplemental Figure S3).
Gigaxonin expression restores the motility of mitochondria in GAN fibroblasts
We previously demonstrated that the expression of wild-type gigaxonin in patient fibroblasts causes the loss of IF aggregates and bundles as vimentin is degraded via proteasomes (Mahammad et al., 2013). Therefore we attempted to rescue the defects in the motile properties of mitochondria by transiently expressing wild-type gigaxonin fused to blue fluorescent protein (BFP-gigaxonin) in GAN fibroblasts. The BFP tag was introduced so that cells expressing gigaxonin could be identified. After 24 h, vimentin IF aggregates and bundles persist in some cells and are frequently associated with BFP-gigaxonin (Figure 4A). In other cells, BFP-gigaxonin is more dispersed, and this coincides with the appearance of smaller vimentin IF aggregates, a reduction in bundles, and a much more dispersed IF network (Figure 4, B and C). We carried out live-cell analyses of the motility of mitochondria in GAN fibroblasts containing the more dispersed pattern of BFP-gigaxonin in transfected cells stained with MitoTracker Red. The results show that BFP-gigaxonin expression restores mitochondrial motility in these GAN cells with respect to net displacements and distance traveled (Figure 4, D and E).
FIGURE 4:

Expression of gigaxonin rescues the motile properties of mitochondria in GAN patient fibroblasts. (A) BFP-gigaxonin (BFP-Gig; white) tends to concentrate in vimentin IF aggregates and bundles (green) at 24 h after transient transfection and processing for immunofluorescence with anti-vimentin. (B) Another cell (asterisk) in the same preparation containing a smaller aggregate and few if any vimentin IF bundles. Note that only one cell in this field was transfected with BFP-gigaxonin (asterisk), which appears more dispersed in the cytoplasm. There is a small, less intensely stained vimentin IF aggregate (VIF; green) and no obvious vimentin IF bundles similar to those seen in the surrounding, nontransfected cells. (C) Higher-magnification view of the same cells; in the one cell expressing BFP-gigaxonin (asterisk), the brightest BFP fluorescence appears to be associated with the remnant of an aggregate. Scale bar, 10 μm. (D) Control and GAN patient cells overexpressing wild-type gigaxonin. The box represents 25th and 75th percentiles, the median is denoted by the horizontal line, the mean is indicated by small dot, and whiskers indicate SD. (E) Bar graph showing that the total distance traveled by mitochondria in control and gigaxonin-overexpressing GAN patient cells is virtually identical. We analyzed 70 mitochondria (stained with MitoTracker Red) in GAN fibroblasts and 62 in control fibroblasts (see Materials and Methods).
Gigaxonin silencing inhibits the motility of mitochondria in normal fibroblasts
We also determined whether knocking down gigaxonin expression in normal fibroblasts could mimic the organizational changes in vimentin IFs seen in GAN fibroblasts and thereby inhibit mitochondrial motility. To this end, we constructed lentiviral vectors encoding three different short hairpin RNA (shRNA) sequences targeting various regions of human gigaxonin and used to them to transduce normal cells. As a control, we transduced normal cells with scrambled RNA sequences (see Materials and Methods). All three silencing vectors reduce gigaxonin levels by >60% at 72 h after transduction as determined by immunoblotting (Figure 5A). At this time, there are no obvious changes in the organization of vimentin IFs. However, by 18 d, vimentin IF aggregates and bundles can be detected in close association with mitochondria (Figure 5B). These cells were stained with Mito-Tracker Red for live-cell image analysis (Figure 5, C–H; see Materials and Methods). These analyses revealed that the translocation of mitochondria is significantly inhibited, as determined by tracking the trajectories of individual mitochondria (Figure 5, C and D). In addition, the net displacement of mitochondria is reduced by >50% in the silenced cells (Figure 5E), and the total distance traveled decreases significantly (from 32.77 ± 0.69 to 18.44 ± 0.28 μm; p < 0.002; Figure 5F). There is also a dramatic decrease in the instantaneous velocities measured for mitochondria in the silenced cells compared with controls (Figure 5, G and H). Therefore gigaxonin silencing significantly inhibits mitochondrial motility. Note that time-lapse imaging involved the random selection of cells in a silenced population, without knowing whether the selected cells had IF bundles or aggregates.
FIGURE 5:

Silencing gigaxonin in normal fibroblasts impairs mitochondrial motility. (A) Three different shRNAs caused a reduction in the protein levels of gigaxonin in control fibroblasts, as demonstrated by immunoblotting with anti-gigaxonin at 72 h after silencing (see Materials and Methods). (B) Left, silencing gigaxonin for longer periods with shRNA #3 (up to 18 d) causes vimentin IF aggregation (green). Right, higher-magnification view of the same aggregate showing mitochondria (red; nucleus, blue). Scale bars, 10 μm. Trajectories of individual mitochondria in (C) control (expressing scrambled sequence) and (D) gigaxonin-silenced cells. (E) Box plot of net displacement of mitochondria in control and gigaxonin-silenced (Gigaxonin KD) cells. The box represents 25th and 75th percentiles, the median is denoted by the horizontal line, the mean is indicated by a small dot, and whiskers indicate SD. (F) Bar graph shows that there are significant differences in the total distance traveled by mitochondria in control and gigaxonin-silenced cells (p < 0.002). (G, H) Instantaneous velocities of individual mitochondria in control and gigaxonin-silenced cells (each color represents a different mitochondrion). We analyzed 68 mitochondria (stained with MitoTracker Red) in GAN fibroblasts and 62 in control fibroblasts (see Materials and Methods).
Lysosomal motility is normal in GAN patient fibroblasts
Mitochondrial motility is powered by motors moving along microtubule and actin filament tracks (Heggeness et al., 1978; Nangaku et al., 1994; Tanaka et al., 1998; Saxton and Hollenbeck, 2012; Lu et al., 2014). Because the overall patterns of microtubules and actin-containing microfilaments appear normal in GAN fibroblasts, even in regions associated with the IF aggregates (Figure 6, A–H; Mahammad et al., 2013), it is somewhat surprising that mitochondrial motility is so significantly inhibited. Given the normal appearance of microtubules and microfilaments, we determined whether the defective transport of mitochondria reflected more general defects in organelle motility in GAN patient cells. To address this, we assayed the motility of lysosomes, another well-studied membrane-bound organelle known to move along both microtubules and microfilaments (Herman and Albertini, 1984; Matteoni and Kreis, 1987; Soni et al., 2005; Semenova et al., 2008). We carried out comparative live-imaging analyses of GAN patient and control fibroblasts stained with LysoTracker Red. The results showed that the motile properties of lysosomes in these two cell types were indistinguishable with respect to trajectories (Figure 6, I and J), net displacements (Figure 6K), total distances traveled (Figure 6L), and instantaneous velocities (Figure 6, M and N, and Supplemental Movie S3; see Materials and Methods). These results demonstrate that the inhibition of mitochondrial motility in GAN patient fibroblasts is not due to global alterations in microtubule or microfilament networks but instead is a consequence of the altered organization of vimentin IFs.
FIGURE 6:

Lysosome motility is normal in GAN patient fibroblasts. (A, B) Double immunofluorescence showing that the overall organization of microtubules (red) appears normal in a GAN fibroblast (vimentin IFs; green). The juxtanuclear aggregate in A is shown at increased magnification, showing that microtubules are in close proximity to the aggregate (E, F). (C, D) The overall distribution of actin (red) appears normal in a GAN fibroblast (vimentin IFs; green). The juxtanuclear aggregate in C is shown at higher magnification to show that actin filaments and stress fibers are also in close proximity to the aggregate (G, H). Images are through-focus confocal stacks. Scale bars, 10 μm. (I, J) Trajectories of individual lysosomes in control and GAN patient fibroblasts show no detectable differences. (K) Box plot of net displacement of mitochondrial motility in control and GAN cells. The box represents 25th and 75th percentiles, the median is denoted by a horizontal line, the mean is indicated by a small dot, and whiskers indicate SD. (L) Bar graph shows the total distance traveled by lysosomes in control and GAN patient cells. (M, N) Instantaneous velocities of individual lysosomes in control and GAN patient cells (different colors represent individual mitochondria). Lysosomes were stained with LysoTracker Red, and their motility was assayed as described in Materials and Methods.
DISCUSSION
In this study, we report a close relationship between mitochondria and the aggregates and bundles of vimentin IFs present in GAN fibroblasts. Of most importance, this close association is correlated with a dramatic decrease in the motility of mitochondria, which behave as if they are tethered or anchored to the IF bundles and aggregates. The coincident changes in IF organization and the positioning and motility of mitochondria detected in GAN cells are likely to reflect alterations in the normal interactions between IFs and these organelles. In normal fibroblasts, the overall distributions of mitochondria and vimentin IFs are very similar (Summerhayes et al., 1983). This association is retained in cells treated with colchicine, for which the majority of mitochondria remain associated with the perinuclear vimentin IF aggregate that forms in response to the disassembly of microtubules (Goldman, 1971; Summerhayes et al., 1983). In vimentin-null mouse fibroblasts, the rate of mitochondrial movement increases by greater than twice compared with wild-type fibroblasts. When vimentin is expressed in these null cells, there is a return to normal rates of motility (Nekrasova et al., 2011). Moreover, when vimentin IF networks in normal mouse fibroblasts are disrupted by a dominant-negative mutant (Vim1-138), mitochondrial motility increases by ∼70% (Nekrasova et al., 2011). The interactions between IFs and mitochondria appear to involve a subdomain of the non–α−helical N-terminus of vimentin (Nekrasova et al., 2011). Whether these interactions are direct or indirect remains to be determined. With respect to this latter possibility, the 1b isoform of plectin, a vimentin IF–binding protein, has been shown to localize to the outer membrane of mitochondria (Winter et al., 2008). Thus plectin could link mitochondria to vimentin IFs (Nekrasova et al., 2011). Taken together, these results demonstrate that vimentin IFs modulate the normal motility of mitochondria, perhaps by intermittently tethering or anchoring them within different regions of the cytoplasm (Nekrasova et al., 2011; Guo et al., 2013, 2014). The latter possibility is further supported by the results of this study demonstrating that the overexpression of wild-type gigaxonin in GAN cells results in the loss of IF bundles and aggregates and the rescue of mitochondrial motility at rates that can be even faster than in control fibroblasts (Figure 4). Conversely, silencing gigaxonin in normal cells recapitulates the disease phenotype: vimentin IFs form bundles and aggregates, and mitochondrial motility is inhibited.
At the present time, we can only speculate about the relationship between defects in the targeting of vimentin subunits for degradation by gigaxonin and the formation of the IF bundles and aggregates. Perhaps in the absence of functional gigaxonin in GAN fibroblasts there is an accumulation of IF protein. Indeed, we previously showed that in some patient fibroblasts, there is an increase in vimentin compared with controls, but this is not a consistent finding, since in cells from other patients, vimentin levels were similar to those in controls (Mahammad et al., 2013). It is also possible that there is an abnormal accumulation of posttranslationally modified vimentin subunits normally destined for degradation. For example, vimentin is an extensively phosphorylated protein, and its state of phosphorylation regulates both its state of polymerization (Chou et al., 1990; Eriksson et al., 2004; Izawa and Inagaki, 2006; Sihag et al., 2007; Snider and Omary, 2014) and its organization with respect to bundling and the formation of juxtanuclear aggregates (Lamb et al., 1989). It is also possible that one or more of the large number of vimentin phosphorylation sites (Izawa and Inagaki, 2006) provide a signal required for the binding of gigaxonin to vimentin subunits (Mahammad et al., 2013; Johnson-Kerner et al., 2015b), targeting them for ubiquitination and degradation by the proteasome. In support of this possibility, it has been shown that another type III IF protein, desmin, is initially phosphorylated and then ubiquitinated before its degradation in skeletal muscle (Cohen et al., 2012). It is also conceivable that the overall deficit in vimentin subunit turnover in GAN fibroblasts causes the accumulation of other phosphorylation sites that regulate the tethering or anchorage of mitochondria to vimentin IF. Of interest, as indicated earlier, the N-terminal non–α-helical domain (residues 2–95) contains a site that is involved in anchoring mitochondria (Nekrasova et al., 2011) and is the most extensively phosphorylated subdomain of vimentin, containing at least 20 identified phosphorylation sites (Izawa and Inagaki, 2006).
Another possible explanation for defective mitochondrial motility is that the expression of mutant gigaxonin somehow alters the normal interactions between the motors known to be responsible for mitochondrial motility and the microtubule and microfilament tracks along which they move (Schwarz, 2013; Korobova et al., 2014; Lu et al., 2014). However, this does not seem to be the case in this study, since both microtubules and microfilaments appear to be normally organized in GAN cells, and the motility of lysosomes, also known to be transported along microtubules and microfilaments (Soni et al., 2005; Semenova et al., 2008; Granger et al., 2014), is indistinguishable between normal and GAN cells.
Because GAN presents primarily as a neurodegenerative disease, it is possible that similar alterations in mitochondrial motility and distribution in neurons involving neuronal IFs could result in a loss of these organelles in synaptic regions and nodes of Ranvier, where they are required for high levels of ATP production, calcium buffering, and signaling (Sheng and Cai, 2012). Furthermore, defects in the neural IF system disrupt the motility and distribution of mitochondria in motor neurons derived from either NFL-knockout mice or mice expressing Charcot–Marie–Tooth disease mutations in NFL (Tradewell et al., 2009; Gentil et al., 2012). Other studies have shown that excess neurofilament heavy chain (NFH) induces neural IF aggregates surrounded by mitochondria that cannot move into neurites (Straube-West et al., 1996). There is also in vitro evidence supporting interactions between mitochondria and neural IFs isolated from spinal cords that appear to be mediated by the non–α-helical tail domain of NFH (Wagner et al., 2003).
Although our studies focused on fibroblasts obtained from GAN patients, the results reported here provide a framework for understanding the defects in the neurons of patients with GAN and should provide new insights into the ongoing National Institutes of Health gene therapy trial involving the intrathecal injection of a gene transfer vector expressing wild-type gigaxonin (https://clinicaltrials.gov/ct2/show/study/NCT02362438#contacts). This possibility is supported by our finding that expression of wild-type gigaxonin not only eliminates IF aggregates and bundles but also rescues mitochondrial motility. Most important, and from a basic science perspective, our results also lend further credence to the roles of the IF cytoskeleton in regulating the motile properties and positioning of mitochondria within cells.
MATERIALS AND METHODS
Mammalian cell culture
BJ-5ta human foreskin fibroblasts (American Type Culture Collection, Manassas, VA) were grown as described (Mahammad et al., 2013) and used as controls in all of our studies. The GAN patient fibroblasts used for all of the quantitative studies of mitochondrial motility described here bear the mutations EXON1c.130C>T and EXON9c.1420G>C (cell repository maintained at Columbia University, New York, NY). Preliminary observations of mitochondrial motility were also carried out on two other GAN patient fibroblasts with the mutations EXON4 (724C>T) and R242X (Columbia University) and IVS6_1g_a and L510X (obtained from R. Van Coster, Ghent University Hospital, Ghent, Belgium; Mahammad et al., 2013). Cells were maintained in MEM with l-glutamine (Invitrogen, Carlsbad, CA) and supplemented with 10% fetal bovine serum (HyClone, Marlborough, MA), 1% BME vitamin solution (Sigma-Aldrich, St. Louis, MO), 1% nonessential amino acids (Corning, Corning, NY), and 0.5% penicillin/streptomycin. All cell cultures were maintained at 37°C in a humidified CO2 incubator.
Antibodies and reagents
Primary antibodies included chicken anti-vimentin (Covance, Dedham, MA), rabbit anti-gigaxonin, and rat anti–α-tubulin (Sigma-Aldrich). Secondary antibodies included Alexa Fluor 488 and Alexa Fluor 633 (Invitrogen). To visualize mitochondria, we used MitoTracker Red CMXROS (MitoTracker Red; Molecular Probes, Eugene, OR); for lysosomes, LysoTracker Red DND-99 (Molecular Probes); and for actin, phalloidin–Alexa 568 (Invitrogen); nuclei were stained with Hoechst 33258 (Invitrogen).
For transient transfection experiments, we used Mirus TransIT-LT1 Transfection Reagent according to the manufacturer’s instructions (Mirus Bio, Madison, WI). At 24 h after transfection, cells were processed for immunofluorescence (see later description).
DNA constructs and cell transfection
Wild-type gigaxonin was cloned into the pTagBFP-C mammalian expression vector as follows. Wild-type gigaxonin was amplified by PCR using primers 5′-GATCCTCGAGCATGGCTAGGGCAGTG-3′ (forward) and 5′-GATCGGATCCACAGGGGAATGAACACG-3′ (reverse). The PCR product was then digested with XhoI and BamHI and cloned into the pTag-BFP-C vector previously digested with the same restriction enzymes. Control cells were transfected with the empty vector.
Gigaxonin-silenced BJ-5ta cell lines were established using shRNA-encoding oligonucleotides with three targeting sequences against gigaxonin: 1) 5’-GAGTGAGTTCCTCCAGTTA-3’; 2) 5’-AGTGAGTTCCTCCAGTTAA-3’; and 3) 5’-GAGAGAGATCCTGGATTAC-3’. These were cloned into pLKO.1 (Addgene, Cambridge, MA), a lentiviral expression vector. Lentiviruses were produced according to the manufacturer’s instructions by cotransfecting lentiviral plasmids, along with the helper plasmids pVSVG and pAX2, into 293FT cells (Invitrogen) using Xfect Transfection Reagent (Clontech, Mountain View, CA). Culture supernatants containing lentivirus particles were collected 48 h later. For lentiviral transductions, target cells were incubated with the viral supernatant supplemented with 8 μg/ml Polybrene (Sigma-Aldrich) for 4–8 h, after which the virus-containing medium was replaced with the growth medium. At 48 h posttransduction, the infected cells were selected by supplementing the cell growth medium with 2 μg/ml puromycin, as previously described (Mahammad et al., 2013). For controls, cells were incubated with viral particles containing scrambled sequences and processed as described.
SDS–PAGE and immunoblotting
Cells grown in 60- or 100-mm culture dishes were lysed in Laemmli sample buffer, and equal amounts of proteins in each lane were separated by SDS–PAGE and immunoblotted as previously described (Helfand et al., 2002, 2011).
Immunofluorescence and electron microscopy
Cells grown on coverslips were processed for indirect immunofluorescence after fixation in methanol as previously described (Prahlad et al., 1998; Yoon et al., 1998). In some cases, live cells were incubated with 100 nM MitoTracker Red for 30 min at 37°C before washing with fresh culture medium and immediately fixed. The fixed, immunostained cells were imaged using a Zeiss confocal LSM510 META (Carl Zeiss, Jena, Germany) microscope with oil immersion objective lenses (Plan-Apochromat, 63×, 1.40 numerical aperture [NA]; Carl Zeiss). Cells were also imaged using the Nikon N-SIM Structured Illumination Super Resolution Microscope (Nikon, Tokyo, Japan) with an oil immersion objective lens (CFI Apochromat, total internal reflection fluorescence (TIRF), 100×, 1.49 NA; Nikon). Electron microscopy was performed as previously described (Starger et al., 1978).
Live-cell imaging
GAN-NHS-NS-08 or BJ-5ta cells were plated in 35-cm no. 1.5 glass-bottom dishes (MatTek, Ashland, MA) at 70% confluency. For labeling mitochondria, cells were incubated with 100 nM MitoTracker Red CMXROS for 30 min at 37°C according to the manufacturer’s instructions (Chen, 1988), rinsed with prewarmed medium, and placed on the microscope stage for immediate observation. The motility of mitochondria was determined by time-lapse imaging at 3-s intervals for 15 min. Images were captured using an Andor (Belfast, Northern Ireland) XDI Revolution spinning-disk confocal system mounted on a Nikon Perfect Focus Ti microscope equipped with an Andor iXon electron-multiplying charge-coupled device camera and an oil immersion objective lens (Apochromat, TIRF, 60×, 1.49 NA; Nikon). Cells were maintained at 37°C on the microscope in an Okolab (Burlingame, CA) CO2 stage incubator. Lysosomes were visualized after incubation in 75 nM LysoTracker Red DND-99 according to the manufacturers’ instructions for 30 min at 37°C, rinsed with prewarmed medium, and immediately analyzed for 15 min as described. Live-imaging analyses were carried out on mitochondria in GAN-NHS-NS-08 and BJ-5ta cells under three experimental conditions: no treatment, overexpression of wild-type gigaxonin, and in cells silenced for gigaxonin expression. In each case, movies were taken of 15–20 cells (both GAN and normal fibroblasts). Specifically, the motility of mitochondria within one randomly chosen cell per coverslip was analyzed. For each condition, five movies were selected for detailed tracking of a total of 60–90 individual mitochondria (Miller and Sheetz, 2004, 2006). The selection of which movies to analyze was based on the ability to track mitochondria over the entire time period without complications such as aggregation with other mitochondria, moving out of the plane of focus, and so on. Identical procedures were used to analyze the motility of lysosomes as described in Results (Figure 6).
Image processing
Mitochondria were segmented using Mytoe software (Lihavainen et al., 2012). After segmentation, individual mitochondria were selected, and centroid positions were tracked to obtain trajectories using custom-written MATLAB (MathWorks, Natick, MA) codes. Net displacement, total distance traveled, and instantaneous velocities were also quantified both for mitochondria and lysosomes using custom-written MATLAB codes. All data were fitted and plotted in Origin software (OriginLab, Northampton, MA). Similar procedures were used to analyze lysosome motility.
Measurement of mitochondrial membrane potential
Cells grown on coverslips were incubated in medium with 100 nM MitoTracker Red CMXROS for 30 min at 37°C, rinsed with phosphate-buffered saline (PBS), and fixed at room temperature in 4% paraformaldehyde in PBS for 10 min. The fixed cells were rinsed in PBS and mounted on slides as previously described (Mahammad et al., 2013). For quantification of MitoTracker Red fluorescence intensity, images of labeled cells were processed using Fiji software (Schindelin et al., 2012). The images were converted to 8-bit grayscale, and mitochondria were thresholded with the autothreshold function using the Rényi entropy algorithm (Kapur et al., 1985). Cells were outlined using Li’s minimum cross entropy thresholding method (Li and Tam, 1998). The fluorescence data are the average fluorescence intensity of mitochondria in the given image minus the background (the average gray level in the cytoplasm outside of mitochondria). The fluorescence intensity measurements were subjected to one-way analysis of variance using Graph-Pad Prism 5 Software (La Jolla, CA). See Supplemental Figure S3.
Supplementary Material
Acknowledgments
We acknowledge the assistance of Kyung Hee Myung in the preparation of cells for light and electron microscopy and Wen Lu for help with the statistical analysis of mitochondrial fluorescence. The Goldman Laboratory is funded by grants from Hannah’s Hope Fund and National Institute of General Medical Sciences Grant PO1GM096971. The Opal Laboratory received funding from Hannah’s Hope Fund and is funded by the National Institute of Neurological Disorders and Stroke (National Institutes of Health Grants R01NS062051 and R01NS082351). The Gelfand Laboratory is supported by National Institute of General Medical Sciences Grants PO1 GM096971 and R01 GM052111.
Abbreviations used:
- GAN
giant axonal neuropathy
- IF
intermediate filament.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-09-0627) on December 23, 2015.
REFERENCES
- Asbury AK, Gale MK, Cox SC, Baringer JR, Berg BO. Giant axonal neuropathy—a unique case with segmental neurofilamentous masses. Acta Neuropathol. 1972;20:237–247. doi: 10.1007/BF00686905. [DOI] [PubMed] [Google Scholar]
- Berg BO, Rosenberg SH, Asbury AK. Giant axonal neuropathy. Pediatrics. 1972;49:894–899. [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- Chernoivanenko IS, Matveeva EA, Gelfand VI, Goldman RD, Minin AA. Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB J. 2015;29:820–827. doi: 10.1096/fj.14-259903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YH, Bischoff JR, Beach D, Goldman RD. Intermediate filament reorganization during mitosis is mediated by p34cdc2 phosphorylation of vimentin. Cell. 1990;62:1063–1071. doi: 10.1016/0092-8674(90)90384-q. [DOI] [PubMed] [Google Scholar]
- Cohen S, Zhai B, Gygi SP, Goldberg AL. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J Cell Biol. 2012;198:575–589. doi: 10.1083/jcb.201110067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JE, He T, Trejo-Skalli AV, Harmala-Brasken AS, Hellman J, Chou YH, Goldman RD. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J Cell Sci. 2004;117:919–932. doi: 10.1242/jcs.00906. [DOI] [PubMed] [Google Scholar]
- Gentil BJ, Minotti S, Beange M, Baloh RH, Julien JP, Durham HD. Normal role of the low-molecular-weight neurofilament protein in mitochondrial dynamics and disruption in Charcot-Marie-Tooth disease. FASEB J. 2012;26:1194–1203. doi: 10.1096/fj.11-196345. [DOI] [PubMed] [Google Scholar]
- Goldman RD. The role of three cytoplasmic fibers in BHK-21 cell motility. I. Microtubules and the effects of colchicine. J Cell Biol. 1971;51:752–762. doi: 10.1083/jcb.51.3.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger E, McNee G, Allan V, Woodman P. The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin Cell Dev Biol. 2014;31:20–29. doi: 10.1016/j.semcdb.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Ehrlicher AJ, Jensen MH, Renz M, Moore JR, Goldman RD, Lippincott-Schwartz J, Mackintosh FC, Weitz DA. Probing the stochastic, motor-driven properties of the cytoplasm using force spectrum microscopy. Cell. 2014;158:822–832. doi: 10.1016/j.cell.2014.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Ehrlicher AJ, Mahammad S, Fabich H, Jensen MH, Moore JR, Fredberg JJ, Goldman RD, Weitz DA. The role of vimentin intermediate filaments in cortical and cytoplasmic mechanics. Biophys J. 2013;105:1562–1568. doi: 10.1016/j.bpj.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggeness MH, Simon M, Singer SJ. Association of mitochondria with microtubules in cultured cells. Proc Natl Acad Sci USA. 1978;75:3863–3866. doi: 10.1073/pnas.75.8.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Mendez MG, Murthy SN, Shumaker DK, Grin B, Mahammad S, Aebi U, Wedig T, Wu YI, Hahn KM, et al. Vimentin organization modulates the formation of lamellipodia. Mol Biol Cell. 2011;22:1274–1289. doi: 10.1091/mbc.E10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Mikami A, Vallee RB, Goldman RD. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J Cell Biol. 2002;157:795–806. doi: 10.1083/jcb.200202027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmke BP, Goldman RD, Davies PF. Rapid displacement of vimentin intermediate filaments in living endothelial cells exposed to flow. Circ Res. 2000;86:745–752. doi: 10.1161/01.res.86.7.745. [DOI] [PubMed] [Google Scholar]
- Herman B, Albertini DF. A time-lapse video image intensification analysis of cytoplasmic organelle movements during endosome translocation. J Cell Biol. 1984;98:565–576. doi: 10.1083/jcb.98.2.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa I, Inagaki M. Regulatory mechanisms and functions of intermediate filaments: a study using site- and phosphorylation state-specific antibodies. Cancer Sci. 2006;97:167–174. doi: 10.1111/j.1349-7006.2006.00161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Kerner BL, Ahmad FS, Diaz AG, Greene JP, Gray SJ, Samulski RJ, Chung WK, Van Coster R, Maertens P, Noggle SA, et al. Intermediate filament protein accumulation in motor neurons derived from giant axonal neuropathy iPSCs rescued by restoration of gigaxonin. Hum Mol Genet. 2015a;24:1420–1431. doi: 10.1093/hmg/ddu556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Kerner BL, Garcia Diaz A, Ekins S, Wichterle H. Kelch domain of gigaxonin interacts with intermediate filament proteins affected in giant axonal neuropathy. PLoS One. 2015b;10:e0140157. doi: 10.1371/journal.pone.0140157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Kerner BL, Roth L, Greene JP, Wichterle H, Sproule DM. Giant axonal neuropathy: an updated perspective on its pathology and pathogenesis. Muscle Nerve. 2014;50:467–476. doi: 10.1002/mus.24321. [DOI] [PubMed] [Google Scholar]
- Kapur JN, Sahoo P, Wong A. A new method for gray-level picture thresholding using the entropy of the histogram. Comput Vis Graph Image Process. 1985;29:273–285. [Google Scholar]
- Korobova F, Gauvin TJ, Higgs HN. A role for myosin II in mammalian mitochondrial fission. Curr Biol. 2014;24:409–414. doi: 10.1016/j.cub.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb NJ, Fernandez A, Feramisco JR, Welch WJ. Modulation of vimentin containing intermediate filament distribution and phosphorylation in living fibroblasts by the cAMP-dependent protein kinase. J Cell Biol. 1989;108:2409–2422. doi: 10.1083/jcb.108.6.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Tam P. An iterative algorithm for minimum cross entropy thresholding. Pattern Recogn Lett. 1998;18:771–776. [Google Scholar]
- Lihavainen E, Makela J, Spelbrink JN, Ribeiro AS. Mytoe: automatic analysis of mitochondrial dynamics. Bioinformatics. 2012;28:1050–1051. doi: 10.1093/bioinformatics/bts073. [DOI] [PubMed] [Google Scholar]
- Lowery J, Kuczmarski ER, Herrmann H, Goldman RD. Intermediate filaments play a pivotal role in regulating cell architecture and function. J Biol Chem. 2015;290:17145–17153. doi: 10.1074/jbc.R115.640359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Ma XN, Zhang HM, Ji HH, Ding H, Zhang J, Luo D, Sun Y, Li XD. Mouse myosin-19 is a plus-end-directed, high-duty ratio molecular motor. J Biol Chem. 2014;289:18535–18548. doi: 10.1074/jbc.M114.569087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahammad S, Murthy SN, Didonna A, Grin B, Israeli E, Perrot R, Bomont P, Julien JP, Kuczmarski E, Opal P, Goldman RD. Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest. 2013;123:1964–1975. doi: 10.1172/JCI66387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoni R, Kreis TE. Translocation and clustering of endosomes and lysosomes depends on microtubules. J Cell Biol. 1987;105:1253–1265. doi: 10.1083/jcb.105.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MG, Restle D, Janmey PA. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys J. 2014;107:314–323. doi: 10.1016/j.bpj.2014.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Direct evidence for coherent low velocity axonal transport of mitochondria. J Cell Biol. 2006;173:373–381. doi: 10.1083/jcb.200510097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray ME, Mendez MG, Janmey PA. Substrate stiffness regulates solubility of cellular vimentin. Mol Biol Cell. 2014;25:87–94. doi: 10.1091/mbc.E13-06-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nekrasova OE, Mendez MG, Chernoivanenko IS, Tyurin-Kuzmin PA, Kuczmarski ER, Gelfand VI, Goldman RD, Minin AA. Vimentin intermediate filaments modulate the motility of mitochondria. Mol Biol Cell. 2011;22:2282–2289. doi: 10.1091/mbc.E10-09-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek G, Wiltz DC, Athanasiou KA. Contribution of the cytoskeleton to the compressive properties and recovery behavior of single cells. Biophys J. 2009;97:1873–1882. doi: 10.1016/j.bpj.2009.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer J, Schlote W, Bischoff A, Boltshauser E, Muller G. Generalized giant axonal neuropathy: a filament-forming disease of neuronal, endothelial, glial, and schwann cells in a patient without kinky hair. Acta Neuropathol. 1977;40:213–218. doi: 10.1007/BF00691956. [DOI] [PubMed] [Google Scholar]
- Pena SD. Giant axonal neuropathy: intermediate filament aggregates in cultured skin fibroblasts. Neurology. 1981;31:1470–1473. doi: 10.1212/wnl.31.11.1470. [DOI] [PubMed] [Google Scholar]
- Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD. Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsh T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5:a011304. doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova I, Burakov A, Berardone N, Zaliapin I, Slepchenko B, Svitkina T, Kashina A, Rodionov V. Actin dynamics is essential for myosin-based transport of membrane organelles. Curr Biol. 2008;18:1581–1586. doi: 10.1016/j.cub.2008.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res. 2007;313:2098–2109. doi: 10.1016/j.yexcr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat Rev Mol Cell Biol. 2014;15:163–177. doi: 10.1038/nrm3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni LE, Warren CM, Bucci C, Orten DJ, Hasson T. The unconventional myosin-VIIa associates with lysosomes. Cell Motil Cytoskeleton. 2005;62:13–26. doi: 10.1002/cm.20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starger JM, Brown WE, Goldman AE, Goldman RD. Biochemical and immunological analysis of rapidly purified 10-nm filaments from baby hamster kidney (BHK-21) cells. J Cell Biol. 1978;78:93–109. doi: 10.1083/jcb.78.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straube-West K, Loomis PA, Opal P, Goldman RD. Alterations in neural intermediate filament organization: functional implications and the induction of pathological changes related to motor neuron disease. J Cell Sci. 1996;109:2319–2329. doi: 10.1242/jcs.109.9.2319. [DOI] [PubMed] [Google Scholar]
- Summerhayes IC, Wong D, Chen LB. Effect of microtubules and intermediate filaments on mitochondrial distribution. J Cell Sci. 1983;61:87–105. doi: 10.1242/jcs.61.1.87. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tang HL, Lung HL, Wu KC, Le AH, Tang HM, Fung MC. Vimentin supports mitochondrial morphology and organization. Biochem J. 2008;410:141–146. doi: 10.1042/BJ20071072. [DOI] [PubMed] [Google Scholar]
- Tradewell ML, Durham HD, Mushynski WE, Gentil BJ. Mitochondrial and axonal abnormalities precede disruption of the neurofilament network in a model of charcot-marie-tooth disease type 2E and are prevented by heat shock proteins in a mutant-specific fashion. J Neuropathol Exp Neurol. 2009;68:642–652. doi: 10.1097/NEN.0b013e3181a5deeb. [DOI] [PubMed] [Google Scholar]
- Wagner OI, Lifshitz J, Janmey PA, Linden M, McIntosh TK, Leterrier JF. Mechanisms of mitochondria-neurofilament interactions. J Neurosci. 2003;23:9046–9058. doi: 10.1523/JNEUROSCI.23-27-09046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter L, Abrahamsberg C, Wiche G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J Cell Biol. 2008;181:903–911. doi: 10.1083/jcb.200710151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Moir RD, Prahlad V, Goldman RD. Motile properties of vimentin intermediate filament networks in living cells. J Cell Biol. 1998;143:147–157. doi: 10.1083/jcb.143.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.