

The cytoplasmic extension of the long-form isoform of BMPRII, unique among TGF-β superfamily receptors, is found to regulate the translation of BMPRII and its clathrin-mediated endocytosis. Both processes reduce its cell surface levels. The higher expression of BMPRII-SF at the plasma membrane results in enhanced activation of Smad signaling.
Abstract
The expression and function of transforming growth factor-β superfamily receptors are regulated by multiple molecular mechanisms. The type II BMP receptor (BMPRII) is expressed as two alternatively spliced forms, a long and a short form (BMPRII-LF and –SF, respectively), which differ by an ∼500 amino acid C-terminal extension, unique among TGF-β superfamily receptors. Whereas this extension was proposed to modulate BMPRII signaling output, its contribution to the regulation of receptor expression was not addressed. To map regulatory determinants of BMPRII expression, we compared synthesis, degradation, distribution, and endocytic trafficking of BMPRII isoforms and mutants. We identified translational regulation of BMPRII expression and the contribution of a 3’ terminal coding sequence to this process. BMPRII-LF and -SF differed also in their steady-state levels, kinetics of degradation, intracellular distribution, and internalization rates. A single dileucine signal in the C-terminal extension of BMPRII-LF accounted for its faster clathrin-mediated endocytosis relative to BMPRII-SF, accompanied by mildly faster degradation. Higher expression of BMPRII-SF at the plasma membrane resulted in enhanced activation of Smad signaling, stressing the potential importance of the multilayered regulation of BMPRII expression at the plasma membrane.
INTRODUCTION
Bone morphogenetic proteins (BMPs) form the most extensive subgroup of the structurally related transforming growth factor-β (TGF-β) superfamily of cytokines (Hinck, 2012). BMPs, originally named for their ability to induce bone growth (Wozney et al., 1988), are now broadly accepted as regulators of multiple processes in health and disease (reviewed in Wang et al., 2014). In embryogenesis, BMPs regulate early events such as gastrulation and mesoderm formation (Mishina et al., 1995). Defects in the expression of BMPs, BMP receptors, or intracellular mediators of BMP signaling result in embryonic lethality through defects in the formation and/or function of essential organs (e.g., heart, lungs, and bone; Wang et al., 2014). Postdevelopment, lack or excess of BMP signals is associated with different diseases, such as fibrodysplasia ossificans progressiva (Shore et al., 2006), osteogenesis imperfecta (Martinez-Glez et al., 2012), pulmonary arterial hypertension (PAH; International PPH Consortium et al., 2000), and cancer (Ehata et al., 2013). In cancer, similar to TGF-βs, BMPs were proposed to mediate both protumorigenic and antitumorigenic activity, with dependence on cellular context (Ehata et al., 2013).
BMPs transduce signals via single-spanning transmembrane serine-threonine kinases, which present structural relatedness and are functionally classified as type I (BMPRIa, BMPRIb, and ACVRIa) and type II (BMPRII, ACVRIIa, and ACVRIIb) receptors (Nickel et al., 2009; Sieber et al., 2009; Miyazono et al., 2010; Ehrlich et al., 2012). Binding of BMPs to these receptors induces and/or modulates the formation of hetero-oligomeric type I/type II receptor complexes (Gilboa et al., 2000; Nohe et al., 2002; Nickel et al., 2009), followed by type II receptor–mediated phosphorylation and activation of type I receptors (Shi and Massague, 2003; Feng and Derynck, 2005). Activated type I receptors relay the signal to the cell interior via canonical (Smad1/5/8) or noncanonical (e.g., mitogen- or stress-activated protein kinases) pathways, resulting in transcriptional regulation of a broad repertoire of genes (Nohe et al., 2004; Sieber et al., 2009; Miyazono et al., 2010). The obligatory requirement for type II receptors in transduction of BMP signals is exemplified in PAH, in which excessive proliferation of vascular smooth muscle cells occurs after lack of expression or intracellular mislocalization of BMPRII (Rudarakanchana et al., 2002; Sobolewski et al., 2008; Frump et al., 2013).
Posttranscriptionally, the expression levels of TGF-β superfamily receptors and their cell-surface levels are regulated by multiple molecular mechanisms, including alternative splicing, secretory and endocytic trafficking, localization to membrane microdomains, degradation, and cleavage (Alexander et al., 1996; Di Guglielmo et al., 2003; Mitchell et al., 2004; Hartung et al., 2006; Itoh and ten Dijke, 2007; Chen, 2009; Shapira et al., 2012; Xu et al., 2012; Hirschhorn et al., 2015). Although endocytosis was reported to play a role in some BMP signaling pathways (Hartung et al., 2006), these earlier studies relied on treatments that result in general inhibition of clathrin-mediated endocytosis (CME) or caveolar endocytosis, treatments whose effects are not limited to the internalization of specific receptors and alter also the trafficking of multiple signaling proteins. We recently used an alternative drug-independent approach to assess the role of the type I TGF-β receptor in signaling by identifying the CME targeting signal on the receptor and eliminating it by site-directed mutagenesis (Shapira et al., 2012). Here we adapted this approach to identify the endocytosis signals on BMPRII and explore their role in surface expression and signaling. BMPRII is expressed as two alternatively spliced isoforms (Rosenzweig et al., 1995): a long form (BMPRII-LF), which contains a C-terminal extension of ∼500 amino acids unique among TGF-β family receptors, and a short form (BMPRII-SF), the molecular weight and structure of which resemble those of the other TGF-β superfamily type II receptors. Alterations to the balance of the expression levels of BMPRII-SF and BMPRII-LF correlate with PAH (Cogan et al., 2012), and mutations within the segment unique to the long form cause disease (Rudarakanchana et al., 2002; Johnson et al., 2012), supporting the notion that this extension regulates specific signaling functions (Rudarakanchana et al., 2002; Foletta et al., 2003). However, the roles of this domain in the regulation of BMPRII expression, intracellular distribution, and/or trafficking are not yet understood.
In the present study, we used epitope-tagged constructs of BMPRII-SF and BMPRII-LF and truncation and alanine substitution mutants of the latter to identify the multilayered and differential regulation of translation, expression, degradation, and endocytosis of alternatively spliced forms of BMPRII.
RESULTS
The C-terminal extension in BMPRII-LF reduces receptor expression
To investigate whether the unique C-terminal extension in BMPRII-LF affects its levels of expression, we measured the steady-state levels of BMPRII-LF, BMPRII-SF, and BMPRII-LF truncation mutants containing shorter versions of the extension (BMPRII-LF mutants TC6, TC7, and TC8; Nohe et al., 2002). The truncation mutants are described in Materials and Methods. To this end, HEK293T cells were transfected with equal molar amounts of vectors encoding myc-tagged constructs of the aforementioned receptors. After 24 h, cells were lysed and subjected to SDS–PAGE, and expression of the myc-tagged receptors was assayed by immunoblotting. The results (Figure 1, A and B) show a major (∼10-fold) difference between the steady-state levels of the two naturally occurring BMPRII alternatively spliced isoforms, BMPRII-SF and BMPRII-LF. Similar results were obtained in COS7 cells expressing the same constructs (7-fold lower expression of BMPRII-LF relative to BMPRII-SF). Dissection of the C-terminal extension unique to BMPRII-LF demonstrates that inclusion of the C-terminal sequence encoding 17 amino acids that differentiates between TC8 and BMPRII-LF (Nohe et al., 2002) is a major determinant of the observed differences in expression, with an additional contribution from the region between TC6 and TC7 (Figure 1, A and B). Of importance, the differences in expression levels are not due to differences in mRNA levels, as shown in Figure 1, C and D, and validated for BMPRII-SF and BMPRII-LF using quantitative real-time PCR (qRT-PCR; Figure 1E).
FIGURE 1:

The steady-state levels of BMPRII expression are regulated by the C-terminal extension in BMPRII-LF. HEK293T cells were transfected with vectors encoding myc-tagged BMPRII-SF, BMPRII-LF, BMPRII-LF truncation mutants (TC6, TC7, TC8), or empty vector (pcDNA3; mock). (A, B) Western blotting to determine steady-state expression levels. At 24 h posttransfection, cells were lysed and subjected to SDS–PAGE and immunoblotting (Materials and Methods). (A) A representative experiment (n = 6). Top, a longer exposure to visualize the lower-expressed myc-BMPRII-LF. LFX6 represents a sixfold higher loading. (B) Quantification of multiple experiments. Results (mean ± SEM) were normalized relative to β-actin (loading control) and taking the expression level of myc-BMPRII-SF as 100%. Asterisks indicate significant differences between the pairs denoted by brackets (*p < 0.02; **p < 10−3; ***p < 10−9; Student’s t test). (C, D) Determination of mRNA levels. At 24 h posttransfection, cells were harvested and subjected to RNA isolation, followed by conversion to cDNA as described in Materials and Methods. The cDNA levels were measured using PCR (Materials and Methods). A representative experiment (n = 4) is shown in C, and quantitative analysis of all experiments is depicted in D. The results (mean ± SEM) were normalized to GAPDH cDNA levels, taking the results for myc-BMPRII-SF as 100%. (E) qRT-PCR quantification of BMPRII-SF and BMPRII-LF mRNA transcripts normalized to GAPDH mRNA. The ratio obtained for BMPRII-SF in each experiment was taken as 1.
Posttranscriptionally, reduction in steady-state protein expression levels may stem from lower synthesis levels or enhanced degradation. To explore the contribution of the former mechanism, we measured the synthesis levels of the foregoing proteins (BMPRII-LF and -SF and TC mutants) by [35S](Met+Cys) incorporation (Figure 2). At 24 h posttransfection, cells were pulse labeled with [35S](Met+Cys)–containing medium (25 min) and subjected to immunoprecipitation using anti-myc antibodies, followed by SDS–PAGE and autoradiography. As shown in Figure 2, A and B, the differences in the syntheses of BMPRII-SF, TC6, TC7, and TC8 were not significant. In contrast, a major and significant difference in [35S](Met+Cys) incorporation was observed between TC8 and BMPRII-LF. The short 35S pulse was designed to measure differences in the synthesis level of the receptors. To explore for a putative contribution by protein degradation within the short time frame of the pulse, we conducted a pulse-chase experiment in which the 25-min 35S pulse was followed by a 3- or 6-h chase in nonradioactive medium (Figure 2, C and D). This experiment revealed that the observed differences in the levels of [35S](Met+Cys)-labeled BMPRII-LF and TC8 cannot be attributed to differences in degradation. This suggests that the region encoding 17 amino acids that differentiates BMPRII-LF from TC8 contributes to the differences in steady-state levels and protein synthesis between these two proteins. However, because the steady-state expression level (unlike 35S incorporation) of TC6 is significantly higher than that of TC7 (Figure 1, A and B), it is still possible that protein degradation plays a role in the differences between the steady-state levels of BMPRII-SF and -LF, as shown later (see later discussion of Figure 8). Furthermore, the differences in synthesis levels of the naturally occurring alternatively spliced forms of BMPRII (SF and LF) may stem from a reduced recruitment of BMPRII-LF mRNA to the ribosomes. To directly assess this possibility, we pelleted denucleated lysates of HEK293T cells transfected with BMPRII-LF or -SF through a 40% sucrose cushion and measured the portion of receptor-encoding mRNA in the ribosome/polysome-enriched pellet relative to the total mRNA levels of the same receptors. The results (Figure 2, E and F) show no reduction in BMPRII-LF mRNA relative to BMPRII-SF in the enriched fraction. This suggests that the observed reduction in synthesis (Figure 2, A and B) is not due to reduced mRNA recruitment and occurs at a later step—for example, translational elongation. Taken together, the foregoing data support the notion that the differences in expression levels of the alternatively spliced forms of BMPRII (BMPRII-LF and BMPRII-SF) stem from differences in translation (readily observed after metabolic pulse labeling) and that the C-terminal portion of BMPRII-LF is an important regulator of its synthesis levels.
FIGURE 2:

Determination of protein synthesis and degradation levels of BMPRII variants. (A–D) HEK293T cells were transfected with myc-BMPRII variants as in Figure 1. After 24 h, cells were washed, starved (30 min), and labeled (25 min) with [35S](Met+Cys). (A, B) Assessment of the synthesis of myc-BMPRII variants. After 35S labeling and lysis, the myc-tagged receptors were immunoprecipitated and subjected to SDS–PAGE, blotting, and autoradiography. (A) A representative experiment (one of four). The receptor appears as a doublet due to glycosylation, as the upper band disappeared upon PNGase F treatment (not shown). (B) Densitometric quantification. For each experiment, the intensity of the specific bands was calibrated relative to that of myc-BMPRII-LF, taken as 1. Bars are mean ± SEM of four experiments. The asterisk indicates a significant difference (p < 0.05) observed between TC8 and myc-BMPRII-LF. (C, D) Pulse-chase measurement of BMP receptor degradation. Cells transfected with the indicated myc-BMPRII constructs were pulse-labeled as described, chased for the indicated periods in nonradioactive complete medium, and subjected to immunoprecipitation, followed by blotting and autoradiography. (C) A representative gel. (D) Densitometric quantification. In each experiment (n = 3), the intensity of the band of each construct at time 0 (end of pulse) was taken as 100%. (E, F) Sucrose cushion assessment of mRNA recruitment to polysomes/ribosomes. HEK293T cells were transfected and processed as described in Sucrose cushion enrichment of polysomal/rRNA fraction. (E) A representative gel (n = 3) depicting the cDNA levels generated from the total mRNA and sucrose cushion–pelleted mRNA fractions. GAPDH cDNA levels in both fractions served as controls. (F) qRT-PCR quantification of pellet/total levels of BMPRII mRNA transcripts normalized to GAPDH mRNA. The ratio obtained for BMPRII-SF in each experiment was taken as 1.
FIGURE 8:

Contribution of the C-terminal extension of BMPRII-LF to receptor degradation depends on the dileucine endocytic signal. HEK293T cells were transfected with myc-BMPRII-LF, myc-BMPRII-LF-AA, or myc-BMPRII-SF and subjected to the CHX-chase receptor degradation assay (Materials and Methods). (A–C) Representative experiments. The immunoblots show myc-BMPRII and β-actin (loading control) before (time 0) or after addition of 300 μM CHX for the indicated times. (D–F) Quantification (mean ± SEM, n = 4) of the intensities of the bands of the specific myc-BMPRII variants after normalization to the respective level at time 0. (G) Effect of degradation inhibitors. At 24 h posttransfection, cells were incubated with either MG132 (25 μM) or chloroquine (25 μg/ml) for another 24 h or left untreated for the same period (vehicle control). Cells were processed and immunoblotted as in Figure 1, and myc-BMPRII receptor levels were analyzed by densitometry. The graph depicts the mean ± SEM (n = 3) receptor levels after normalization to β-actin, taking the control sample as 100%. Asterisks indicate a significant increase in the receptor level relative to the untreated sample (*p < 0.05).
To explore further the regulatory role of the C-terminal sequence of BMPRII-LF, we initially examined the degree of conservation of the last C-terminal 98 nucleotides (numbers 4182–4279 in the human BMPRII-LF sequence) between different species at both protein (unpublished data) and transcript levels (Figure 3A). This analysis revealed a high degree of conservation. Moreover, prediction of the secondary structure of this region (nucleotides 4182–4279) in the coding sequence of BMPRII-LF with the Mfold server (Zuker and Jacobson, 1998; Zuker, 2003; Waugh et al., 2002) revealed a conserved tendency to form a stem-loop–based structure with predicted ΔG (free energy difference) ranging from −28 to −35 kcal/mol (−32.7 kcal/mol for the predicted structure shown in Figure 3B). This tendency to form secondary stem-loop–based structures was also observed in BMPRII sequences showing less conservation (the wit receptor of Drosophila melanogaster and the BMPRII receptor of Xenopus laevis; unpublished data). On the basis of these data, we opted to add the last 99 nucleotides of the coding sequence of BMPRII-LF to BMPRII-SF, generating in this manner a BMPRII-SF-modified (BMPRII-SFM) construct that includes this potential translational regulator sequence of BMPRII-LF. Comparative analysis of the expression levels of BMPRII-SF and BMPRII-SFM by immunoblotting revealed significantly lower levels of BMPR-SFM than BMPRII-SF (Figure 3C), confirming the expression-attenuating potential of the added C-terminal sequence. Of note, addition of the same nucleotide sequence to the 3’ end of the coding sequence of an unrelated protein (green fluorescent protein [GFP]) also resulted in reduced expression levels (Figure 3, E and F). These findings suggest that the terminal 99-nucleotide sequence of BMPRII-LF has expression-reducing potential over a broad spectrum of proteins. Furthermore, within the 99-nucleotide structure (Figure 3B), both 5’ and 3’ nucleotides contribute to the stabilization of the stem. Thus elimination of 51 nucleotides from this sequence (the TC8 mutant compared with BMPRII-LF) is predicted to abrogate the structure as a whole, interfering with its attenuating capability. Indeed, TC8 is expressed to a significantly higher level than BMPRII-LF (Figures 1 and 2).
FIGURE 3:

Addition of the 3’-sequence of the coding region of BMPRII-LF to BMPRII-SF decreases its levels of expression. (A) Sequence alignment of the 3’-sequence (98 nucleotides, numbers 4182–4279 in human BMPRII-LF) of the coding region of BMPRII from different species. (B) Mfold prediction (Zuker and Jacobson, 1998; Zuker, 2003; Waugh et al., 2002) of the secondary structure of the same 3’-sequence of the coding region of human BMPRII-LF. (C, D) Immunoblot-based detection of the expression levels of BMPRII-SF and the extended BMPRII-SF mutant (BMPRII-SFM). HEK293T cells were transfected with vectors encoding myc-tagged BMPRII-SF, BMPRII-SFM (the BMPRII-SF mutant extended by addition of 99 coding nucleotides, numbers 4181–4279, from the 3’ end of BMPRII-LF), or pcDNA3 (mock). At 24 h posttransfection, cells were lysed and subjected to SDS–PAGE and immunoblotting. (C) A representative experiment (n = 3); experiments were done in triplicates of independently transfected samples. (D) Quantification of multiple experiments. Results (mean ± SEM) were normalized relative to β-actin (loading control) and taking the expression level of myc-BMPRII-SF as 100%. Asterisk indicates significant difference between BMPRII-SF and BMPRII-SFM (p < 0.001; Student’s t test). (E) Immunoblot of GFP and a GFP mutant (GFP-C) extended at the 3’ end of the coding region by the aforementioned 99-nucleotide sequence. HEK293T cells were transfected by GFP- or GFP-C–encoding vectors. At 6 h posttransfection, they were processed as described in Materials and Methods. (F) A densitometric quantification of three independent experiments, showing a significant difference between GFP and GFP-C (*p < 0.001).
Because receptor signaling is usually initiated by ligand binding to cell-surface receptors, we proceeded to investigate whether the differences between the BMPRII isoforms and mutants are also reflected in their cell surface levels. To this end, we opted for a proteinase K degradation approach, which allows for simultaneous calculation of the levels of the receptor at the plasma membrane and the portion of the receptor exposed at the plasma membrane relative to its entire cell content. The cell surface receptors were exposed to digestion by proteinase K at 4°C (to avoid endocytosis of the enzyme). Under these conditions, only the receptors residing at the cell surface are exposed to the digesting enzyme, whereas intracellular receptors are protected. The difference between the levels of myc-tagged BMPRII isoforms and mutants in untreated versus proteinase K–treated pairs of the same transfected sample yield the cell surface levels of the receptors (Figure 4B), enabling the calculation of the cell surface/total ratio (Figure 4C). Our results show that BMPRII-LF, which exhibits the lowest synthesis and steady-state levels (Figures 1–3), shows a concomitant low level of expression at the cell surface (Figure 4B). Of note, all BMPRII constructs, and especially BMPRII-LF, are mainly localized to the plasma membrane at steady state (Figure 4C). This finding was confirmed by measurement of endoglucosidase H (EndoH)–resistant fractions, for which 95% ± 3 of BMPRII-LF and 78% ± 4 of BMPRII-SF were EndoH resistant. The highest difference in the cell surface level was measured between the two naturally occurring isoforms, BMPRII-SF and BMPRII-LF. However, a clear difference emerged also between the cell surface levels of TC6 and TC7. Such a difference, albeit smaller, was observed also between the steady-state levels of these truncation mutants (Figure 1). These differences cannot be attributed to protein synthesis, as the latter did not differ significantly between these two mutants (Figure 2), suggesting that additional processes might be involved in the differential regulation of BMPRII isoforms.
FIGURE 4:

The length of the C-terminal extension of BMPRII-LF determines its steady-state cell surface level. HEK293T cells were transfected as in Figure 1 with the indicated myc-BMPRII constructs. At 24 h posttransfection, the cell surface receptors (exposed to externally added enzyme) were digested (or not) with proteinase K at 4°C (see Materials and Methods). Cells were lysed and subjected to SDS–PAGE and immunoblotting and probing with anti-myc antibodies (A, top) or anti-β-actin (A, bottom; loading control). (A) A representative gel. (B) Quantification of the levels of proteinase K–sensitive myc-BMPRII. Data (mean ± SEM, n = 6) were calibrated relative to the value obtained for myc-BMPRII-SF, taken as 100%. Asterisks indicate significant differences between the bracketed pairs. (C) Ratio of cell surface–localized to total BMPRII levels. The cell surface receptor levels were calculated from the difference between proteinase K–treated and untreated samples, yielding the proteinase K–sensitive fraction. Asterisks indicate significant differences between the bracketed pairs (*p < 0.05; **p < 0.001; Student’s t test).
Regulation of BMPRII cell surface levels and signaling by endocytosis
Differences in cell surface levels can arise from different endocytosis rates, as previously shown for influenza hemagglutinin variants carrying distinct endocytosis targeting sequences (Keren et al., 2001). We first examined whether the two native isoforms, BMPRII-SF and BMPRII-LF, which present the largest difference in cell surface levels, are characterized by different endocytosis rates. To this end, we expressed myc-BMPRII-SF or myc-BMPRII-LF in either HEK293T or COS7 cells; experiments with the latter cell line were added to allow direct comparison to former results obtained in our lab in endocytosis studies (Ehrlich et al., 2001; Hartung et al., 2006; Shapira et al., 2012). After immunofluorescence labeling of the myc-tagged cell surface receptors in the cold (Materials and Methods), the cells were incubated at 37°C for defined periods and subjected to endocytosis measurements of the fluorescence-labeled receptors by the point-confocal assay described by us earlier (Ehrlich et al., 2001). Typical images of cells subjected to this assay, which show a much stronger shift to a vesicular pattern for BMPRII-LF than for BMPRII-SF, are depicted in Figure 5A. Quantitative time-dependent point-confocal measurements of the labeled myc-BMPRII-LF or myc-BMPRII–SF remaining at the cell surface (Figure 5, B and C) reveal a markedly faster endocytosis of myc-BMPRII-LF than myc-BMPRII-SF in both cell types. Similar results were obtained in the presence of ligand (10 nM BMP2). Of note, not only was the rate of myc-BMPRII-LF faster, but the percentage internalized after prolonged incubation at 37°C was also higher. The half-time for internalization of BMPRII-LF (the low endocytosis rate of BMPRII-SF precluded an accurate measurement) was 15–20 min, in the same time range encountered for type I and type II TGF-β receptors (Ehrlich et al., 2001; Shapira et al., 2012). Most of the internalization of myc-BMPRII-LF was blocked by treatments that interfere with CME, such as incubation in sucrose-containing hypertonic medium (Heuser and Anderson, 1989) or treatment with PitStop (von Kleist et al., 2011). In contrast, nystatin (an inhibitor of caveolar endocytosis) had only a minor effect (Figure 5, D and E). These findings suggest that CME is the main internalization pathway of BMPRII-LF under these conditions. This is in line with the lack of caveolin-1 in HEK293T cells (Hartung et al., 2006), although the residual internalization of BMPRII-SF in COS7 cells (but not in HEK293T cells) could represent some contribution by caveolar-like endocytosis, as reported earlier (Hartung et al., 2006).
FIGURE 5:

The C-terminal extension of BMPRII-LF directs it to clathrin-mediated endocytosis. HEK293T or COS7 cells were transfected with myc-BMPRII-LF or myc-BMPRII-SF. After 24 h, they were left untreated or subjected to internalization-inhibiting treatments (hypertonic sucrose–supplemented medium, PitStop, or nystatin). The receptors at the cell surface were then labeled at 4°C (time 0) with anti-myc, followed by Alexa 546–GαM Fab’, incubated for defined intervals at 37°C, and fixed at 4°C (Materials and Methods). In experiments conducted in the presence of ligand, BMP2 (10 nM) was added together with the secondary Fab’ and retained throughout the experiment. (A) Typical images. Bar, 10 μm. (B, C) Quantitative endocytosis measurements in HEK293T (B) and COS7 (C) cells. The fluorescence intensity remaining at the cell surface was measured by the point-confocal method (Materials and Methods). Results are mean ± SEM of 200 cells/time point, taking for each sample the intensity at time 0 as 100%. (D, E) Endocytosis of myc-BMPRII-LF is inhibited by CME inhibitors but not by nystatin. For each treatment, the fluorescence intensity at the cell surface was measured at time 0 and after 20 min of incubation at 37°C in medium containing inhibitors (where indicated). Two hundred cells were measured for each condition, and the intensity of the same sample at time 0 was taken as 100%. Treatments that inhibit CME (sucrose and PitStop) induced a significant reduction in myc-BMPRII-LF endocytosis (*p < 0.01).
These findings localize the molecular determinants directing BMPRII-LF to CME to the unique C-terminal extension differentiating BMPRII-LF from BMPRII-SF. To identify the endocytosis signal(s) involved, we studied the endocytosis of the different-length myc-BMPRII-LF truncation mutants (TC5–TC8). As shown in Figure 6, A and B, myc-BMPRII-SF and the shorter myc-BMPRII-LF truncation mutants (TC5 and TC6) exhibited only marginal endocytosis in 20 min, whereas myc-BMPRII-LF and the longer-truncation mutants (TC7 and TC8) exhibited similar and significant endocytosis. It therefore follows that the endocytosis signal(s) found in BMPRII-LF reside in the segment between TC6 and TC7. Examination of this sequence for potential CME consensus motifs revealed one such motif from the dileucine family, L870 I871. We therefore mutated these two residues in BMPRII-LF to Ala, generating the myc-BMPRII-LF-AA mutant. Indeed, this mutant failed to undergo significant internalization in both HEK293T and COS7 cells, and addition of ligand (10 nM BMP2) had no effect on its internalization (Figure 6, C–E). We conclude that BMPRII-LF is directed to CME by a single endocytosis signal, L870 I871. To examine whether the different endocytosis rates of BMPRII-LF and BMPRII-LF-AA affect the cell surface expression levels, we measured by immunoblotting the steady-state expression levels of BMPRII-LF and BMPRII-LF-AA (Figure 7, A and B), as well as their sensitivity to proteinase K digestion in the cold (Figure 7, C and D). Quantification of the blots (Figure 7, B and D) revealed a mild difference between their surface levels (nearly twofold higher level for myc-BMPRII-LF-AA), with a somewhat smaller difference in the total expression level. Of note, a similar approximately twofold decrease in the cell surface expression levels was also observed between TC6 (which lost the endocytosis signal) and TC7 (which retained this signal; Figure 4).
FIGURE 6:

Endocytosis of BMPRII-SF, BMPRII-LF and its truncation and alanine replacement mutants. HEK293T or COS7 cells were transfected with the indicated myc-BMPRII variants, followed by immunofluorescence labeling of the cell surface receptors at 4°C as in Figure 4. Samples were then shifted to 37°C for 20 min (A, B) or for the indicated times (C–E) to allow endocytosis. (A, B) The internalization determinant of BMPRII localizes to the region between TC6 and TC7. For each construct, fluorescence intensity at the cell surface was measured by the point-confocal method (200 cells/sample) at time 0 and after a 20-min incubation at 37°C, and the intensity of the same sample at time 0 was taken as 100%. The asterisk denotes a significant difference between TC6 and TC7 (*p < 0.05). (C) Typical images of cells expressing the endocytosis-defective BMPRII-LF-AA mutant before and after internalization for the indicated periods (10–90 min). Bar, 10 μm. Note the lack of internalization (very little vesicular staining) even at long incubation periods. (D, E) Quantification of myc-BMPRII-LF-AA endocytosis relative to myc-BMPRII-LF in HEK293T (D) and in COS7 (E) cells. The fluorescence intensity remaining at the cell surface was measured by the point-confocal method (Materials and Methods). Results are mean ± SEM of 200 cells/time point, taking for each sample the intensity at time 0 as 100%. As shown in D and E, addition of ligand (10 nM BMP2) as in Figure 5 had no effect on the internalization of BMPRII-LF-AA in either cell line.
FIGURE 7:
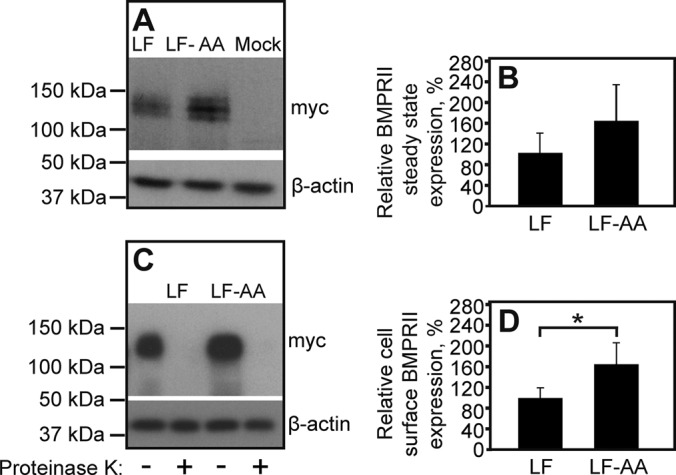
Clathrin-mediated endocytosis attenuates the cell surface expression of BMPRII-LF. HEK293T cells were transfected with myc-BMPRII-LF or myc-BMPRII-LF-AA. At 24 h posttransfection, the steady-state expression of the transfected receptors (A, B) and their expression levels at the plasma membrane (C, D) were measured as in Figures 1 and 4, respectively. (A) A representative gel. (B) Quantification of the steady-state expression levels of myc-BMPRII-LF and myc-BMPRII-LF-AA from multiple experiments (n = 6). Results (mean ± SEM) were normalized relative to β-actin, taking the expression level of myc-BMPRII-LF as 100%. (C) A representative immunoblot with or without proteinase K digestion. (D) Quantification of cell surface–localized myc-BMPRII-LF and myc-BMPRII-LF-AA. Results (mean ± SEM, n = 6) were derived from the difference between proteinase K–treated and untreated samples, as in Figure 4. The asterisk indicates a significant increase (p < 0.05) in cell surface level of myc-BMPRII-LF-AA relative to that of myc-BMPRII-LF (taken as 100%).
To explore the potential contribution of the C-terminal extension in BMPRII-LF and of the dileucine endocytosis signal in this domain to BMPRII degradation, we compared the degradation rates of BMPRII-LF, BMPRII-LF-AA, and BMPRII-SF after cycloheximide (CHX) addition (Figure 8). Because the major fraction of the BMPRII constructs is localized to the cell surface (Figure 4), the experiment measures mainly the degradation of the cell surface receptors. The degradation rate of BMPRII-LF, derived from the initial slope (up to 2 h), was faster than those of BMPRII-SF or BMPRII-LF-AA (∼20%/h for BMPRII-LF vs. ∼8%/h for BMPRII-SF and 11%/h for BMPRII-LF-AA). To probe for the cellular machinery involved in the observed degradation, we treated cells transfected with BMRII-LF, LF-AA, or –SF with chloroquine (an inhibitor of lysosomal degradation) or MG132 (a proteasomal inhibitor). As shown in Figure 8G, both chloroquine and MG132 significantly increased the accumulation of BMRII-LF. Minor accumulation of BMRII-SF was observed with MG132. The exclusive protective effect of chloroquine on BMPRII-LF is in accord with its significantly faster endocytosis relative to the other variants. Taken together, elimination of the dileucine endocytosis signal (e.g., in BMPRII-LF-AA or in the shorter-truncation mutant TC6 relative to TC7) has a mild effect on BMPRII degradation and/or cell surface levels, whereas a considerable contribution to these differences between BMPRII-LF and BMPRII–SF is due to their differential synthesis (Figure 2).
To address the roles of the C-terminal extension in BMPRII-LF and its endocytosis motif in signaling to the canonical Smad 1/5/8 pathway, we compared the levels of phospho-Smad (pSmad) in cells expressing BMPRII-LF, BMPRII-LF-AA, or BMPRII–SF. COS7 cells were cotransfected with yellow fluorescent protein (YFP)–Smad1 together with myc-BMPRII (LF, LF-AA, or SF) or β-Gal (mock) and stimulated (or not) by BMP2 (10 nM, 30 min). As shown in Figure 9A, the mock-transfected cells exhibited clear BMP2-stimulated increase in both endogenous phospho-Smad1/5/8 (pSmad1/5/8) and phospho-YFP-Smad1, indicating that the transfection procedure did not interfere with the capability of the cells to respond to the ligand. Because transfection with YFP-Smad1 resulted in its enhanced phosphorylation already in the absence of ligand and the ligand-induced stimulation was nearly lost (Figure 9A), we focused our analysis on the effects of BMPRII variants on the phosphorylation of endogenous Smad1/5/8. Expression of myc-BMPRII-LF did not affect significantly endogenous pSmad1/5/8 formation (either without or with BMP2), in accord with its low expression at the plasma membrane. Expression of the endocytosis-defective myc-BMPRII-LF-AA, whose surface expression level is only somewhat higher than that of myc-BMPRII-LF, had similar effects on the ligand-dependent activation of endogenous Smad1/5/8. Transfection with myc-BMPRII-SF, whose surface expression level is much higher than that of myc-BMPRII-LF and is also endocytosis defective, resulted in a small increase in pSmad1/5/8 formation already before ligand addition and in increased level of pSmad1/5/8 upon BMP2 stimulation. To assess directly whether CME is dispensable for Smad1/5/8 phosphorylation, we treated untransfected COS7 cells with the specific CME inhibitor PitStop and measured pSmad1/5/8 levels upon BMP2 stimulation. As shown in Figure 9, B–D, there was no significant inhibition of pSmad1/5/8 formation in response to BMP2 in the presence of PitStop under conditions that block BMPRII-LF endocytosis. Taken together, these findings indicate that endocytosis of BMPRII is not required for activation of signaling to the Smad1/5/8 pathway and that the surface levels of the receptors correlate with their signaling capability.
FIGURE 9:

BMPRII-dependent phosphorylation of BMP-responsive Smads correlates with BMPRII surface expression levels and does not require its endocytosis. (A) COS7 cells were cotransfected with YFP-Smad1 together with β-galactosidase (mock), myc-BMPRII-LF, myc-BMPRII-LF-AA, or myc-BMPRII-SF. After 24 h, cells were serum starved and stimulated (or not) with 10 nM BMP2 for 30 min. Panels depict representative immunoblots (n = 3), which were probed with anti-pSmad1/5/8, anti-myc, or anti-GAPDH. Numbers under specific lanes indicate the level of YFP-pSmad1 or endogenous (Endo) pSmad1/5/8 after calibration to GAPDH and relative to unstimulated mock-transfected cells in the same experiment. (B, C) Untransfected COS7 cells were serum starved, treated or not with PitStop, and subjected to BMP2 stimulation and Western blotting as described to probe for endogenous total Smad1/5/8 or pSmad1/5/8. (B) A representative experiment. (C) Quantification of the mean ± SEM (n = 4) of pSmad1/5/8 to tSmad1/5/8 ratio. Asterisks indicate a significant increase (p < 0.01) in the ratio. (D) Control experiment showing that PitStop blocks the internalization of BMPRII-LF in the presence of ligand (conditions similar to those of the signaling assay). The internalization experiment was performed as in Figure 5.
DISCUSSION
BMPRII-LF is unique among the TGF-β superfamily receptors due to a C-terminal extension of 512 amino acids in its cytoplasmic domain. The functional significance of this extension was supported by demonstration of its binding to diverse cellular factors, including the endocytic protein EPS15R (Hartung et al., 2006), the dynein light chain Tctex-1 (Machado et al., 2003), the kinases cGKI (Schwappacher et al., 2009) and LIMK (Lee-Hoeflich et al., 2004), and Trb3, a regulator of Smurf1 stability and Smad-dependent signaling output (Chan et al., 2007). In different cellular contexts, such interactions were proposed to influence BMPRII Smad-dependent and Smad-independent signaling, BMPRII trafficking, and BMP-induced differentiation of distinct cell types. Moreover, the lethality of mice homozygous for BMPRII-SF but lacking BMPRII-LF (Leyton et al., 2013), the presence of disease-causing mutations within this C-terminal extension (exon 12) in PAH (Thomson et al., 2000; Machado et al., 2001), and the proposed role for differences in the expression ratio between BMPRII-SF and BMPRII-LF in determining the penetrance of PAH (Cogan et al., 2012) further demonstrate the importance of this molecular domain. However, the role(s) of this C-terminal extension (either at the level of coding sequence or protein) in the synthesis, degradation, and trafficking of BMPRII had not been addressed. Here we show that molecular determinants within the mRNA sequence and/or encoded by exon 12 of BMPRII regulate its synthesis and clathrin-mediated internalization. This differential regulation of alternatively spliced forms of BMPRII has direct implications for the overall and plasma membrane–localized steady-state levels of the BMPRII receptor forms, the kinetics of their degradation, and the intensity of their ability to activate the Smad1/5/8 pathway in response to ligand.
Our studies on the steady-state expression levels of BMPRII-SF, BMPRII-LF, and their mutants demonstrate that the C-terminal extension unique to BMPRII-LF has two elements that reduce its expression relative to BMPRII-SF. The major effects are contributed by the very C-terminal end of BMPRII-LF, accompanied by a contribution from the region between TC6 and TC7 (Figures 1 and 2). These differences are detected also at the level of the cell surface expression of the receptors, for which the contribution of the region between TC6 and TC7 is even more accentuated (Figure 4). To delineate further the mechanisms involved, we used an experimental setup based on metabolic pulse labeling of exogenously expressed isoforms and mutants of BMPRII (at equimolar levels) under the same promoter and with the same 5’-untranslated region (UTR) and 3’-UTR regions (Figure 2). This allowed us to identify the regulation of the synthesis of BMPRII isoforms at the level of translation. We show that the most-3’ region of exon 12 (99 nucleotides, numbers 4181–4279, encoding 32 amino acids and a stop codon), which is unique to BMPRII-LF and is predicted to fold into a stem-loop–based secondary structure (Figure 3B), attenuates the expression of BMPRII (Figures 2 and 3) on a translational level (Figure 2). Multiple molecular mechanisms have been proposed to regulate protein translation, including adaptation to the tRNA pool (codon usage), charge of amino acids that are incorporated in the polypeptide chain, mRNA folding energy, and different activities of RNA-binding proteins (Kozak, 1986; Wells, 2006; Ingolia et al., 2011; Tuller et al., 2011; Pop et al., 2014). Here we show that the reduced expression of BMPRII-LF and BMPRII-SFM (the BMPRII-SF mutant extended by addition of 99 coding nucleotides, numbers 4181–4279, from the 3’ end of BMPRII-LF) correlates with the presence of an RNA sequence with structure-forming tendencies, which can also attenuate the expression of unrelated proteins such as GFP (Figure 3, E and F). Of note, such secondary RNA structures are the basis of recognition by numerous RNA-binding proteins (Draper, 1995). In addition, when translation is carried out in endoplasmic reticulum-tethered polysomes, negative regulation of elongation would be expected to result in an overall decrease in the level of the protein being synthesized (e.g., BMPRII-LF). Within the TGF-β superfamily of receptors and ligands, TGF-β1 and TGF-β3 have been suggested to be regulated at the level of translation (Arrick et al., 1991; Fraser et al., 2002). To our knowledge, the present study is the first to report on such regulation of a receptor from this superfamily. Note that the differences in expression of BMPRII-SF and BMPRII-LF are greater than those between BMPRII-SF and BMPRII-SFM, attesting to the additional regulatory element(s) (e.g., the endocytosis signal that enhances BMPRII-LF degradation; Figure 8).
Numerous studies on the endocytosis of receptors of the TGF-β superfamily suggested CME as the major internalization pathway; a potential contribution by caveolar endocytosis has been contentious (Ehrlich et al., 2001; Yao et al., 2002; Di Guglielmo et al., 2003; Mitchell et al., 2004; Hartung et al., 2006; Chen, 2009; Hirschhorn et al., 2012; Shapira et al., 2012). Moreover, conflicting results were obtained when addressing the roles for endocytosis of the receptors in the regulation of activation of Smad signaling pathways (Hayes et al., 2002; Penheiter et al., 2002; Di Guglielmo et al., 2003; Hartung et al., 2006; Chen et al., 2009; Chen, 2009; Hirschhorn et al., 2012; Kim et al., 2012; Shapira et al., 2012, 2014; Umasankar et al., 2012). Such conflicts may reflect the reliance on treatments (chemical or genetic) that alter/inhibit altogether CME and/or caveolar endocytosis, since such general treatments affect not only the endocytosis of the respective receptors, but also the endocytosis, distribution, and trafficking of numerous cellular factors. In the present study, we identified CME as the major endocytosis pathway of BMPRII and reported marked differences in the endocytic potential of the alternatively spliced forms of BMPRII (i.e., lack of endocytosis of BMPRII-SF; Figure 5). Moreover, we identified a previously unknown CME-targeting signal, of the dileucine class of endocytic motifs, localized at the C-terminal of BMPRII-LF (Figure 6). On the basis of this finding, we generated an endocytosis-defective BMPRII-LF mutant (BMPRII-LF-AA) and used it, together with the naturally alternatively spliced forms of BMPRII, to investigate the relationship between BMPRII endocytosis, expression level, degradation, and signaling (Figures 6–9).
Note that the different endocytosis rates of BMPRII-LF and BMPRII-LF-AA affect their cell surface expression levels, as measured by the proteinase K digestion assay (Figure 7). The approximately twofold difference between their surface expression levels correlates with a similarly higher degradation rate for the endocytosis-capable BMPRII-LF (Figure 8). The slower degradation of BMPRII-LF-AA appears to be due to the fact that it does not undergo endocytosis, since it is similar to that of BMPRII-SF, which also lacks the endocytosis-targeting motif (Figure 8). This conclusion is in line with the very similar decrease in the cell surface expression level between TC6 (which lacks the endocytosis motif) and TC7 (Figure 4). Moreover, in contrast to the endocytosis-defective BMPRII-SF and BMPRII-LF-AA, the degradation of BMPRII-LF is sensitive to both proteasomal and lysosomal inhibitors, in line with its significantly faster endocytosis (Figure 8G). These results are in line with a report that chloroquine increases cell surface BMPRII-LF levels and restores BMP9 signaling in endothelial cells harboring PAH-related BMPRII mutations (Dunmore et al., 2013). The notion of a positive correlation between the cell surface expression levels of BMPRII and the activation intensity of pSmad1/5/8 by BMP is also supported by the results depicted in Figure 9A, where BMPRII-SF higher surface expression correlated with increased levels of pSmad1/5/8. Because this BMPRII variant is hardly endocytosed, these findings may imply that endocytosis of BMPRII is dispensable for Smad1/5/8 activation. This notion is validated by the insensitivity of endogenous Smad1/5/8 activation by BMP2 to the CME inhibitor PitStop (Figure 9, B–D).
Taken together, the data in the present study support the notion that the expression levels and plasma membrane levels of BMPRII are determined by two molecular processes—translational regulation of protein synthesis (which provides the major contribution) and endocytosis/degradation (mild modulatory effect). Both mechanisms enhance the expression of BMPRII-SF relative to BMPRII-LF at the cell surface (where the receptors are exposed to stimulation by exogenous ligands), resulting in activation of the Smad1/5/8 pathway at elevated intensities. Of interest, BMPRII-SF was reported to be incapable of associating with and activating at least a subset of non-Smad BMP signals (Foletta et al., 2003; Lee-Hoeflich et al., 2004), implying that the alternative splicing of BMPRII may be an important regulator of the balance of activation of canonical versus noncanonical signals by BMPs.
MATERIALS AND METHODS
Reagents
DMEM, fetal calf serum (FCS), l-glutamine, penicillin-streptomycin (25 and 40 μg/ml, respectively), Hanks’ balanced salt solution (HBSS), and nystatin were from Biological Industries (Beit HaEmek, Israel). Recombinant human BMP2 was a gift from W. Sebald (University of Wurzburg, Wurzburg, Germany), and Opti-MEM was from Life Technologies (Carlsbad, CA). Phosphate-buffered saline (PBS), DMEM without l-methionine and l-cystine, protein G–Sepharose, fatty acid–free bovine serum albumin (BSA; fraction V), protease inhibitor cocktail (P8340; PIC), Na3VO4, CHX, phenylmethanesulfonyl fluoride (PMSF), dithiothreitol, Phosphatase Inhibitor Cocktail 2, Phosphatase Inhibitor Cocktail 3, chloroquine, MG132, and sucrose were from Sigma-Aldrich (St. Louis, MO). Anti–myc tag 9E10 mouse ascites (Evan et al., 1985) was from Covance Research Products (Denver, PA), and anti-GFP (FL) was from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-mouse (GαM) F(ab′)2 conjugated to Alexa 546 was from Invitrogen-Molecular Probes (Eugene, OR). Anti–myc F(ab′)2 (prepared as in Gilboa et al., 1998) and fluorescent GαM F(ab′)2 were converted to monovalent Fab′ as described (Henis et al., 1994). Normal goat γ-globulin and peroxidase-conjugated GαM and goat anti-rabbit (GαR) immunoglobulin Gs (IgGs) were from Jackson ImmunoResearch (West Grove, PA). Monoclonal rabbit IgG anti-pSmad1/5/8 (Ser-463/Ser-465) and monoclonal rabbit IgG against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were from Cell Signaling (Danvers, MA), and mouse anti–β-actin was from MP Biomedicals (Solon, OH). PitStop 2 was obtained from Abcam (Cambridge, United Kingdom). Promix cell labeling mix [35S-(Met+Cys), >1000 Ci/mmol] was from PerkinElmer (Boston, MA). Other standard materials used throughout were from Sigma-Aldrich.
Cell culture and transfection
COS7 and HEK293T cells (American Type Culture Collection, Manassas, VA) were grown in DMEM supplemented with 10% FCS, penicillin, streptomycin, and l-glutamine as described (Gilboa et al., 2000; Hartung et al., 2006). Transient transfections of HEK293T or COS7 cells were carried out using TransIT-LT1 Mir2300 (Mirus, Madison, WI) according to the manufacturer’s instructions. Cells were assayed 24 h after transfection.
Plasmids
Expression vectors (in pcDNA1) encoding human BMPRII-SF, BMPRII-LF, and BMPRII-LF truncation mutants containing shorter segments of the extension unique to BMPRII-LF (all carrying an extracellular myc epitope tag) were described by us earlier (Nohe et al., 2002). Briefly, PCR mutagenesis was used to introduce stop codons at the indicated nucleotide position, with nucleotide numbers according to Kawabata et al. (1995) (TC6 at 2238, TC7 at 2946, and TC8 at 3064). The protein products are 746 (TC6), 982 (TC7), and 1021 amino acids (TC8), compared with 529–amino acid–long BMPRII-SF and 1038–amino acid–long BMPRII-LF. They were subcloned into pcDNA3 by PCR, followed by restriction digest and religation, and verified by sequencing. YFP-Smad1 in pEYFP-C1 was generated by in-frame fusion of enhanced YFP N-terminal to Smad1. The pcDNA3 plasmid was from Invitrogen (Waltham, MA), and β-Gal in pcDNA1 was a gift from H. F. Lodish (Whitehead Institute, Cambridge, MA).
Mutagenesis
The alanine substitution mutant of human myc-BMPRII (BMPRII-LF-AA) was generated by PCR using the QuikChange Mutagenesis Kit from Stratagene (Cedar Creek, TX), with the myc-BMPRII-LF plasmid serving as a template. The forward mutagenesis primer was 5’-AATTCCAGTCCTGATGAGCATGAGCCTGCTGCGAGACGAGAGCAACAAGCTGGCC-3’. The complementary sequence that served as the reverse primer was 5’-GGCCAGCTTGTTGCTCTCGTTCTCGCAGCAGGCTCATGCTCATCAGGACTGGAATT-3’. BMPRII-SFM, a BMPRII-SF construct containing a C-terminal extension of the last 99 nucleotides of the coding sequence of BMPRII-LF (nucleotides 4181–4279), was prepared by overlapping PCR using the mutagenesis-introducing primers 5’-CTATGCAGAACGAGCGCAGAAGGGCAGTTCATTC-CAAATCC-3’ and 5’-AATGAACTGCCCTTCTGCGCTCGTTCTGCATAGCAGTAGAC-3’. All constructs were verified by sequencing.
To generate GFP-C, a mutant of GFP extended at the 3’ coding region by addition of the 99-nucleotide BMPRII-LF–derived sequence, we used overlapping PCR using pEGFP-N1 (Clontech, Mountain View, CA) as a template and the following set of primers: GFP forward, TTTTCCTTTAGGATCCACCATGGTGAGCAAGGGCGAGGAG; GFP-C extension reverse, GGATTTGGAATGAACTGCCCTCTTGTACAGCTCGTCCATG; GFP-C extension forward, ATGGACGAGCTGTACAAGAGGGCAGTTCATTCCAAATCCAGCAC; and GFP-C reverse, TTTTCCTTTTGCGGCCGCTCACAGACAGTTCATTCCTATATC. To generate a similar wild-type (wt) GFP construct (without the extension), we used the same GFP forward primer, with the wt GFP–only reverse, TTTTAATTTTGCGGCCGCCTTACTTGTACAGCTCGTCC. Both GFP-C and GFP constructs were inserted into the BamH1-Not1 sites of pcDNA3 and validated by sequencing.
Immunoblotting
HEK293T or COS7 cells were transfected in suspension (1 μg DNA/600,000 cells) with vectors encoding myc-BMPRII-SF, TC6, TC7, TC8, BMPRII-LF, BMPRII-LF-AA, GFP, GFP-C, or pcDNA3 (mock) and seeded in six-well plates. After 24 h, cells were washed twice with cold PBS, and equal numbers of cells were lysed on ice (30 min) with lysis buffer (150 mM NaCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 0.5% IGEPAL CA-630, 1% Triton X-100, PIC, and 0.1 mM Na3VO4). After low-speed centrifugation to remove nuclei and cell debris, the lysates were subjected to SDS–PAGE (10% polyacrylamide), loading 300,000 cells/lane, followed by immunoblotting as described (Kfir et al., 2005). The blots were probed (12 h, 4°C) by primary antibodies (1:1000 anti-myc, 1:5000 anti-GFP, 1:50,000 anti-β-actin), followed by peroxidase-coupled GαM IgG (1:5000 for 1 h at 22°C). The bands were visualized by enhanced chemiluminescence (Western Bright; Advansta, Santa Monica, CA) and quantified by densitometry using TINA software (version 2.10 g; Raytest Isotopenmessgeraete, Straubenhardt, Germany).
Protein synthesis and protein degradation pulse-chase assays
Suspended HEK293T cells were transfected as described by myc-BMPRII-SF, TC6, TC7, TC8, BMPRII-LF, or pcDNA3 (mock) vectors and seeded in six-well plates. After 24 h, cells were washed twice, incubated (30 min, 37°C) in l-methionine– and l-cystine–free DMEM, and labeled for 25 min with [35S](Met+Cys) (70 μCi/well), followed by three washes with PBS. Degradation was measured by chasing (3–6 h, 37°C) in complete DMEM supplemented with 10% FCS. Cells were lysed on ice (45 min) with immunoprecipitation buffer (420 mM NaCl, 50 mM HEPES, 5 mM EDTA, 1% IGEPAL CA-630, 3 mM dithiothreitol, PIC [1:100], Phosphatase Inhibitor Cocktail 2 [1:100], and Phosphatase Inhibitor Cocktail 3 [1:100]). After low-speed centrifugation to remove nuclei and cell debris, lysates (600,000 cells for each lysed sample) were immunoprecipitated with 3 μg of mouse anti-myc antibody for 16 h at 4°C. Protein G–Sepharose beads were blocked in PBS containing 3% BSA for 16 h at 4°C, and 100 μl of beads was added to the cell lysates for 2 h at 4°C. The complexes were washed three times in immunoprecipitation buffer; bound material was solubilized in 60 μl of SDS sample buffer, subjected to SDS–PAGE (10% polyacrylamide), electrotransferred onto nitrocellulose, and subjected to autoradiography (Fluoro Image Analyzer FLA-5000; Fuji Photo Film, Tokyo, Japan).
Measurements of mRNA levels
Suspended HEK293T or COS7 cells were transfected as described by myc-BMPRII-SF, TC6, TC7, TC8, myc-BMPRII-LF, or pcDNA3 vectors and seeded in six-well plates. After 24 h, cells were washed twice with PBS and harvested, and their RNA was isolated using EZ-RNA total RNA isolation kit (Biological Industries). To remove DNA contaminations, the extracted RNA was isolated again using acidic phenol:chloroform:isoamyl-alcohol wash (125:24:1). After centrifugation at 7000 × g (3 min, 22°C), the RNA phase was purified using the RNeasy minikit (Qiagen, Valencia, CA). The purified RNA was incubated (30 min, 37°C) with RNase-free DNase I (New England BioLabs, Ipswich, MA), followed by inactivation of DNase I with 50 mM EDTA and incubation at 75°C for 10 min. The RNA was then converted to cDNA using Verso cDNA synthesis kit (Thermo Scientific, Waltham, MA). For the reverse transcriptase reaction, anchored oligo dT primers were added, and PCR was applied according to manufacturer’s instructions. The cDNA levels of each BMPRII construct were determined by PCR using the forward primer 5’-TGCCCGCTTTATAGTTGGAG-3’ and the reverse primer 5’-AGAATGAGCAAGACGGCAAG-3’. Results were normalized to GAPDH cDNA levels, determined by PCR using the forward primer 5’-TGAGCACCAGGTGGTCTCC-3’ and the reverse primer 5’-TAGCCAAATTCGTTGTCATACCAG-3’. PCR was conducted using Taq ready mix from Hy-Labs (Rehovot, Israel) according to the manufacturer’s recommendations. For each reaction, 200 ng of cDNA served as template. Quantification of band intensity in DNA gels was by densitometry using TINA software.
qRT-PCR was carried out with the Rotor Gene 6000 system (Corbett-Qiagen), using Absolute Blue SYBER Green ROX (Thermo Scientific) in duplicate. Nontemplate controls (NTCs) and quantitative standards (GAPDH) were included. Analysis was with the Rotor Gene 6000 system series software. The BMPRII forward primer was 5’-ATGACTTCCTCGCTGCAGCGG-3’, and the reverse primer was 5’-TCTGCGAAGCAGCCGC-3’. For GAPDH, the primers used were 5’-CGGAGTCAACGGATTTGGTC-3’ (forward) and 5’-GAATTTGCCATGGGTGGAAT-3’ (reverse).
Sucrose cushion enrichment of polysomal/rRNA fraction
HEK293T cells were transfected with myc-BMPRII-SF or -LF (3 × 10-cm plates/construct; 20 μg DNA/plate, calcium phosphate transfection). After 24 h, cells were treated with 100 μg/ml cycloheximide for 5 min. We subjected 10% of the cells to total RNA extraction using the EZ-RNA kit. We lysed 90% of the cells in 1.5 ml of lysis buffer (10 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 0.5 mM CaCl2, 130 mM KCl, and 250 mM sucrose, supplemented with 200 U of RNase inhibitor, 0.1 mg/ml heparin, 0.5% IGEPAL, and 0.5% sodium deoxycholate) for 10 min on ice. Cells were then centrifuged at 1800 × g (10 min, 4°C), and the supernatant was loaded on 10 ml of 40% sucrose cushion (in lysis buffer, without supplements). After centrifugation (25,000 × g, 17 h, 4°C), the pellet was resuspended with 110 μl of RNase- and DNase-free water. RNA pellet and total RNA were treated with 2 U of TURBO DNase (Ambion-Thermo Scientific, Waltham, MA; 37°C, 20 min) and purified from DNA residues with acidic phenol:chloroform, followed by RNeasy minikit (Qiagen). cDNA was generated with poly-dT primers using Moloney murine leukemia virus enzyme reverse transcriptase (Promega, Madison, WI) according to the manufacturer’s instructions. After reverse transcription, samples were digested overnight with DPN1 restriction enzyme to eliminate residual plasmid DNA.
Degradation measurements by CHX chase
Suspended HEK293T cells were transfected as described by myc-BMPRII-SF or myc-BMPRII-LF and seeded in 60-mm plates. After 24 h, cells were incubated with 300 μM CHX for 1–6 h at 37°C, lysed, and subjected to SDS–PAGE (loading lysates equivalent to 300,000 cells/lane) and immunoblotting.
Treatment with degradation inhibitors
COS7 or HEK293T cells were transfected in suspension (1 μg DNA/600,000 cells) by myc-BMPRII-SF, BMPRII-LF, or BMPRII-LF-AA and seeded in six-well plates. After 24 h, cells were either left untreated (control) or treated with 25 μM MG132 or 25 μg/ml chloroquine for 24 h at 37°C and subjected to SDS–PAGE and immunoblotting using anti-myc.
Measurement of cell surface expression by proteinase K treatment
Suspended HEK293T cells were transfected as described by myc-BMPRII-SF, TC6, TC7 myc-BMPRII-LF, or myc-BMPRII-LF-AA and seeded in 60-mm plates. After 24 h, cells were washed twice with cold PBS and harvested on ice by pipetting. The cells were pelleted at low speed and suspended in serum-free DMEM. Proteinase K (300 μg/ml; dissolved in 50 mM Tris, 10 mM CaCl2, pH 8) was added for 15 min at 4°C, whereas control cells were incubated in buffer only. The reaction was stopped with 5 mM PMSF (5 min, 4°C). After centrifugation (16,000 × g, 10 s, 4°C), the cells were washed in PBS containing 5 mM PMSF, lysed, and subjected to SDS–PAGE (loading 300,000 cells/lane) and immunoblotting.
Immunofluorescence labeling of myc-tagged BMPRII variants at the cell surface
COS7 or HEK293T cells grown on glass coverslips in six-well plates were transfected with 1 μg of DNA of myc-BMPRII-SF, myc-BMPRII-LF, or myc-BMPRII-LF-AA. After 24 h, cells were incubated (30 min, 37°C) in serum-free DMEM, washed with cold HBSS/HEPES/BSA (20 mM HEPES, pH 7.2, 2% BSA), blocked with normal goat γ-globulin (200 μg/ml, 30 min, 4°C), and labeled with anti-myc (20 μg/ml, 1 h, 4°C), followed by Alexa 546 GαM Fab′ (40 μg/ml, 30 min, 4°C), all in HBSS/HEPES/BSA. After washing at 4°C, cells were fixed with 4% paraformaldehyde in PBS (30 min, 22°C). Labeled slides were mounted with ProLong antifade reagent (Life Technologies). Fluorescence images were recorded with a CoolSNAP HQ-M camera (Photometrics, Tucson, AZ) using a 63×/1.4 numerical aperture oil immersion objective mounted on an AxioImager D.1 microscope (Carl Zeiss Microimaging, Jena, Germany).
Internalization measurements
COS7 or HEK293T cells were grown on glass coverslips in six-well plates and transfected using 1 μg of DNA of myc-BMPRII-SF, myc-BMPRII-LF, or myc-BMPRII-LF-AA per well. After 24 h, the internalization of the myc-tagged receptors was quantified by the point-confocal method, using a fluorescence recovery after photobleaching setup under nonbleaching illumination conditions as described (Ehrlich et al., 2001). Briefly, the cell surface receptors were labeled at 4°C as described for immunofluorescence labeling of myc-tagged receptors. After labeling, the cells were either fixed immediately with 4% paraformaldehyde (first at 4°C, then at 22°C; time 0) or incubated in HBSS/HEPES/BSA at 37°C for the indicated periods to allow endocytosis before fixation and mounting for immunofluorescence. Endocytosis was quantified by measuring the reduction in the fluorescence intensity levels at the plasma membrane, focusing the laser beam through the 63× objective at defined spots (1.86 μm2) in the focal plane of the plasma membrane away from vesicular staining and passing the fluorescence through a pinhole in the image plane to make it a true confocal measurement (Ehrlich et al., 2001). At each time point, at least 200 cells were measured.
Treatments affecting internalization
Endocytosis assays were conducted in HBSS/HEPES/BSA; all treatments were initiated by a 15-min preincubation (37°C) with the inhibitory drug/medium. The cells were kept under the inhibitory condition throughout the labeling and internalization measurement. Hypertonic treatment to disrupt the structure of clathrin-coated pits (Heuser and Anderson, 1989) was conducted in HBSS/HEPES/BSA supplemented with 0.45 M sucrose (Heuser and Anderson, 1989; Ehrlich et al., 2001). Nystatin treatment to inhibit caveolar endocytosis (Schnitzer et al., 1994; Di Guglielmo et al., 2003; Mitchell et al., 2004) used 25 μg/ml drug. Treatment with the clathrin inhibitor PitStop was at 30 μM (von Kleist et al., 2011).
Smad phosphorylation assay
COS7 cells were seeded and grown for 24 h in six-well plates. They were cotransfected with 1 μg of DNA of YFP-Smad1 and 1 μg of DNA of myc-BMPRII-LF, myc-BMPRII-LF-AA, myc-BMPRII-SF, or β-Gal (mock). At 24 h posttransfection, cells were starved for 3 h in serum-free DMEM and stimulated or not with 10 nM BMP2 for 30 min. Cells were lysed and subjected to SDS–PAGE (loading 300,000 cells/lane), followed by immunoblotting as described in Immunoblotting. The blots were probed by anti-pSmad1/5/8 (1:1000), anti-myc (0.6 μg/ml), or anti-GAPDH (1:20,000), followed by peroxidase-GαR or -GαM IgG (1:7500). Bands were visualized by enhanced chemiluminescence and quantified by densitometry.
Acknowledgments
This work was supported by grants from the Israel Science Foundation to Y.I.H. (148/13) and M.E. (1529/11). Y.I.H. is an incumbent of the Zalman Weinberg Chair in Cell Biology at the George S. Wise Faculty of Life Sciences, Tel Aviv University.
Abbreviations used:
- BMP
bone morphogenetic protein
- BMPRII
type II BMP receptor
- BMPRII-LF
BMPRII long form
- BMPRII-SF
BMPRII short form
- BSA
bovine serum albumin
- CME
clathrin-mediated endocytosis
- FCS
fetal calf serum
- GαM
goat anti-mouse
- GαR
goat anti-rabbit
- HBSS
Hanks’ balanced salt solution
- PAH
pulmonary arterial hypertension
- PIC
protease inhibitor cocktail
- TGF-β
transforming growth factor-β
- YFP
yellow fluorescent protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-08-0547) on January 6, 2016.
REFERENCES
- Alexander JM, Bikkal HA, Zervas NT, Laws ER, Jr, Klibanski A. Tumor-specific expression and alternate splicing of messenger ribonucleic acid encoding activin/transforming growth factor-b receptors in human pituitary adenomas. J Clin Endocrinol Metab. 1996;81:783–790. doi: 10.1210/jcem.81.2.8636304. [DOI] [PubMed] [Google Scholar]
- Arrick BA, Lee AL, Grendell RL, Derynck R. Inhibition of translation of transforming growth factor-b 3 mRNA by its 5’ untranslated region. Mol Cell Biol. 1991;11:4306–4313. doi: 10.1128/mcb.11.9.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MC, Nguyen PH, Davis BN, Ohoka N, Hayashi H, Du K, Lagna G, Hata A. A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol Cell Biol. 2007;27:5776–5789. doi: 10.1128/MCB.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Hou WH, Liu IH, Hsiao G, Huang SS, Huang JS. Inhibitors of clathrin-dependent endocytosis enhance TGFb signaling and responses. J Cell Sci. 2009;122:1863–1871. doi: 10.1242/jcs.038729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG. Endocytic regulation of TGF-b signaling. Cell Res. 2009;19:58–70. doi: 10.1038/cr.2008.315. [DOI] [PubMed] [Google Scholar]
- Cogan J, Austin E, Hedges L, Womack B, West J, Loyd J, Hamid R. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation. 2012;126:1907–1916. doi: 10.1161/CIRCULATIONAHA.112.106245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-b receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- Draper DE. Protein-RNA recognition. Annu Rev Biochem. 1995;64:593–620. doi: 10.1146/annurev.bi.64.070195.003113. [DOI] [PubMed] [Google Scholar]
- Dunmore BJ, Drake KM, Upton PD, Toshner MR, Aldred MA, Morrell NW. The lysosomal inhibitor, chloroquine, increases cell surface BMPR-II levels and restores BMP9 signalling in endothelial cells harbouring BMPR-II mutations. Hum Mol Genet. 2013;22:3667–3679. doi: 10.1093/hmg/ddt216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehata S, Yokoyama Y, Takahashi K, Miyazono K. Bi-directional roles of bone morphogenetic proteins in cancer: another molecular Jekyll and Hyde. Pathol Int. 2013;63:287–296. doi: 10.1111/pin.12067. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Gutman O, Knaus P, Henis YI. Oligomeric interactions of TGF-b and BMP receptors. FEBS Lett. 2012;586:1885–1896. doi: 10.1016/j.febslet.2012.01.040. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Shmuely A, Henis YI. A single internalization signal from the di-leucine family is critical for constitutive endocytosis of the type II TGF-b receptor. J Cell Sci. 2001;114:1777–1786. doi: 10.1242/jcs.114.9.1777. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in TGF-b signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol. 2003;162:1089–1098. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser D, Wakefield L, Phillips A. Independent regulation of transforming growth factor-b1 transcription and translation by glucose and platelet-derived growth factor. Am J Pathol. 2002;161:1039–1049. doi: 10.1016/s0002-9440(10)64265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frump AL, Lowery JW, Hamid R, Austin ED, de Caestecker M. Abnormal trafficking of endogenously expressed BMPR2 mutant allelic products in patients with heritable pulmonary arterial hypertension. PLoS One. 2013;8:e80319. doi: 10.1371/journal.pone.0080319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis YI, Knaus P. Bone morphogenetic protein receptor complexes on the surface of live cells: A new oligomerization mode for serine/threonine kinase receptors. Mol Biol Cell. 2000;11:1023–1035. doi: 10.1091/mbc.11.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa L, Wells RG, Lodish HF, Henis YI. Oligomeric structure of type I and type II transforming growth factor b receptors: homodimers form in the ER and persist at the plasma membrane. J Cell Biol. 1998;140:767–777. doi: 10.1083/jcb.140.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S, Henis YI, Knaus P. Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol. 2006;26:7791–7805. doi: 10.1128/MCB.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes S, Chawla A, Corvera S. TGF b receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158:1239–1249. doi: 10.1083/jcb.200204088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis YI, Moustakas A, Lin HY, Lodish HF. The types II and III transforming growth factor-b receptors form homo-oligomers. J Cell Biol. 1994;126:139–154. doi: 10.1083/jcb.126.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck AP. Structural studies of the TGF-bs and their receptors - insights into evolution of the TGF-b superfamily. FEBS Lett. 2012;586:1860–1870. doi: 10.1016/j.febslet.2012.05.028. [DOI] [PubMed] [Google Scholar]
- Hirschhorn T, Barizilay L, Smorodinsky NI, Ehrlich M. Differential regulation of Smad3 and of the type II transforming growth factor-b receptor in mitosis: Implications for signaling. PLoS One. 2012;7:e43459. doi: 10.1371/journal.pone.0043459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn T, di Clemente N, Amsalem AR, Pepinsky RB, Picard JY, Smorodinsky NI, Cate RL, Ehrlich M. Constitutive negative regulation in the processing of the anti-Mullerian hormone receptor II. J Cell Sci. 2015;128:1352–1364. doi: 10.1242/jcs.160143. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International PPH Consortium. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-b receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- Itoh S, ten Dijke P. Negative regulation of TGF-b receptor/Smad signal transduction. Curr Opin Cell Biol. 2007;19:176–184. doi: 10.1016/j.ceb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Hemnes AR, Perrien DS, Schuster M, Robinson LJ, Gladson S, Loibner H, Bai S, Blackwell TR, Tada Y, et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302:L474–484. doi: 10.1152/ajplung.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata M, Chytil A, Moses HL. Cloning of a novel type II serine/threonine kinase receptor through interaction with the type I transforming growth factor-b receptor. J Biol Chem. 1995;270:5625–5630. doi: 10.1074/jbc.270.10.5625. [DOI] [PubMed] [Google Scholar]
- Keren T, Roth MG, Henis YI. Internalization-competent influenza hemagglutinin mutants form complexes with clathrin-deficient multivalent AP-2 oligomers in live cells. J Biol Chem. 2001;276:28356–28363. doi: 10.1074/jbc.M102235200. [DOI] [PubMed] [Google Scholar]
- Kfir S, Ehrlich M, Goldshmid A, Liu X, Kloog Y, Henis YI. Pathway- and expression level-dependent effects of oncogenic N-Ras: p27Kip1 mislocalization by the Ral-GEF pathway and Erk-mediated interference with Smad signaling. Mol Cell Biol. 2005;25:8239–8250. doi: 10.1128/MCB.25.18.8239-8250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JD, Kang H, Larrivee B, Lee MY, Mettlen M, Schmid SL, Roman BL, Qyang Y, Eichmann A, Jin SW. Context-dependent proangiogenic function of bone morphogenetic protein signaling is mediated by disabled homolog 2. Dev Cell. 2012;23:441–448. doi: 10.1016/j.devcel.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Influences of mRNA secondary structure on initiation by eukaryotic ribosomes. Proc Natl Acad Sci USA. 1986;83:2850–2854. doi: 10.1073/pnas.83.9.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Hoeflich ST, Causing CG, Podkowa M, Zhao X, Wrana JL, Attisano L. Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 2004;23:4792–4801. doi: 10.1038/sj.emboj.7600418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyton PA, Beppu H, Pappas A, Martyn TM, Derwall M, Baron DM, Galdos R, Bloch DB, Bloch KD. Deletion of the sequence encoding the tail domain of the bone morphogenetic protein type 2 receptor reveals a bone morphogenetic protein 7-specific gain of function. PLoS One. 2013;8:e76947. doi: 10.1371/journal.pone.0076947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RD, Rudarakanchana N, Atkinson C, Flanagan JA, Harrison R, Morrell NW, Trembath RC. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2003;12:3277–3286. doi: 10.1093/hmg/ddg365. [DOI] [PubMed] [Google Scholar]
- Martinez-Glez V, Valencia M, Caparros-Martin JA, Aglan M, Temtamy S, Tenorio J, Pulido V, Lindert U, Rohrbach M, Eyre D, et al. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012;33:343–350. doi: 10.1002/humu.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- Mitchell H, Choudhury A, Pagano RE, Leof EB. Ligand-dependent and -independent TGF-b receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell. 2004;15:4166–4178. doi: 10.1091/mbc.E04-03-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- Nickel J, Sebald W, Groppe JC, Mueller TD. Intricacies of BMP receptor assembly. Cytokine Growth Factor Rev. 2009;20:367–377. doi: 10.1016/j.cytogfr.2009.10.022. [DOI] [PubMed] [Google Scholar]
- Nohe A, Hassel S, Ehrlich M, Neubauer F, Sebald W, Henis YI, Knaus P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem. 2002;277:5330–5338. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- Nohe A, Keating E, Knaus P, Petersen NO. Signal transduction of bone morphogenetic protein receptors. Cell Signal. 2004;16:291–299. doi: 10.1016/j.cellsig.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Penheiter SG, Mitchell H, Garamszegi N, Edens M, Dore JJ, Jr, Leof EB. Internalization-dependent and -independent requirements for transforming growth factor b receptor signaling via the Smad pathway. Mol Cell Biol. 2002;22:4750–4759. doi: 10.1128/MCB.22.13.4750-4759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Rouskin S, Ingolia NT, Han L, Phizicky EM, Weissman JS, Koller D. Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol Syst Biol. 2014;10:770. doi: 10.15252/msb.20145524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig BL, Imamura T, Okadome T, Cox GN, Yamashita H, ten Dijke P, Heldin CH, Miyazono K. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc Natl Acad Sci USA. 1995;92:7632–7636. doi: 10.1073/pnas.92.17.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–1525. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwappacher R, Weiske J, Heining E, Ezerski V, Marom B, Henis YI, Huber O, Knaus P. Novel crosstalk to BMP signalling: cGMP-dependent kinase I modulates BMP receptor and Smad activity. EMBO J. 2009;28:1537–1550. doi: 10.1038/emboj.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira KE, Gross A, Ehrlich M, Henis YI. Coated pit-mediated endocytosis of the type I transforming growth factor-b (TGF-b) receptor depends on a di-leucine family signal and is not required for signaling. J Biol Chem. 2012;287:26876–26889. doi: 10.1074/jbc.M112.362848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira KE, Hirschhorn T, Barzilay L, Smorodinsky NI, Henis YI, Ehrlich M. Dab2 inhibits the cholesterol-dependent activation of JNK by TGF-b. Mol Biol Cell. 2014;25:1620–1628. doi: 10.1091/mbc.E13-09-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-b signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Sieber C, Kopf J, Hiepen C, Knaus P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 2009;20:343–355. doi: 10.1016/j.cytogfr.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Sobolewski A, Rudarakanchana N, Upton PD, Yang J, Crilley TK, Trembath RC, Morrell NW. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: potential for rescue. Hum Mol Genet. 2008;17:3180–3190. doi: 10.1093/hmg/ddn214. [DOI] [PubMed] [Google Scholar]
- Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-b family. J Med Genet. 2000;37:741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Veksler-Lublinsky I, Gazit N, Kupiec M, Ruppin E, Ziv-Ukelson M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011;12:R110. doi: 10.1186/gb-2011-12-11-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umasankar PK, Sanker S, Thieman JR, Chakraborty S, Wendland B, Tsang M, Traub LM. Distinct and separable activities of the endocytic clathrin-coat components Fcho1/2 and AP-2 in developmental patterning. Nat Cell Biol. 2012;14:488–501. doi: 10.1038/ncb2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, Zhang Q, Ye J, Yan Z, Denduluri S, et al. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis. 2014;1:87–105. doi: 10.1016/j.gendis.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh A, Gendron P, Altman R, Brown JW, Case D, Gautheret D, Harvey SC, Leontis N, Westbrook J, Westhof E, et al. RNAML: a standard syntax for exchanging RNA information. RNA. 2002;8:707–717. doi: 10.1017/s1355838202028017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DG. RNA-binding proteins: a lesson in repression. J Neurosci. 2006;26:7135–7138. doi: 10.1523/JNEUROSCI.1795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- Xu P, Liu J, Derynck R. Post-translational regulation of TGF-beta receptor and Smad signaling. FEBS Lett. 2012;586:1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Ehrlich M, Henis YI, Leof EB. Transforming growth factor-b receptors interact with AP2 by direct binding to b2 subunit. Mol Biol Cell. 2002;13:4001–4012. doi: 10.1091/mbc.02-07-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M, Jacobson AB. Using reliability information to annotate RNA secondary structures. RNA. 1998;4:669–679. doi: 10.1017/s1355838298980116. [DOI] [PMC free article] [PubMed] [Google Scholar]