

Abstract
Transactive response DNA binding protein 43 (TDP‐43) proteinopathy is the major hallmark of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. It is also present in a subset of Alzheimer's disease cases. Recently, few reports showed TDP‐43 changes in cognitively normal elderly. In Caucasians, TDP‐43 proteinopathy independently correlate with cognitive decline. However, it is challenging to establish direct links between cognitive and/or neuropsychiatric symptoms and protein inclusions in neurodegenerative diseases because individual cognitive reserves modify the threshold for clinical disease expression. Cognitive reserve is influenced by demographic, environmental and genetic factors. We investigated the relationships between demographic, clinical and neuropathological variables and TDP‐43 proteinopathy in a large multiethnic sample of cognitively normal elderly. TDP‐43 proteinopathy was identified in 10.5%, independently associated with older age (P = 0.03) and Asian ethnicity (P = 0.002). Asians showed a higher prevalence of TDP‐43 proteinopathy than Caucasians, even after adjustment for sex, age, Braak stage and schooling (odds ratio = 3.50, confidence interval 1.41–8.69, P = 0.007). These findings suggested that Asian older adults may be protected from the clinical manifestation of brain TDP‐43 proteinopathy. Future studies are needed to identify possible race‐related protective factors against clinical expression of TDP‐43 proteinopathies.
Keywords: Asian, autopsy, cognitively normal elderly, dementia, postmortem, race, TDP‐43 proteinopathy
Introduction
Transactive response DNA binding protein 43 (TDP‐43) plays multiple roles in RNA processing, maturation and transport 11, 38. TDP‐43 protein is normally found in cell nuclei. However, in subjects with frontotemporal lobar degeneration with TDP‐43 inclusions (FTLD‐TDP), amyotrophic lateral sclerosis (ALS) and in a subset of Alzheimer's disease (AD) cases, ubiquitinated, phosphorylated and truncated forms of TDP‐43 aggregate in the cytoplasm and in the processes of neurons and glial cells 2, 13, 33, 51. In Caucasian subjects, TDP‐43 proteinopathy was found to independently associate with cognitive decline, and with episodic and working memory impairment 60. However, it is challenging to establish a direct link between cognitive and neuropsychiatric symptoms, and the protein inclusion burden in neurodegenerative diseases 49. A few studies of cognitively normal elderly subjects identified TDP‐43 proteinopathy in up to one‐third of the individuals included, especially in the amygdala 5, 25, 59. Resilience to neurodegenerative changes has been identified by several studies 1, 5, 7, 9, 15, 21, 25, 34, 36, 54. Evidence suggests that such individuals suffer a milder neuronal loss than symptomatic cases 3. In the case of TDP‐43, it is unclear why these inclusions were not associated with clinical symptoms. Demographic, environmental and genetic factors influence cognitive reserve in other neurodegenerative diseases 18, 23, 44. For example, higher education attainment is associated with a milder clinical manifestation of AD 6, 22.
Aiming to expand the understanding about TDP‐43 proteinopathy in cognitively healthy older adults, we took advantage of a large, population‐based and multiethnic sample of individuals from Brazil to conduct an extensive clinicopathological study investigating the associations between demographic, clinical and neuropathological variables and TDP‐43 proteinopathy.
Methods
Participants
All of the fully characterized 983 cases procured by the Brain Bank of the Brazilian Aging Brain Study Group (BBBABSG) at the University of São Paulo from February 2004 to December 2013 were eligible to participate in this study. The University of São Paulo research committee approved the study. Written informed consent for brain donation and provision of clinical information were obtained from the next‐of‐kin 20, 26, 27. The inclusion criteria were age >50 years at the time of death, normal cognitive function [according to the Clinical Dementia Rating scale (CDR)—informant section only 46 —and the Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) 32 ], the absence of sufficient neuropathological changes to fulfill the criteria for any neurodegenerative disease or vascular dementia and no family history of FTLD. Educational attainment was given by years of formal education. Race was self‐declared and confirmed by the interviewer by visual inspection. Race was classified as Caucasian, black/admixed and Asians.
Clinical evaluation
The subject's cognitive function status was obtained as a part of a validated semi‐structured interview administered by trained nurses to a knowledgeable informant who had at least weekly contact with the subject over the 6 months prior to death 20. Cognitive function was assessed using the CDR (informant section only) 46 and IQCODE 32. Participants with CDR = 0 and IQCODE ≤3.41 were considered cognitively normal. Neuropsychiatric symptoms were evaluated using the Neuropsychiatric Inventory (NPI) 17, 19. We considered both the total score and the individual item scores for delusions, hallucinations, agitation, dysphoria, anxiety, apathy, irritability, euphoria, disinhibition, aberrant motor behavior, nighttime disturbances and appetite and eating abnormalities. The interview also included demographic and socioeconomic status information. Autopsy service files were consulted to identify the place of birth and filiation of the participants.
Neuropathological evaluation
For BBBABSG cases, autopsies were performed within 20 h after death. The left hemisphere was fixed in 4% buffered formaldehyde. The right hemisphere was sectioned coronally and snap‐frozen. A neurodegeneration‐directed dissection was conducted, producing samples of the following regions: middle frontal gyrus, middle and superior temporal gyri, angular gyrus, superior anterior cingulate gyrus, visual cortex, hippocampal formation at the level of lateral geniculate body, amygdala, basal ganglia at the level of the anterior commissure, thalamus, midbrain, pons, medulla oblongata and cerebellum. Samples were embedded in paraffin and sectioned at 8 μm. All areas were stained with hematoxylin and eosin and selected sections were immunostained with antibodies against β‐amyloid (4G8, 1:10 000; Signet Pathology Systems, Dedham, MA, USA), phosphorylated tau (PHF‐1, 1:2000; gift of Peter Davies, NY, USA) and α‐synuclein after peroxidase K treatment of the tissue (EQV‐1, 1:10 000; gift of Kenji Ueda, Tokyo, Japan) (Table 1). We used internationally accepted neuropathological criteria and guidelines for neuropathological diagnosing and staging 8, 12, 40, 42, 47. AD‐type pathology was scored using the Braak and Braak staging system 8 and the Consortium to Establish a Registry for AD (CERAD) criteria 47. A neuropathological diagnosis of AD was made when individuals met the criteria for intermediate AD neuropathologic change, according to the latest National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of AD (NIA‐AA criteria) 45. Assessment of cerebrovascular changes was performed as described elsewhere 27. Hippocampal sclerosis was defined as severe loss of neurons and gliosis in the CA1 sector of the hippocampus fulfilling criteria for types 3 or 4 53. Acute ischemic/hypoxic changes or localized hippocampal infarcts were not criteria for diagnosis of hippocampal sclerosis.
Table 1.
Standard tissue staining protocol—Brain Bank of the Brazilian Aging Brain Study group and additional brain regions stained for transactive response DNA binding protein 43 (TDP‐43). Abbreviations: H&E = hematoxylin and eosin staining; phospho‐tau = hyperphosphorylated tau; TDP‐43 = hyperphosphorylated TDP‐43 (ser409/410); X = immunolabeled region
| Brain region | H&E | β‐Amyloid | Phospho‐tau | α‐Synuclein | TDP‐43 |
|---|---|---|---|---|---|
| Middle and inferior frontal gyri | X | X | X | X | |
| Middle and superior temporal gyri | X | X | X | X* | |
| Angular gyrus | X | X | X | X | |
| Calcarine cortex | X | X | X | X | |
| Cingulate gyrus and superior frontal gyrus (same block) | X | X | X | X | X† |
| Hippocampal formation at the level of lateral geniculate body | X | X | X | X | X* |
| Amygdala | X | X | X | X* | |
| Basal ganglia at the level of the anterior commissure | X | ||||
| Thalamus | X | X | |||
| Midbrain | X | X | X | X | X† |
| Pons | X | X | X | ||
| Medulla oblongata | X | X | |||
| Cerebellum | X | X | X | ||
| Olfactory bulb | X |
*All cases.†TDP‐43‐positive cases only.
Investigation of TDP‐43‐positive inclusions
In cases meeting criteria for this study, we used immunohistochemistry to detect phosphorylated TDP‐43 (mouse monoclonal MAb 409/410, 1:1000; Cosmo Bio, Tokyo, Japan) in the amygdala, hippocampal formation and inferior/middle temporal gyrus. In individuals showing abnormal deposits of phosphorylated TDP‐43 in at least one of these regions, three supplementary brain regions were immunostained for TDP‐43: the anterior cingulate gyrus, superior frontal gyrus (same block) and the superior colliculus. TDP‐43 positivity was stratified by region and by the type of inclusion. The rationale for choosing these regions of interest was based on the distribution of TDP‐43 proteinopathy in normal controls 5 and on a previous finding from our group showing that the superior colliculus was affected in all FTLD‐TDP cases 28. Positivity was considered when a TDP‐43 inclusion was visualized in any part of the region of interest, including the subpial zone 5, 25. TDP‐43 immunostaining was assessed by investigators who were blinded to the clinical, demographic and neuropathological data. CN read all the slides; ATDLA and RDR reviewed the cases; finally, LTG reviewed all of the positive cases and the few cases where there was disagreement between the first observers.
Statistical analysis
We investigated the association between TDP‐43 positivity and sociodemographic, clinical and neuropathological variables. TDP‐43, CERAD, Lewy bodies (LB), vascular lesions, hippocampal sclerosis and NPI were analyzed as binary outcomes. TDP‐43 was categorized as positive or negative, whereas CERAD was categorized as “absence of neuritic plaques” (CERAD = 0) or “presence of neuritic plaques” (CERAD > 0). LB, vascular lesions and hippocampal sclerosis were classified as “absence of lesions” or “presence of any lesion.” Braak staging was classified in three groups (0, I/II or ≥III—all CERAD ≤ scarce). NPI sub‐items were categorized as “altered” (NPI score ≥1) or “normal” (NPI score = 0) because NPI was a skewed variable in our sample. In the univariate analyses, unpaired t‐tests were used to evaluate the association between TDP‐43 positivity and continuous variables. One‐way analysis of variance was used to investigate racial group differences in age at death and years of education using the Bonferroni post‐hoc correction for two by two comparisons between groups. Chi‐square or Fisher's exact tests were used to determine the association between TDP‐43 and categorical variables. A multivariate logistic regression models adjusted for age, educational attainment, Braak stage and CERAD classification were used to investigate the association between TDP‐43 and ethnicity. In these multivariate analyses, Caucasian ethnicity was used as the reference. We also used multivariate logistic regression to examine the association between Braak stage and TDP‐43, adjusted for age, sex and years of education. The significance level was set at 0.05 for two‐tailed tests. Statistical analyses were performed using SAS 9.3 (Cary, NC, USA).
Results
Sociodemographic and clinical characteristics of participants
We analyzed 323 cognitively normal older adults in order to determine whether they showed TDP‐43 proteinopathy. The mean age at death was 69.9 ± 11.7 years, the mean education attainment was 5.1 ± 4.1 years and 52.3% were females. An analysis of ethnicity indicated that 61.6% were Caucasian, 26.9% were black or admixed and 11.5% were Asian.
Thirty‐four (10.5%) of the study subjects showed TDP‐43 proteinopathy. Univariate analyses showed that participants with TDP‐43 were older than those without TDP‐43 (74.0 ± 12.3 vs. 69.3 ± 11.4 years, respectively; P = 0.03; Table 2). Asians were more likely to show TDP‐43 proteinopathy than the other races (Asian: 9.3%; non‐Asian 29.4%, P = 0.002), although they (Asians: 72.7 ± 12.3) did not significantly differ in age at death from the other race groups (Caucasian: 70.2 ± 11.5 years, P = 0.66; Black/admixed: 67.9 ± 11.4, P = 0.10). Within the Asian group, subjects born in Japan and China were significantly more prone to exhibit TDP‐43 deposits (P = 0.04). Moreover, compared with other race groups, Asians had a higher proportion of females and showed higher education attainment (Table 3).
Table 2.
Characteristics of transactive response DNA binding protein 43 (TDP‐43)‐positive and ‐negative groups (n = 323). Abbreviations: CERAD = Consortium to Establish a Registry for AD; IQCODE = Informant Questionnaire on Cognitive Decline in the Elderly; NFT = neurofibrillary tangles; NPI = Neuropsychiatric Inventory; SD = standard deviation
| Characteristic | TDP‐negative (n = 289) | TDP‐positive (n = 34) | P |
|---|---|---|---|
| Sociodemographic | |||
| Age (years), mean (SD) | 69.4 (11.3) | 74.0 (13.3) | 0.03* |
| Sex, female (%) | 51.6 | 58.8 | 0.42† |
| Race (%) | |||
| Caucasian | 62.6 | 52.9 | 0.002‡ |
| Black and admixed | 28.0 | 17.7 | |
| Asian | 9.3 | 29.4 | |
| Place of birth of Asians (%) | |||
| Born in Brazil | 76.9 | 40.0 | 0.04‡ |
| Born in Japan/China | 23.1 | 60.0 | |
| Schooling (years), mean (SD) | 5.1 (4.1) | 5.3 (4.7) | 0.76* |
| Neuropathological lesions | |||
| Braak NFT stage (%) | |||
| 0 | 32.5 | 26.5 | 0.03‡ |
| I–II | 55 | 44.1 | |
| >III | 12.5 | 29.4 | |
| CERAD | |||
| 0 | 84.1 | 73.5 | 0.14‡ |
| ≠0 | 15.9 | 26.5 | |
| Lewy bodies (%) | |||
| 0 | 98.2 | 94.1 | 0.12‡ |
| ≥1 | 1.8 | 5.9 | |
| Vascular lesions (%) | 4.1 | 0 | 0.25‡ |
| Hippocampal sclerosis (%) | 2.8 | 8.8 | 0.16‡ |
| Clinical | |||
| IQCODE, mean (SD) | 3.00 (0.03) | 3.00 (0.02) | 0.96* |
| Neuropsychiatric symptoms | |||
| NPI ≥ 1 (%) | 48.3 | 38.2 | 0.26† |
| Delusions (%) | 2.1 | 2.9 | 1.00‡ |
| Hallucinations (%) | 2.8 | 5.9 | 0.60‡ |
| Dysphoria (%) | 15.3 | 11.8 | 0.62† |
| Anxiety (%) | 19.9 | 17.6 | 0.82† |
| Agitation/aggression (%) | 9.8 | 5.9 | 0.55† |
| Euphoria (%) | 1.3 | 5.9 | 0.18‡ |
| Disinhibition (%) | 2.4 | 2.9 | 1.00‡ |
| Irritability/lability (%) | 9.7 | 11.8 | 0.76† |
| Apathy (%) | 10.8 | 11.8 | 1.00† |
| Aberrant motor activity (%) | 3.1 | 5.9 | 0.61‡ |
| Nighttime behaviors (%) | 14.6 | 5.9 | 0.12† |
| Appetite/eating changes (%) | 26.4 | 11.8 | 0.09† |
*Unpaired t‐test.†Chi‐square test.‡Fisher's exact test.
Table 3.
Characteristics of transactive response DNA binding protein 43 (TDP‐43)‐positive and ‐negative groups (n = 323). Abbreviations: SD = standard deviation
| Characteristic | White (n = 199) | Black and admixed (n = 87) | Asian (n = 37) | P |
|---|---|---|---|---|
| Sociodemographic | ||||
| Age (years), mean (SD)* | 70.2 (11.5) | 67.9 (11.5) | 72.7 (12.3) | 0.09 |
| Sex, female (%)† | 54.8 | 41.4 | 64.9 | 0.03 |
| Schooling (years), mean (SD)‡ | 5.2 (4.2) | 4.1 (3.5) | 7.4 (4.3) | 0.0004 |
*Unpaired t‐test.†Chi‐square test.‡Kruskal–Wallis test.
Because the Asian group had a significantly higher educational attainment (7.5 ± 4.3 years) than the Caucasian (5.2 ± 4.2 years, P = 0.005) or black/admixed (4.1 ± 3.5 years, P < 0.0001) groups, we tested whether the association between TDP‐43 and race remained significant in a multivariate analysis adjusting for education, age, sex, Braak stage and CERAD classification. Asians still showed a significantly greater prevalence of TDP‐43 proteinopathy, even after adjustment for education and age [odds ratio (OR) = 3.54, confidence interval (CI) = 1.43–8.78, P = 0.006; Table 4 ]. The association between TDP‐43 proteinopathy and Asian race remained significant (OR = 3.27, 95% CI = 1.29–8.31, P = 0.01), even after adjustment for age, education Braak stage and CERAD classification (Table 4). TDP‐43‐negative and ‐positive groups did not differ in terms of their total NPI score (P = 0.20) or any NPI sub‐item (P > 0.05 for all). IQCODE scores were also similar between groups (P = 0.96).
Table 4.
Association between transactive response DNA binding protein 43 (TDP‐43) proteinopathy and race (n = 323). Abbreviations: OR = odds ratio; CI = confidence interval
| Model | Crude | Model 1* | Model 2† | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |
| Black and admixed | 0.74 (0.29–1.95) | 0.55 | 0.80 (0.30–2.10) | 0.64 | 0.78 (0.29–2.07) | 0.62 |
| Asian | 3.72 (1.56–8.91) | 0.003 | 3.54 (1.43–8.78) | 0.006 | 3.27 (1.29–8.31) | 0.01 |
Reference: Caucasian group.*Model 1: logistic regression adjusted for age and years of education.†Model 2: logistic regression adjusted for age, years of education, Braak and CERAD stage.
Neuropathological description of participants
The great majority of the cases, 202 subjects (66.2%) did not present AD neuropathologic changes (not ADRC) according to the current NIA‐AA criteria 45. Another 96 (31.5%) had low levels of AD‐type chances (low ADNC). In few cases, not enough data on diffuse plaques were available to discriminate between low and not ADRC categories. Very few showed overlapping disease: 2.2% presented LB restricted to the brainstem, 3.7% showed microvascular lesions and 3.4% had hippocampal sclerosis. Univariate analysis showed that abnormal TDP‐43 proteinopathy was associated with a higher Braak stage (P = 0.03; Table 1). However, this association was not significant in a multivariate analysis that adjusted for age, sex and education (Braak I–II: OR = 0.82, 95% CI = 0.33–2.05; Braak III: OR = 1.94, 95% CI = 0.63–5.98, P = 0.28). The TDP‐43‐negative and ‐positive groups were similar with respect to the presence of neuritic plaques, synucleinopathies (restricted to lower brainstem), vascular lesions and hippocampal sclerosis (P > 0.05 for all).
In the 34 participants who showed TDP‐43 proteinopathy, the most frequently affected region was the amygdala (n = 31, 91.2%), followed by the entorhinal cortex (n = 17, 50%), dentate gyrus (n = 12, 35.2%), subiculum (n = 8, 23.5%), the CA1 region of the hippocampus (n = 7, 20.5%) and the inferior/middle temporal gyrus (n = 6, 17.6%) (Figure 1). We occasionally found TDP‐43 proteinopathy in the anterior cingulate gyrus (n = 2, 5.8%) or the superior frontal gyrus or superior colliculus (n = 1, 2.9%). Table 5 shows the localization of the TDP‐43 proteinopathy identified in each of the positive cases. All types of pathological TDP‐43 proteinopathy were found in our sample (Figure 1). Neuronal cytoplasmic inclusions were present in 79.4% of the TDP‐43‐positive cases, dystrophic neurites in 58.8% and glial cytoplasmic inclusions in 52.9%. Intranuclear neuronal inclusions and TDP‐43 threads were found in five cases each.
Figure 1.
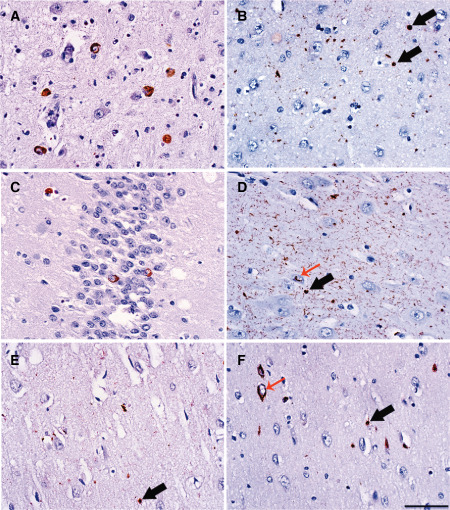
Examples of transactive response DNA binding protein 43 proteinopathy in vulnerable brain regions of older cognitively normal adults. A. Neuronal intracytoplasmic inclusions (NCIs) in amygdala. B. Glial intracytoplasmic inclusions (GCIs) in the entorhinal cortex (black arrows). C. NCIs in the dentate gyrus. D. Numerous short dystrophic neurites, GCIs (black arrow) and neuronal intranuclear inclusions cat's‐eye type (red arrow) in the subiculum. E. GCIs (black arrow) in the CA1. F. NCIs (red arrow) and GCIs (black arrow) in the temporal cortex. Scale bar: 50 μm.
Table 5.
Brain regions affected by transactive response DNA binding protein 43 (TDP‐43) proteinopathy in the positive cases (n = 34). Abbreviation: AMYG = Amygdala; CG = cingulate gyrus; DG = dentate gyrus; EC = entorhinal cortex; SC = superior colliculus; SFG = superior frontal gyrus; SUB = subiculum; TC = temporal cortex; X = TDP‐43 proteinopathy present
| Case | AMYG | EC | DG | SUB | CA1 | TC | CG | SFG | SC |
|---|---|---|---|---|---|---|---|---|---|
| 1 | X | X | X | X | X | X | |||
| 2 | X | X | X | ||||||
| 3 | X | X | |||||||
| 4 | X | X | |||||||
| 5 | X | ||||||||
| 6 | X | X | |||||||
| 7 | X | ||||||||
| 8 | X | ||||||||
| 9 | X | X | X | X | X | ||||
| 10 | X | X | |||||||
| 11 | X | ||||||||
| 12 | X | X | X | ||||||
| 13 | X | ||||||||
| 14 | X | X | |||||||
| 15 | X | X | X | ||||||
| 16 | X | X | X | ||||||
| 17 | X | X | X | X | X | X | X | X | X |
| 18 | X | ||||||||
| 19 | X | ||||||||
| 20 | X | ||||||||
| 21 | X | X | X | X | |||||
| 23 | X | X | X | ||||||
| 24 | X | X | X | ||||||
| 25 | X | X | X | X | |||||
| 26 | X | ||||||||
| 27 | X | X | X | X | X | X | |||
| 28 | X | X | X | ||||||
| 29 | X | X | X | ||||||
| 30 | X | ||||||||
| 31 | X | ||||||||
| 32 | X | ||||||||
| 33 | X | ||||||||
| 33 | X | X | X | ||||||
| 34 | X |
Discussion
We investigated pathological TDP‐43 proteinopathy in the brains of cognitively normal subjects sourced from a multiethnic sample, with a wide range of educational attainment. TDP‐43 proteinopathy was present in 10.5% of the study participants. Consistent with previous reports, TDP‐43 proteinopathy was associated with older age and the amygdala was the most commonly affected brain region. The main novel finding of this study was the higher prevalence of TDP‐43 proteinopathy in subjects of Asian ethnicity.
TDP‐43 protein is emerging as a comorbidity of neurodegenerative diseases. TDP‐43 proteinopathy is the most common pathological finding in FTLD 12. Moreover, a growing body of evidence demonstrates that TDP‐43 proteinopathy is present in a substantial percentage of patients with AD and chronic traumatic encephalopathy 33, 41. TDP‐43 proteinopathy is deemed to be pathological, but their role in neurodegenerative processes is still being debated. Because of the shortage of well‐structured population‐based clinicopathological series focusing on brain aging and neurodegenerative diseases, only a few studies have investigated TDP‐43 proteinopathy in cognitively normal elderly individuals 5, 25, 59. These studies focused mainly on highly educated Caucasian subjects with an average age of death of over 70 years. The study participants investigated in the present study were relatively younger, had a greater range of exposure to education 22 and came from an ethnically diverse city 55. The slightly lower prevalence of TDP‐43 proteinopathy observed in our series may reflect the lower age of the study subjects because TDP‐43 pathology is more prevalent in older individuals 5, 25.
To our knowledge, this is the first study to report ethnicity‐related differences in the prevalence of TDP‐43 proteinopathy. The population base from which the study subjects were drawn included all of the inhabitants of São Paulo, a city of 11 million people (5.6% of Brazilian population). Our study sample did not deviate significantly from census data regarding age, sex, race, years of schooling or socioeconomic status 20. The São Paulo population is highly ethnically mixed, with more than 90% of its ancestry derived from former African slaves and European immigrants. Moreover, the São Paulo region is unique within Brazil in that it received a large number of Japanese immigrants during the early 20th century and currently hosts the largest ethnic Japanese population outside Japan 30. In this study, participants of Asian descent were mostly born in Japan or were first‐generation Japanese immigrants, and a small minority came from China. The Brazilian law mandates that autopsies must be performed in all persons without a death certificate, which provides a large potential recruitment base for population‐based neuropathological studies, without bias towards individuals with dementia. All autopsies are conducted at the University of São Paulo; this significantly reduced the inter‐rater variation in both interviews and pathology assessments.
The higher prevalence of TDP‐43 proteinopathy in Asians is intriguing. A recent published abstract also suggested high prevalence of TDP‐43 proteinopathy in cognitive healthy older Japanese cohort 57. However, no other ethnical groups were included in this study for comparison. Unfortunately, we are not aware of other reports that we could use to compare our findings. In fact, cultural beliefs tend to limit autopsies in Asians 39. Several factors could explain the higher prevalence of TDP‐43 proteinopathy in the Asian individuals in our series. Higher educational attainment can protect against some neurodegenerative diseases, such as AD 6, 22. However, our results remained unchanged by multivariate analysis adjusted for education. This was not unexpected as a clinical study identified a similar prevalence of dementia in Asians who immigrated to Brazil before World War II and the rest of the Asian population 43. TDP‐43 proteinopathy is age‐dependent, but average age of death was similar among different racial groups in this study. Asians could have a higher tolerance for harboring TDP‐43 proteinopathy without cognitive consequences. In fact, epidemiological studies showed lower incidences of ALS and early onset FTLD in Asians, as compared with Caucasians 16, 31. Furthermore, as evidenced in AD, the correlation between brain proteinopathy and neuronal loss—the best predictor of cognitive decline—is not linear in a great proportion of individuals 3. Regarding TDP‐43 proteinopathy, it might be that this lack of correlation is even more pronounced in Asians. Ethnicity‐related genomic variant differences may explain our results. Autopsy‐proven, ethnicity‐related vulnerability to neurodegenerative changes in this same cohort have been demonstrated before. In a previous investigation we showed ancestry‐related differences in the susceptibility to β‐amyloid neuritic plaques 55. Immigrants who live in São Paulo show high levels of acculturation, including adopting a Brazilian diet 14. This presumably reduces the influence of environmental factors in the results. A genomic variant of the TMEM106B locus (rs1990622 [A]) was associated with TDP‐43 proteinopathy in older adults without FTLD. Genetic studies of genomic variants and its modifiers in ethnic groups of cognitively normal individuals with TDP‐43 proteinopathy may identify targets for the prevention of TDP‐43‐associated dementia.
One approach to testing whether Asians tolerate TDP‐43 proteinopathy more effectively than the other ethnicities would be to include participants with cognitive decline in the study; this could facilitate analysis of whether the impaired Caucasian group showed a higher prevalence of TDP‐43 proteinopathy than, for example, an impaired Asian group. Therefore, we plan to test this hypothesis in future studies.
Our neuropathological analyses corroborated the findings of other studies showing that the amygdala was the brain region most vulnerable to TDP‐43 pathology in cognitively normal subjects, followed by the hippocampus 5, 25. Preferential involvement of the amygdala by TDP‐43 pathology was also found in AD 33 and other neurodegenerative diseases 37. Curiously, this early involvement of the amygdala/hippocampus is similar to what is observed for AD and old age, but differs from the progression pattern hypothesized for TDP‐43 pathology spreading in FTLD/ALS 10. This suggests that TDP‐43 proteinopathy in older age and FTD may have different etiologies and possibly different risk factors. In one case (#17) only, we found TDP‐43 proteinopathy in all brain areas analyzed. Although this case could be interpreted as an asymptomatic FTLD‐TDP, the TDP‐43 burden was much higher in hippocampus and amygdala than in cortical areas, suggesting a spread pattern more similar to the one described for AD. The development of TDP‐43 pathology driven by tau‐positive neurofibrillary pathology could explain the preferential limbic pattern of TDP‐43 proteinopathy in non‐FTLD older adults. In our univariate analysis, inclusions were associated with neurofibrillary tangles. However, this association was lost after adjustment for age, sex and education.
A previous study suggested a stereotypical spread of TDP‐43 changes in older adults 25. Although we did not observe this in all cases, a high percentage of the subjects showed a pattern of pathology that was consistent with this scheme. Therefore, our findings supported both limbic 4, 29 and multisystemic 24 theories for the progression of TDP‐43 pathology. TDP‐43 proteinopathy in the hippocampus was associated with hippocampal sclerosis in another series 48, 52. In the present study, although individuals with TDP‐43 proteinopathy showed higher proportion of hippocampal sclerosis (8.8%) than TDP‐43‐negative cases (2.8%), this difference was not statistically significant. This may be due to small sample size of TDP‐43‐positive cases or lack of cognitively impaired subjects in our series. The small fraction of individuals without TDP‐43 proteinopathy who showed hippocampal sclerosis could be explained by the high incidence of vascular brain pathology in this series 27; it is possible that in such cases remote brain ischemia was the trigger. Also, Brazilian guidelines for the treatment of common illnesses such as diabetes could partially explain this result, because sulfonylurea usage has been associated with hippocampal sclerosis in older adults 50.
Abnormalities of the amygdala may manifest as neuropsychiatric symptoms. Although 91% of our TDP‐43‐positive cases showed TDP‐43 inclusions in the amygdala, TDP‐43‐positive and ‐negative groups showed similar levels of neuropsychiatric symptoms. The mild burden of TDP‐43 proteinopathy in positive cases or a lack of sensitivity of the NPI could explain this discrepancy.
This study was limited by the lack of antemortem clinical data. We attempted to compensate for this using trained interviewers to obtain information from informants who were close to the subjects using a semi‐structured protocol and validated questionnaires 20. The CDR informant scores have been demonstrated to show close agreement with clinician scores 58. In a previous study 20, we applied this informant‐based protocol to living patients and assessed its sensitivity and specificity using a diagnosis established by full assessment of the patients. This analysis found that the informant‐based approach showed 93.7% specificity for a diagnosis of normal cognition. Another limitation of the study was imposed by the small available sample size of older Asian adults, although the number of Asian individuals was relatively large when compared with other multiethnic series. Finally, neurodegenerative disease changes may be present in an asymmetric fashion, including TDP‐43 35, 56. In this study, we only had access to one hemisphere for neuropathological examination that could potentially result in a false‐negative result in some cases. We tried to minimize this limitation across the groups by focusing on the left hemisphere for the neuropathological analysis. The left hemisphere represents the dominant hemisphere in over 95% of the cases in all ethnic groups. In conclusion, this clinicopathological study of an ethnically diverse series of cognitively normal older adults indicated that those with Asian ancestry had a higher prevalence of TDP‐43 proteinopathy, as compared with Caucasian subjects. These findings should serve as a basis for future studies of the role of TDP‐43 proteinopathy in older individuals with normal cognitive function and provide a reminder that information obtained from one ethnic group may not always be generalizable across other groups.
Acknowledgments
We thank the families of the brain donors, the physicians and staff of the São Paulo Autopsy service for their unconditional support and the Brazilian Aging Brain Study Group for their assistance with data collection. Financial support was provided by grant from the São Paulo Research Foundation, Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grant number: 2011/19833‐7). RDR received a research grant from FAPESP (PhD fellowship, 2012/07526‐5) and from CAPES (99999.012331/2013‐09); RPL was supported by FAPESP award (2010/06521‐4); LTG was funded by institutional NIH grants (P50AG023501, P01AG019724 and R01 AG040311).
References
- 1. Adler CH, Connor DJ, Hentz JG, Sabbagh MN, Caviness JN, Shill HA et al (2010) Incidental Lewy body disease: clinical comparison to a control cohort. Mov Disord 25:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amador‐Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R et al (2007) TDP‐43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol 61:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrade‐Moraes CH, Oliveira‐Pinto AV, Castro‐Fonseca E, da Silva CG, Guimaraes DM, Szczupak D et al (2013) Cell number changes in Alzheimer's disease relate to dementia, not to plaques and tangles. Brain 136 (Pt 12):3738–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K et al (2009) Phosphorylated TDP‐43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol 117:125–136. [DOI] [PubMed] [Google Scholar]
- 5. Arnold SJ, Dugger BN, Beach TG (2013) TDP‐43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol 126:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bennett DA, Wilson RS, Schneider JA, Evans DA, Mendes de Leon CF, Arnold SE et al (2003) Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology 60:1909–1915. [DOI] [PubMed] [Google Scholar]
- 7. Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH (1994) Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one‐year autopsy population from a geriatric hospital. Cereb Cortex 4:138–150. [DOI] [PubMed] [Google Scholar]
- 8. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Braak E (1997) Frequency of stages of Alzheimer‐related lesions in different age categories. Neurobiol Aging 18:351–357. [DOI] [PubMed] [Google Scholar]
- 10. Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al (2013) Stages of pTDP‐43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buratti E, Baralle FE (2010) The multiple roles of TDP‐43 in pre‐mRNA processing and gene expression regulation. RNA Biol 7:420–429. [DOI] [PubMed] [Google Scholar]
- 12. Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ et al (2007) TDP‐43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cardoso MA, Hamada GS, de Souza JM, Tsugane S, Tokudome S (1997) Dietary patterns in Japanese migrants to southeastern Brazil and their descendants. J Epidemiol 7:198–204. [DOI] [PubMed] [Google Scholar]
- 15. Caselli RJ, Walker D, Sue L, Sabbagh M, Beach T (2010) Amyloid load in nondemented brains correlates with APOE e4. Neurosci Lett 473:168–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cronin S, Hardiman O, Traynor BJ (2007) Ethnic variation in the incidence of ALS: a systematic review. Neurology 68:1002–1007. [DOI] [PubMed] [Google Scholar]
- 17. Cummings JL (1997) The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology 48 (5 Suppl. 6):S10–S16. [DOI] [PubMed] [Google Scholar]
- 18. Cummings JL, Altman J (2005) Genotype, proteotype, phenotype relationships in neurodegenerative diseases. Highlights from the 21st Ipsen Foundation Alzheimer's Disease Symposium, September 13, 2004, Paris, France. Rev Neurol Dis 2:80–84. [PubMed] [Google Scholar]
- 19. Cummings JL, Mega M, Gray K, Rosenberg‐Thompson S, Carusi DA, Gornbein J (1994) The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 44:2308–2314. [DOI] [PubMed] [Google Scholar]
- 20. de Lucena Ferretti RE, Damim AE, Dozzi Brucki SM, Schafirovits Morillo L, Rilho Perroco T, Campora F et al (2010) Post‐mortem diagnosis of dementia by informant interview. Dement Neuropsychol 4:138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P et al (1992) Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 13:179–189. [DOI] [PubMed] [Google Scholar]
- 22. Farfel JM, Nitrini R, Suemoto CK, Grinberg LT, Ferretti RE, Leite RE et al (2013) Very low levels of education and cognitive reserve: a clinicopathologic study. Neurology 81:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Froehlich TE, Bogardus ST Jr, Inouye SK (2001) Dementia and race: are there differences between African Americans and Caucasians? J Am Geriatr Soc 49:477–484. [DOI] [PubMed] [Google Scholar]
- 24. Geser F, Martinez‐Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ et al (2009) Clinical and pathological continuum of multisystem TDP‐43 proteinopathies. Arch Neurol 66:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geser F, Robinson JL, Malunda JA, Xie SX, Clark CM, Kwong LK et al (2010) Pathological 43‐kDa transactivation response DNA‐binding protein in older adults with and without severe mental illness. Arch Neurol 67:1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grinberg LT, Ferretti RE, Farfel JM, Leite R, Pasqualucci CA, Rosemberg S et al (2007) Brain bank of the Brazilian aging brain study group—a milestone reached and more than 1,600 collected brains. Cell Tissue Bank 8:151–162. [DOI] [PubMed] [Google Scholar]
- 27. Grinberg LT, Nitrini R, Suemoto CK, Lucena Ferretti‐Rebustini RE, Leite RE, Farfel JM et al (2013) Prevalence of dementia subtypes in a developing country: a clinicopathological study. Clinics 68:1140–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grinberg LT, Rueb U, Heinsen H (2011) Brainstem: neglected locus in neurodegenerative diseases. Front Neurol 2:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC, Parisi JE (2008) Temporal lobar predominance of TDP‐43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 116:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. IBGE (2010) Síntese de Indicadores Sociais: Uma Análise das Condições de Vida da População Brasileira. Estudos e Pesquisas—Informação Demográfica e Socioeconômica: Rio de Janeiro, RJ, Brazil. [Google Scholar]
- 31. Ikejima C, Yasuno F, Mizukami K, Sasaki M, Tanimukai S, Asada T (2009) Prevalence and causes of early‐onset dementia in Japan: a population‐based study. Stroke 40:2709–2714. [DOI] [PubMed] [Google Scholar]
- 32. Jorm AF (1994) A short form of the Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE): development and cross‐validation. Psychol Med 24:145–153. [DOI] [PubMed] [Google Scholar]
- 33. Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR et al (2014) Staging TDP‐43 pathology in Alzheimer's disease. Acta Neuropathol 127:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Josephs KA, Whitwell JL, Parisi JE, Knopman DS, Boeve BF, Geda YE et al (2008) Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol Aging 29:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. King A, Bodi I, Nolan M, Troakes C, Al‐Sarraj S (2015) Assessment of the degree of asymmetry of pathological features in neurodegenerative diseases. What is the significance for brain banks? J Neural Transm doi: 10.1007/s00702-015-1410-8 (Advance online publication). [DOI] [PubMed] [Google Scholar]
- 36. Klos KJ, Ahlskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF et al (2006) Alpha‐synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology 66:1100–1102. [DOI] [PubMed] [Google Scholar]
- 37. Kovacs GG, Milenkovic I, Wohrer A, Hoftberger R, Gelpi E, Haberler C et al (2013) Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol 126:365–384. [DOI] [PubMed] [Google Scholar]
- 38. Lagier‐Tourenne C, Polymenidou M, Cleveland DW (2010) TDP‐43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19 (R1):R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lipson JG (1996) Nursing among diverse cultures. Come all peoples of the earth. Reflections 22:9–10. [PubMed] [Google Scholar]
- 40. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136 (Pt 1):43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA et al (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47:1113–1124. [DOI] [PubMed] [Google Scholar]
- 43. Meguro K, Meguro M, Caramelli P, Ishizaki J, Ambo H, Chubaci RY et al (2001) Elderly Japanese emigrants to Brazil before World War II: II. Prevalence of senile dementia. Int J Geriatr Psychiatry 16:775–779. [DOI] [PubMed] [Google Scholar]
- 44. Miles TP (2001) Dementia, race and education: a cautionary note for clinicians and researchers. J Am Geriatr Soc 49:490. [DOI] [PubMed] [Google Scholar]
- 45. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morris JC (1993) The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 47. Morris JC, Edland S, Clark C, Galasko D, Koss E, Mohs R et al (1993) The consortium to establish a registry for Alzheimer's disease (CERAD). Part IV. Rates of cognitive change in the longitudinal assessment of probable Alzheimer's disease. Neurology 43:2457–2465. [DOI] [PubMed] [Google Scholar]
- 48. Nag S, Yu L, Capuano AW, Wilson RS, Leurgans SE, Bennett DA, Schneider JA (2015) Hippocampal sclerosis and TDP‐43 pathology in aging and Alzheimer's disease. Ann Neurol 77:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nelson PT, Braak H, Markesbery WR (2009) Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 68:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nelson PT, Estus S, Abner EL, Parikh I, Malik M, Neltner JH et al (2014) ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol 127:825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 52. Probst A, Taylor KI, Tolnay M (2007) Hippocampal sclerosis dementia: a reappraisal. Acta Neuropathol 114:335–345. [DOI] [PubMed] [Google Scholar]
- 53. Rauramaa T, Pikkarainen M, Englund E, Ince PG, Jellinger K, Paetau A, Alafuzoff I (2013) Consensus recommendations on pathologic changes in the hippocampus: a postmortem multicenter inter‐rater study. J Neuropathol Exp Neurol 72:452–461. [DOI] [PubMed] [Google Scholar]
- 54. Sabbagh MN, Sandhu SS, Farlow MR, Vedders L, Shill HA, Caviness JN et al (2009) Correlation of clinical features with argyrophilic grains at autopsy. Alzheimer Dis Assoc Disord 23:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schlesinger D, Grinberg LT, Alba JG, Naslavsky MS, Licinio L, Farfel JM et al (2013) African ancestry protects against Alzheimer's disease‐related neuropathology. Mol Psychiatry 18:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stefanits H, Budka H, Kovacs GG (2012) Asymmetry of neurodegenerative disease‐related pathologies: a cautionary note. Acta Neuropathol 123:449–452. [DOI] [PubMed] [Google Scholar]
- 57. Uchino A, Takao M, Saito Y, Sumikura H, Nakano Y, Hatsuta H et al (2014) Incidence and extent of TDP‐43 proteinopathy in aging human brain. Brain Pathol 24 (Suppl. S1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Waite L, Grayson D, Jorm AF, Creasey H, Cullen J, Bennett H et al (1999) Informant‐based staging of dementia using the clinical dementia rating. Alzheimer Dis Assoc Disord 13:34–37. [DOI] [PubMed] [Google Scholar]
- 59. Wilson AC, Dugger BN, Dickson DW, Wang DS (2011) TDP‐43 in aging and Alzheimer's disease—a review. Int J Clin Exp Pathol 4:147–155. [PMC free article] [PubMed] [Google Scholar]
- 60. Wilson RS, Yu L, Trojanowski JQ, Chen EY, Boyle PA, Bennett DA, Schneider JA (2013) TDP‐43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 70:1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]