

Abstract
We have synthesized a novel derivative of Digitoxin, termed “MonoD”, which demonstrates cytotoxic effects in lung cancer cells with much higher potency as compared to Digitoxin. Our data show that within 1 h of MonoD treatment, H460 cells showed increased oxidative stress, increased formation of autophagic vacuoles, and increased expression of pro-autophagic markers Beclin-1 and LC3-II. Cells pretreated with MnTBAP, a superoxide scavenger not only lowered superoxide production, but also had lower levels of LC3-II and Beclin-1. Prolonged treatment with MonoD-induced apoptosis in lung cancer cells. We investigated MonoD-dependent regulation of Akt and Bcl2, proteins that are known regulators of both autophagy and apoptosis. Molecular and pharmacologic inhibitors of Bcl2 and Akt, when combined with MonoD, led to higher expression of LC3-II and Beclin-1 as compared to MonoD alone, suggesting a repressive effect for these proteins in MonoD-dependent autophagy. Pretreatment of cells with an autophagy inhibitor repressed the apoptotic potential of MonoD, confirming that early autophagic flux is important to drive apoptosis. Therapeutic entities such as MonoD that target multiple pathways such as autophagy and apoptosis may prove advantageous over current therapies that have unimodal basis for action and may drive sustained tumor regression, which is highly desirable.
Lung cancer is a leading cause of cancer mortality, accounting for approximately 28% of all cancer deaths, which is more than that from the next three common cancers (colon, breast, and prostate) combined. Almost 75% of lung cancers are of the non-small cell lung cancer (NSCLC) type, and exhibit intrinsic resistance to anticancer drugs, with limited response to platinum-based therapy (Manish Shanker et al., 2010; Jemal et al., 2011; Gatti et al., 2013). Furthermore, most molecular therapies including inhibitors of epidermal growth factor receptor lead to acquired resistance in NSCLC. Such challenges have prompted the evaluation of novel drugs that may be able to concomitantly target multiple mechanisms for sustained tumor regression.
Sensitization of cancers through apoptotic mechanisms represents the most successful approach to development of chemotherapeutic agents. However, cancer cells may either be intrinsically resistant to apoptotic signaling through canonical pathways or may acquire resistance through a variety of genetic and epigenetic alterations. This has led to evaluation of alternate pathways of cell death that can be targeted independently or concomitantly with apoptotic signaling mechanisms in an effort to elicit effective and sustained inhibition of cancer (Johnstone et al., 2002). From this perspective, autophagy has received much attention in recent years as a potent mechanism to overcoming cancer resistance. Autophagy is a self-digestive process wherein misfolded and aggregated proteins along with damaged organelles are sequestered by double-membraned vesicles termed autophagosomes, and delivered to the lysosome for subsequent degradation and recycling. Interestingly, the impact of autophagy on eventual cellular fate in the context of cancer is dichotomous, and has been a topic of intense debate (Hippert et al., 2006; Marx, 2006; Lerena et al., 2008; Altman and Rathmell, 2009; Bhutia et al., 2013). One school of thought suggests that since autophagy rids the cell of defective constituents and supplies the cell with nutrients through recycling (which tumors need), tumor cells may exploit autophagy in order to survive (Townsend et al., 2012). However, data in our study support the alternate mechanism of autophagy wherein sustained autophagic flux eventually leads to excessive self-degradation of cellular components that are essential for survival (such as mitochondria), eventually driving cell death. Recent studies have similarly emphasized the development of novel drugs that induce initial autophagy eventually leading to apoptotic cell death.
Digitoxin, a cardenolide used to treat congestive heart failure and atrial fibrillation, has demonstrated considerable anti-tumor efficacy in breast, brain and cervical cancers. However, concerns related to cardiotoxic side effects arising from to its narrow therapeutic index dampened investigative efforts of its cytotoxic potential. To alleviate this problem, we designed a novel set of cardenolide analogs that can mimic the anticancer effects of Digitoxin but at lower doses, thereby recapitulating the therapeutic benefits of Digitoxin signaling while overcoming Digitoxin-associated toxicity. Our preliminary study demonstrated potent anti-tumorigenic effects against several forms of cancer (Iyer et al., 2010). Of the synthesized analogs, the β-D-Digitoxose (henceforth abbreviated as MonoD) form was identified as having the greatest anti-tumor potential in our lung cancer model.
The present study assessed the role of autophagy in promoting tumor cell death in NSCLC NCI-H460 (hence referred to as H460) cell line by MonoD, and the signaling pathways associated with such autophagy-mediated cell death. Our study demonstrates that both Digitoxin and MonoD led to increased autophagic flux in H460 cells within 1 h of drug exposure. Prolonged exposure to the drugs potentiates apoptosis through the intrinsic apoptotic pathway. We document that combination therapy involving either Digitoxin or MonoD and small molecule activators of apoptosis creates greater autophagic flux, leading to accelerated cell death. To our knowledge, this is the first study that presents detailed mechanistic investigations of Digitoxin and its analogs in relation to autophagy and apoptosis in a lung cancer model.
Materials and Methods
Synthesis of MonoD
The β-D-Digitoxose (Mono D) form of Digitoxin was synthesized as described previously by O’Doherty et al. (Iyer et al., 2010).
Chemicals and reagents
Amino guanidine (AG), Thiazolyl blue tetrazolium bromide (MTT), Cisplatin, Rapamycin, Bafilomycin A1, Sodium dichromate (Na2Cr2O7.2H2O) (Cr[VI]), Pan-caspase inhibitor zVAD.FMK, and Akt-inhibitor LY294002 were obtained from Sigma-Aldrich (St Louis, MO). Mn(III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) and Bcl-2 inhibitor VI (ABT-737) were purchased from Calbiochem (La Jolla, CA). The oxidative probes, 4,5-Diaminofluorescein diacetate (DAF-2DA), Dichlorofluorescein (DCF) diacetate, Dihydroethidium (DHE), and the apoptosis dye Hoechst 33342 were from Molecular Probes (Eugene, OR). Antibodies for Beclin-1, p38 MAPK, GAPDH, p-Akt Ser 473, total-Akt, NF-κB, and peroxidase-labeled secondary rabbit and mouse antibodies were from Cell Signaling Technology. Antibody for superoxide dismutase (SOD) was procured from Sigma Aldrich. Antibodies for Bcl-2 and LC3 were from Santa Cruz Biotechnology and Novus Biologicals, respectively. Cyto-ID® autophagy detection kit was purchased from Enzo Life Sciences. Bicinchoninic acid (BCA) was obtained from Thermo Scientific and chemiluminescent substrate from Pierce (Supersignal® West Pico, Pierce).
Cell culture
Both human lung epithelial NCI-H460 cancer cell line and nontumorigenic human bronchial epithelial Beas-2B cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured as described earlier at in 5% CO2 environment at 37°C (Medan et al., 2012).
MTT assay
Cells (~2,500 cells per well) were plated in 96 well cell-culture microplates (Costar, USA) and incubated overnight in cell culture medium to allow them to adhere. Cells were then exposed to the appropriate concentration of drug or vehicle for up to 72 h. Cell viability/proliferation was evaluated by the MTT (Sigma-Aldrich, St. Louis, MT) assay. The absorbance of solubilized formazan was read at 570 nm using ELISA reader (Bio-TEK, Synergy-1).
ROS/RNS detection
Intracellular nitric oxide (NO), hydrogen peroxide (H2O2), and superoxide (·o2 −)− production was determined by fluorometric analysis using specific probes DAF, DCF, and DHE, respectively, as described earlier (Azad et al., 2013).
Apoptosis measurements
Sub-confluent (80%) densities of cells were treated with test drugs and incubated with 10 μg/ml Hoechst 33,342 nuclear stain for 30 min at 37°C. The apoptotic cells having condensed chromatin and/or fragmented nuclei were visualized and scored under the EVOS® FL Cell Imaging System. Apoptotic cells were quantified by creating a grid using ImageJ and using a cell counter.
Autophagy detection
Qualitative detection of autophagy was performed using Cyto-ID® autophagy detection kit. Approximately 1 × 105 cells were stained with 0.25 μl/ml dye and incubated at 37°C for 30 min. Cells were visualized under a fluorescence microscope and images were obtained. Background subtraction was performed using ImageJ with a 50-pixel rolling ball radius. Corrected total cell fluorescence was calculated by subtracting mean fluorescence of the background from the total integrated density of the field.
Western blotting
After specific treatments, H460 cells were incubated in lysis buffer containing 20 mM Tris–HCl (pH 7.5), 1% Triton X-100, 150 mM NaCl, 10% glycerol, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 100 mM phenylmethylsulfonyl fluoride (PMSF), and a protease inhibitor mixture for 20 min on ice. After precipitating insoluble debris by centrifugation at 14,000g for 15 min at 4°C, the supernatants were collected and assayed for protein content using bicinchoninic acid (BCA) method. Equal amount of protein per sample (40 μg) were resolved on a 10% SDS–PAGE and transferred onto a 0.45-μm nitrocellulose membrane (Pierce) using Trans-Blot® Turbo™ Transfer System from Bio-Rad. The transferred membranes were blocked for 1 h in 5% non-fat dry milk in TBST (0.05% Tween-20 in TBS) and incubated with the appropriate primary antibodies and horseradish peroxidase-conjugated isotype specific secondary antibodies. The immune complexes were detected by chemiluminescence imaged using myECL Imager from Thermo Fisher Scientific and quantified by imaging densitometry, using ImageJ. Mean densitometry data from independent experiments were normalized to the control where indicated.
Statistical analysis
The data represent mean ± SD from three or more independent experiments. Statistical analysis was performed by Student’s t-test at a significance level of P < 0.05 for all experiments for the indicated data sets.
Results
Synthesis and characterization of MonoD
MonoD is the β-D digitoxose analog of Digitoxin, which was synthesized using a palladium-based de-novo method that has been developed by our group as described previously (Iyer et al., 2010). Both Digitoxin and MonoD consist of identical aglycone regions, but vary in the sugar region. While Digitoxin contains a trisaccharide, β-D-Digitoxose (MonoD) has only one sugar unit that is modified such that hydroxyl groups are on the third and fourth carbon (Supplementary Fig. 1). Our preliminary assessment of growth inhibition in the normal Beas-2B cells and H460 cancer cells indicated that MonoD is much more efficacious on H460 cells as compared to Beas-2B cells (Supplementary Fig. 2). We therefore proceeded to assess the efficacy of MonoD in lung cancer cells and delineate intracellular pathways that drive its anti-tumor effects.
MonoD reduces cell viability/proliferation and induces autophagy in lung cancer cells
We first assessed for the cytostatic and autophagic effects of Digitoxin and MonoD on H460 cells. The cells were treated with varying doses of Digitoxin and MonoD over 24 h, and showed a dose-dependent decrease in viability/proliferation (Fig. 1A). Since we observed sustained effects of MonoD and Digitoxin over 72 h for 50 nM of drug, (Fig. 1B), we chose this dose of the drug for all subsequent experiments.
Fig. 1.
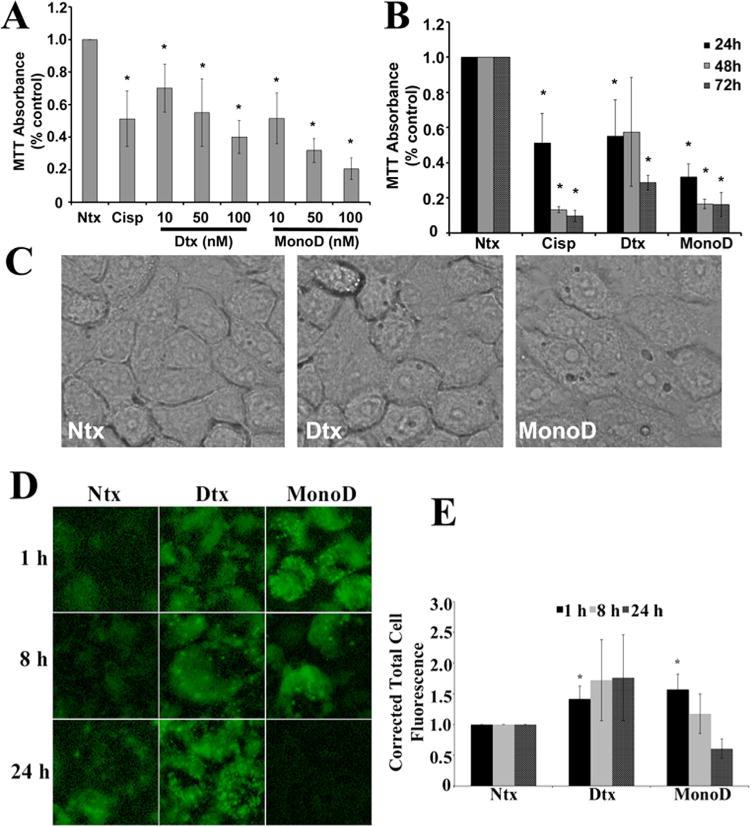
Digitoxin and MonoD reduce cell viability/proliferation and induce autophagy in H460 lung cancer cells. A: H460 Cells were treated with increasing concentrations of Digitoxin and MonoD (10, 50, and 100 nM) for 24 h and assessed for viability/proliferation using MTT (5 mg/ml). Cisplatin (300 μM) was used as a positive control. MTT absorbance at 570 nm was measured and normalized relative to no-drug control (referred to as “Ntx” here and henceforth in the figures). B: Time course of the MTT viability/proliferation assay performed over 72 h with 50 nM Digitoxin and MonoD. C: Cells treated for 1 h with 50 nM Digitoxin and MonoD and imaged using EVOS® FL Cell Imaging System under transmitted light at 40× resolution. D: Visualization of autophagic vacuole accumulation and flux performed using Cyto-ID®, a green fluorescent dye (0.25 μl/ml) that selectively labels the LC3-II protein in autophagic vacuoles imaged using EVOS® FL Cell Imaging System at 40× resolution. E: Autophagic vacuoles were quantified using ImageJ analysis software after performing background subtraction and plotted as Corrected Total Cell Fluorescence.
We next assessed for initiation of autophagy by assessing for the formation of autophagic vacuoles, which is indicative of the formation of the autophagosome. Phase-contrast microscopy clearly demonstrated formation of cytoplasmic vacuoles and numerous punctate structures in drug-treated cells in as little as 1 h; both the size and the number of vacuoles were much higher in MonoD-treated cells as compared to Digitoxin treatment (Fig. 1C). The hallmark of autophagy induction is the formation of cellular autophagosome punctae containing the microtubule-associated protein 1A/1B-light chain 3 (LC3). Translocation of LC3 protein from the cytosol (LC3-I form) to the autophagosome membrane (LC3-II form) is currently a standard method to monitor autophagy (Kabeya et al., 2000). Cyto-ID®, a green fluorescent dye that selectively labels the LC3-II protein in autophagic vacuoles, showed increased accumulation with both Digitoxin and MonoD treatment as compared to the untreated control—however, temporal differences were noted in MonoD- versus Digitoxin-treated cells (Fig. 1D). Maximal autophagic flux was observed within 1 h of MonoD treatment, which decreased at 8 h and disappeared at 24 h. On the other hand, cells treated with Digitoxin demonstrated a delayed onset of autophagic flux, but with relatively sustained levels of punctate staining observed even at 24 h (Fig. 1E).
Although H460 cells showed some basal level of autophagy, the autophagic flux increased significantly with MonoD treatment as indicated by the LC3-II band intensity (Fig. 2A and B), which correlated with the extent of autophagosome formation. Beclin1, a well-established mediator of autophagy that is recruited early in the autophagic cascade, showed upregulation with Digitoxin and MonoD (P < 0.05) treatment. Both NF-κB, a positive modulator of Beclin1 transcription and p38 MAPK, which has been reported to induce autophagy via Beclin-1 overexpression, (Cui et al., 2007; Kim et al., 2010) showed significant upregulation with MonoD treatment (P < 0.05) (Fig. 2C and D). Immunoblotting for key regulators of autophagy revealed that both Digitoxin and MonoD induced expression levels of Beclin 1, NF-κB, and p38 MAPK, which peaked around 1 h before reducing at 6 h (Fig. 2E). Therefore, in order to better understand the intracellular mechanism that drives Digitoxin- and MonoD-mediated early autophagy, we probed for autophagic proteins after 1 h of drug treatment. The dynamic nature of autophagic flux is evident from the decrease in LC3-II at 6 h as compared to 1 h with MonoD treatment. Furthermore, pre-treatment with the autophagy inhibitor Bafilomycin A1 causes accumulation of p62 SQSTM1 over time, indicating the presence of autophagic flux (Supplementary Fig. 3). All these results taken together strongly indicate that both Digitoxin and MonoD initiate autophagy in H460 cells.
Fig. 2.
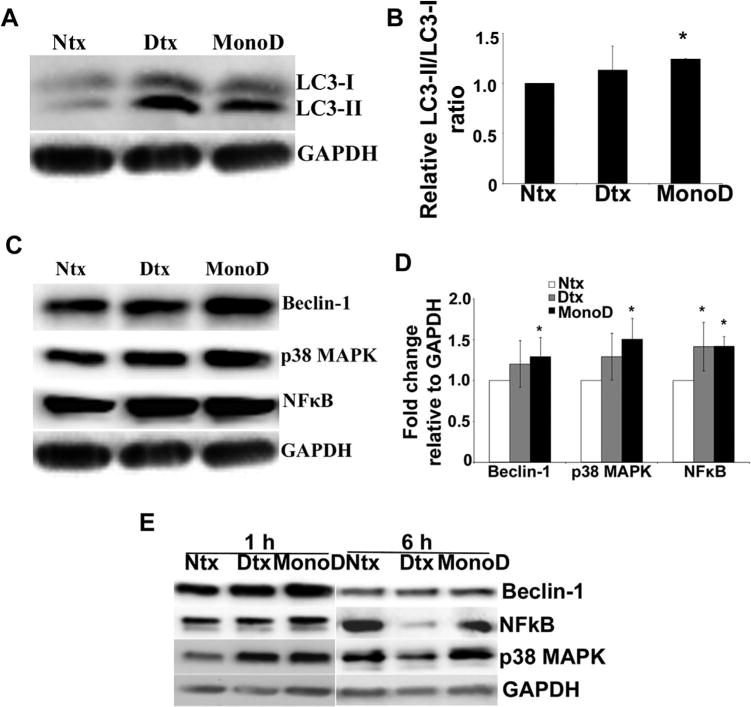
Digitoxin and MonoD treatment modulate levels of autophagy markers and autophagy regulatory proteins. A: H460 cells were treated with 50 nM Digitoxin and MonoD for 1 h, lysed and 40 μg lysate was separated on a 10% SDS-PAGE and assayed for autophagic marker LC3-II levels by immunoblot analysis. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. B: Densitometric quantification of LC3-II/LC3-I levels in response to drug treatment. C: H460 cells treated with 50 nM Digitoxin and MonoD for 1 h were lysed and assayed for autophagy regulatory proteins such as Beclin-1, NF-κB and p38 MAPK levels by immunoblot analysis. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. D: Beclin-1, NF-κB and p38 MAPK blots were quantified with densitometry using ImageJ. (*P < 0.05 for each drug treated data point as compared to non-treated control). E: H460 cells were treated with 50 nM Digitoxin and MonoD for 1 and 6 h, lysed and 40 μg lysate was separated on a 10% SDS–PAGE and assayed for autophagic marker Beclin-1, p38 MAPK, and NFkB levels by immunoblot analysis to demonstrate the temporal change in autophagy regulatory proteins. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples.
Digitoxin and MonoD modulate intracellular levels of reactive radicals
Reactive radicals, including reactive oxygen species (ROS) and reactive nitrogen species (RNS), serve as signaling molecules in a variety of cellular processes including autophagy (Azad et al., 2008; Tripathi et al., 2013). Therefore we assessed for cellular ROS-RNS levels in both Digitoxin- and MonoD-treated H460 cells. Both MonoD and Digitoxin induced a significant increase in •O2- (P < 0.01) as compared to control; moreover, the effect of MonoD on induction of superoxide radicals was significantly higher as compared to Digitoxin-treated cells (P < 0.05) (Fig. 3A). Changes in intracellular levels of nitric oxide (assayed using DAF-DA) and peroxide (assayed using DCF-DA) were not significant. We also observed a down-regulation of superoxide dismutase (SOD) upon treatment with Digitoxin and MonoD, which suggests an effect of these drugs on upstream modulators of reactive radicals (Fig. 3B).
Fig. 3.

Digitoxin and MonoD modulate intracellular levels of reactive species. A: Cells were treated with 50 nM Digitoxin and MonoD and assayed for nitric oxide, hydrogen peroxide, and superoxide production using DAF, DCF and DHE fluorescent probes. Cr(VI) (100 μM) was used as a positive control. Fluorescence intensity was recorded using a multi-mode fluorescence plate reader. Plots show relative fluorescence intensity over no drug treated control at the peak response time of 15 min. B: Cells treated with Digitoxin and MonoD were lysed and lysates separated on a SDS–PAGE and probed for SOD. C: Cells were pre-treated with 300 μM aminoguanidine for 1 h prior to treatment with 50 nM Digitoxin and MonoD. Superoxide production was assayed using DHE (Fig. 2D) and fluorescence intensity was measured at the peak response time of 15 min post-drug treatment. D: Cells were pre-treated with 300 μM aminoguanidine for 1 h prior to treatment with 50 nM Digitoxin and MonoD. Following this, cells were treated with 50 nM Digitoxin and MonoD for 1 h and lysed and assayed for autophagy regulatory proteins such as Beclin-1, NF-κB, and p38 MAPK levels by immunoblot analysis. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. E: Cells pre-treated for 1 h with 300 μM aminoguanidine prior to 1 h treatment with 50 nM Digitoxin and MonoD were analyzed for autophagosome formation by Cyto-ID®. Rapamycin (2 nM) was used as a positive control. Following treatment with test drugs, cells were incubated with Cyto-ID® for 30 min at 37°C and intracellular Cyto-ID® fluorescence imaged using EVOS® FL Cell Imaging System at 40× resolution. F: Autophagic vacuoles were quantified using ImageJ after performing background subtraction and plotted as Corrected Total Cell Fluorescence. (*P < 0.05 for each drug treated data point as compared to non-treated control; #P < 0.05 versus Digitoxin- and MonoD-treated datasets).
The potential role of ROS and RNS in Digitoxin- and MonoD-induced autophagy was further examined by evaluating the effects in the presence of ROS/RNS modulators. NO synthase inhibitor aminoguanidine (AG) drove significant scavenging of drug-induced •O2- (Fig. 3C) and NO (Supplementary Fig. 4). In order to assess whether changes in reactive radicals affected intracellular mediators of autophagy, we investigated the potential regulation of autophagic proteins by NO (Fig. 3D). Surprisingly, short-term modulation of NO had no significant effects on the expression levels of Beclin-1, NF-κB, or p38 MAPK. Furthermore, qualitative analysis of punctate containing LC3-II using Cyto-ID® staining showed no significant effect on vacuole formation mediated by NO modulation (through AG) as compared to the control groups (Fig. 3E and F). These observations indicate that although Digitoxin and MonoD induce both oxidative and nitrosative stress, the autophagic effects of MonoD may not be mediated directly through NO.
Regulation of autophagic flux and vacuole formation in response to MonoD is mediated by superoxide
Since we did not observe any change in autophagic flux in response to NO modulators, we wanted to establish whether superoxide played a predominant role in mediating autophagy induced by Digitoxin and MonoD. We therefore tested for drug-mediated ROS generation in the presence of MnTBAP, a superoxide dismutase mimetic and •O2-scavenger, which completely inhibited •O2- production (P < 0.01) in the presence of both Digitoxin and MonoD (Fig. 4A). H460 cells that were pretreated with MnTBAP also showed complete inhibition of LC3-II levels and greater accumulation of LC3-I. Such LC3 localization to the cytosolic fraction (LC3-I) confirmed that pretreatment with MnTBAP led to an inhibition of Digitoxin- and MonoD-induced autophagy (Fig. 4B). Further, the increase in levels of Beclin-1, NF-κB, and p38 MAPK that was observed with MonoD treatment was countered significantly (P < 0.05) in cells pretreated with MnTBAP (Fig. 4C and D). MnTBAP-mediated inhibition of MonoD-induced autophagy was further confirmed qualitatively using Cyto-ID® staining. Cells pretreated with MnTBAP showed reduced translocation of LC3 to the autophagosome membrane when treated with the positive control rapamycin (P < 0.05) and MonoD (P = 0.06) as compared to the drug treated controls (Fig. 4E and F). These data suggest that MonoD-induced autophagy in H460 cells is mediated by the production of •O2-.
Fig. 4.
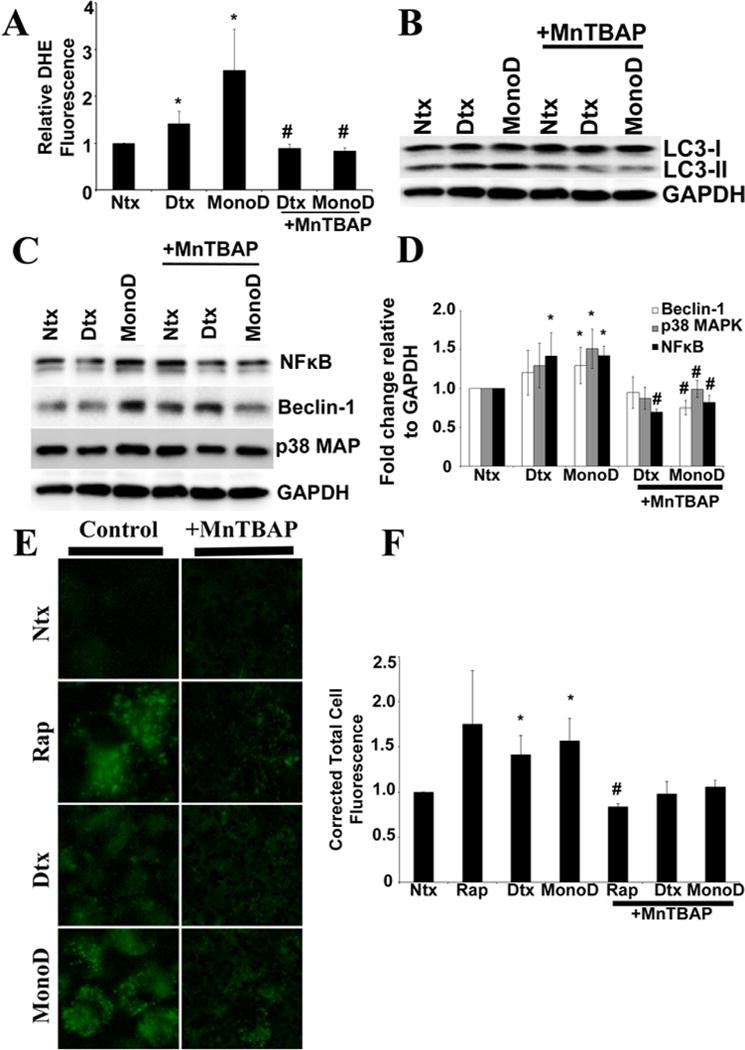
Regulation of autophagic flux and vacuole formation in response to MonoD is mediated by superoxide. A: Cells were pre-treated with 100 μM MnTBAP for 1 h prior to treatment with 50 nM Digitoxin and MonoD. Superoxide production was assayed using DHE fluorescence intensity, respectively, at the peak response time of 15 min post-drug treatment. B: Cells pretreated for 1 h with MnTBAP (100 μM) were treated with 50 nM Digitoxin and MonoD for 1 h and lysed and assayed for detection of autophagy marker LC3-II levels. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. C: Lysate obtained from cells treated for 1 h with 50 nM Digitoxin and MonoD following 1 h pre-treatment with 100 μM MnTBAP was assayed for autophagy regulatory proteins Beclin-1, NFkB, and p38 MAPK using immunoblotting. D: Beclin-1, NF-κB, and p38 MAPK blots were quantified with densitometry using ImageJ. E: Cells pretreated for 1 h with MnTBAP (100 μM) were treated with 50 nM Digitoxin and MonoD for 1 h and assayed for intracellular autophagic vacuole formation using Cyto-ID®. Following treatment with test drugs, cells were incubated with Cyto-ID® for 30 min at 37°C and intracellular Cyto-ID® fluorescence imaged using EVOS® FL Cell Imaging System at 40× resolution. F: Autophagic vacuoles were quantified using ImageJ after performing background subtraction and plotted as Corrected Total Cell Fluorescence. (*P < 0.05 for each drug treated data point as compared to non-treated control; #P < 0.05 versus Digitoxin- and MonoD-treated datasets).
Bcl-2 inhibits MonoD-induced autophagy
Given that Digitoxin and MonoD treatment in H460 cells reduced overall cell viability/proliferation, we assessed the effect of MonoD and Digitoxin on apoptosis in order to evaluate whether the early autophagic flux observed had any effect on cell death. Prolonged treatment with Digitoxin and MonoD-induced apoptosis at 8 h in a dose-dependent manner (Fig. 5A and B). We then assessed for possible mechanistic crosstalk between autophagy and apoptosis to identify putative regulatory proteins that may direct this interaction. Beclin-1, a key modulator of MonoD-induced autophagy in our cellular model is regulated via its inhibitory interaction with pro-survival factor Bcl-2 (Pattingre et al., 2005). Additionally, Beclin-1 has been shown to be a target of protein kinase Akt, where Akt mediated Beclin-1 phosphorylation inhibited autophagy and increased oncogenic transformation (Wang et al., 2012).
Fig. 5.
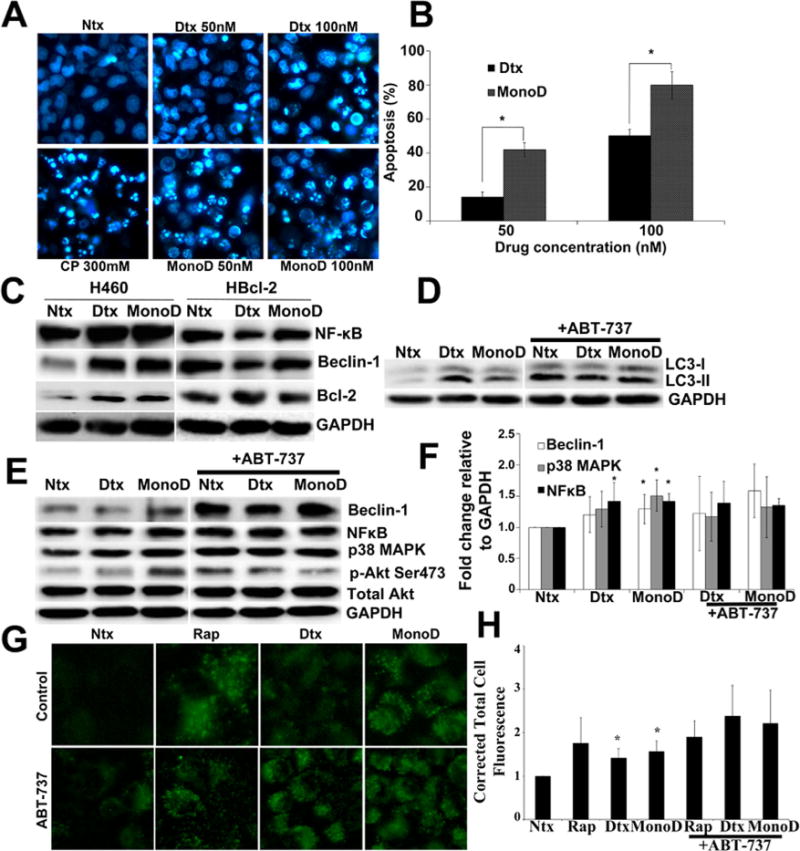
Prolonged Digitoxin and MonoD treatment eventually leads to apoptosis and Bcl-2 inhibits autophagic flux in H460 cells. A: Digitoxin and MonoD activate apoptosis in H460 cells which can be inhibited using a pan-caspase inhibitor zVAD-FMK. Cells were treated with respective drugs for 8 h following 1 h pre-treatment with 20 μM zVAD-FMK. Cisplatin was used as a positive control. Following treatment, cells were incubated at 37°C with 5 μM Hoechst 33,342 for 30 min and imaged using EVOS® FL Cell Imaging System at 40× resolution. B: Apoptosis quantification was done using ImageJ by creating a grid and initializing a cell counter and counting the apoptotic and live cells. C: To understand the regulatory effect of Bcl-2 on autophagy regulatory proteins, H460 cells overexpressing Bcl-2 (H-Bcl2) were treated for 1 h with 50 nM Digitoxin and MonoD in parallel with H460 cells. Lysates were obtained and probed for autophagy regulatory proteins Beclin-1, NFkB, and p38 MAPK. Blots were reprobed for GAPDH to confirm equal loading. D: Cells were pretreated for 1 h with Bcl-2 inhibitor ABT-737 (1 μM), followed by 50 nM Digitoxin and MonoD for 1 h. Forty micro gram cell lysate was separated on a 10% SDS–PAGE followed by immunoblotting and probed for autophagy marker LC3-II. E: Lysates obtained as described in Figure 4D were probed for autophagy regulatory proteins Beclin-1, NF-κB, and p38 MAPK. F: Blots were quantified using ImageJ after normalizing to GAPDH as a loading control. G: Cells were pretreated for 1 h with Bcl-2 inhibitor ABT-737 (1 μM), followed by 50 nM Digitoxin and MonoD for 1 h. Autophagic fluorescence was visualized using Cyto-ID® autophagy detection kit. H: Autophagic vacuoles were quantified for corrected total cell fluorescence as described earlier. (*P < 0.05 versus Digitoxin- and MonoD-treated datasets).
In order to clarify the role played by Bcl-2 in mediating Digitoxin- and MonoD-induced regulation of autophagic proteins, we tested the effect of these drugs on a previously generated H460 cell line that has stable overexpression of Bcl-2 (H-Bcl2) (Azad et al., 2006). Treatment with both Digitoxin and MonoD failed to induce up-regulation of autophagy regulators Beclin-1 and NF-κB in H-Bcl2 as compared to H460 cells (Fig. 5C). This result validated the antagonistic role of Bcl2 in driving Beclin-1-dependent autophagy in response to Digitoxin and MonoD in H460 cells.
We also assessed for the importance of Bcl-2 activity in MonoD-induced autophagy by pretreating H460 cells with ABT-737, a BH3 mimetic that selectively inhibits Bcl-2. Cells treated with MonoD in the presence of Bcl-2 inhibitor showed higher accumulation of autophagosome membrane bound LC3-II as compared to the cells treated with MonoD alone (Fig. 5D). In addition to the increased accumulation of autophagy marker LC3-II, Bcl-2 inhibition in combination with MonoD resulted in an increased expression of Beclin-1, though the increase was not significant (Fig. 5E and F). Microscopic evaluation of the cells treated with Digitoxin and MonoD with and without Bcl-2 inhibitor exhibited greater uptake of Cyto-ID® when pretreated with the inhibitor, indicating the increased presence of autophagosomal moieties (Fig. 5G and H). The increase in expression of autophagy regulatory proteins and fluorescent intensity of cells in the ABT-737-pretreated group were not found to be significant due to the loss of membrane integrity of these cells in as little as 1 h, suggesting a highly accelerated rate of activation of proteins in the autophagic cascade that pushed the cells rapidly towards apoptosis (Supplementary Fig. 5).
Akt inhibits MonoD-induced autophagy
The early autophagic flux and induction of apoptosis with prolonged exposure to drug seems to suggest that autophagy may play a pro-death role in our system. Akt has been shown to inhibit autophagy and drive oncogenic transformation (Wang et al., 2012). Therefore, we assessed for the role of Akt in MonoD-induced autophagy in our cellular model by using LY294002, a highly selective reversible inhibitor of PI3 kinase. Pretreatment of cells with 20 μM LY294002 increased LC3-II levels as compared to cells treated with MonoD alone (Fig. 6A). Additionally, co-treatment of MonoD with LY294002 further increased the expression level of autophagy regulator Beclin-1, though the increase was not significant (Fig. 6B and C). A statistically significant increase in the Cyto-ID® staining in the cells treated with MonoD in the presence of LY294002 (P < 0.01) as compared to MonoD alone (P < 0.05) was also observed (Fig. 6D and E). Cells treated concomitantly with Akt inhibitor in the presence of Digitoxin and MonoD had vacuoles that appeared much larger in size than cells treated with test drugs alone (Supplementary Fig. 5). Overall, the data suggested anti-autophagic role for Akt, driven by inhibition of Beclin-1 and other autophagic mediators.
Fig. 6.
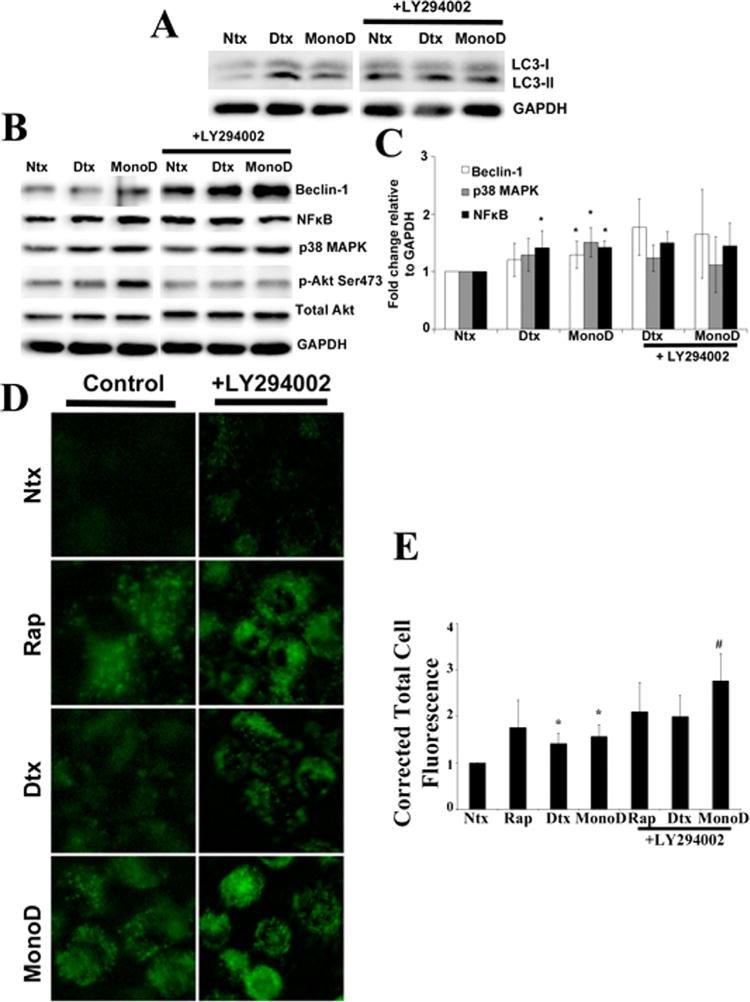
Akt inhibits MonoD-induced autophagy. A: Cells were pretreated for 1 h with Akt inhibitor LY294002 (20 μM), followed by 50 nM Digitoxin and MonoD for 1 h. Forty micro gram cell lysate was separated on a 10% SDS–PAGE followed by immunoblotting and probed for autophagy marker LC3-II. Please note the control lanes Ntx,Dtx, and MonoD were a part of a combined experiment with Bcl2-inhibitor and are the same as in Figure 4E. B: Lysates obtained as described in Figure 5A were probed for autophagy regulatory proteins Beclin-1, NFkB, and p38 MAPK and quantified as described earlier. Please note the control lanes Ntx, Dtx, and MonoD were a part of a combined experiment with Bcl2-inhibitor and are the same as in Figure 4F. C: Beclin-1, NF-κB, and p38 MAPK blots were quantified with densitometry using ImageJ. D: Cells were pretreated for 1 h with Akt inhibitor LY294002 (20 μM), followed by 50 nM Digitoxin and MonoD for 1 h. Autophagic fluorescence was visualized using Cyto-ID1 autophagy detection kit. E: Autophagic vacuoles were quantified using ImageJ analysis software after performing background subtraction and plotted as Corrected Total Cell Fluorescence. (*P < 0.05 versus untreated control; #P < 0.05 versus MonoD-treated dataset)
Crosstalk between MonoD-induced autophagy and apoptosis
We finally wanted to address whether the early autophagic flux induced by both MonoD and Digitoxin had a direct influence on the apoptosis observed after prolonged exposure to drug (Fig. 5A). We examined the effect of MonoD and Digitoxin on induction of apoptosis in the presence of both Rapamycin and Bafilomycin A1 (Fig. 7A). Bafilomycin A1 inhibits autophagy by preventing the maturation of autophagic vacuoles by interfering with the fusion of autophagosomes with the lysosomes (Yamamoto et al., 1998). Cells pretreated with Rapamycin showed increased sensitivity to apoptosis, whereas pretreatment with Bafilomycin A1 significantly decreased the overall apoptosis in MonoD treated cells (Fig. 7B). This data suggests a clear relation between autophagy and apoptosis, and posits a pro-death role for autophagy wherein early autophagic flux driving cells to undergo apoptosis in response to MonoD and Digitoxin.
Fig. 7.
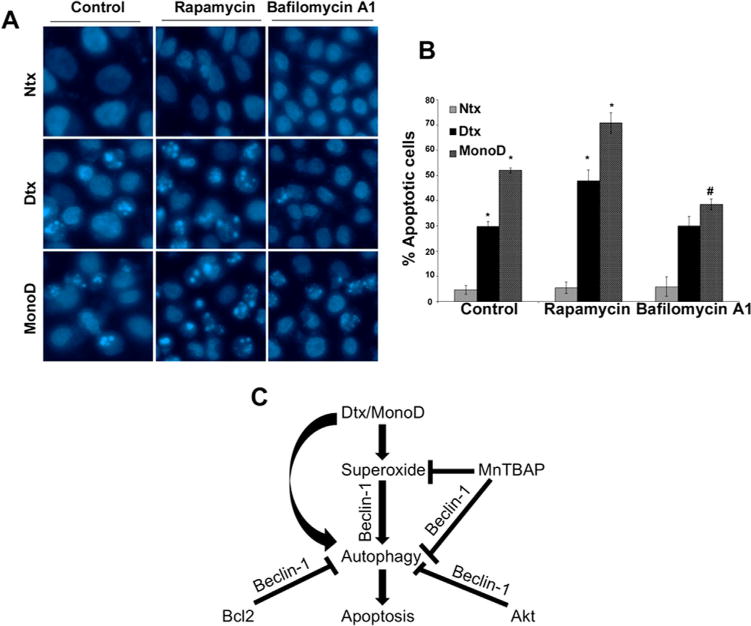
Schematic of Digitoxin and MonoD-induced autophagy. A: To assess the interplay between autophagy and apoptosis, cells were pretreated for 1 h with 2 μM Rapamycin, an autophagy inducer and 10 nM Bafilomycin A1, an autophagy inhibitor. Following this, the cells were treated with Digitoxin and MonoD for 24 h and assessed for apoptosis by staining with 5 μM Hoechst 33342. (*P < 0.05 versus untreated control; #P < 0.05 versus MonoD-treated dataset). B: Quantification of the apoptosis data. C: Digitoxin- and MonoD-driven autophagy is regulated through superoxide, which is mediated via the up-regulation of Beclin-1. MnTBAP scavenges superoxide, which leads to Beclin-1 down-regulation resulting in inhibition of autophagic response. Pro-survival factors Bcl-2 and Akt inhibit autophagy via Beclin-1, which is reversible in the presence of Bcl-2 and Akt inhibitors leading to up-regulation of Beclin-1 expression and increased autophagic response. Prolonged treatment with Digitoxin and MonoD cause sustained autophagy, which eventually sensitizes H460 cells to apoptosis.
Discussion
As a result of de novo or acquired resistance to molecularly targeted lung cancer therapies, there is a pressing need to discover and design therapies that can concomitantly target multiple pathways of cell death, thereby leading to sustained tumor regression. In this context, Digitoxin and its monosaccharide synthetic analog MonoD, have shown considerable promise as anti-cancer therapeutics (Pongrakhananon et al., 2013).
We found MonoD to exert higher cytotoxic activity as compared to Digitoxin. This is evident from the cell viability/proliferation data where a decrease in cell viability/proliferation with 50 nM MonoD is comparable to 100 nM Digitoxin (Fig. 1A). Additionally, MonoD caused accelerated loss in cell viability/proliferation—reduction in viability/proliferation with 50 nM MonoD at 24 h is comparable to that with 50 nM Digitoxin at 72 h (Fig. 1B). These results are also validated from quantification of apoptotic cells, which showed comparable levels of apoptosis using 50 nM MonoD and 100 nM Digitoxin (Fig. 5B). This effect seems to be more specific to H460 cells as normal Beas-2B cells do not show the same level of inhibition (Supplementary Fig. 2). Taken together, these data support the higher cytotoxic potential of MonoD preferentially in lung cancer cells.
In this study, we identified a novel mode of action for Digitoxin and MonoD whereby they induce both autophagic and apoptotic cell death in H460 lung cancer cells. Short-term exposure of H460 cells with Digitoxin and MonoD led to the formation of reactive free radicals including superoxide and nitric oxide, promoted the formation of cytoplasmic vacuoles, and activated several autophagy regulators such as Beclin-1, NF-κB, and p38 MAPK. On the other hand, sustained exposure to drug led to the activation of apoptotic cell death, which was facilitated by early autophagic flux. A dynamic interplay seems to exist between autophagy in the short term and apoptosis over the longer term, which is mediated by important intracellular mediators including Akt and Bcl2. Inhibition of Akt and Bcl2 activity intensified the effects of MonoD, resulting in an accelerated rate of autophagic flux and apoptotic death.
Formation of autophagic vacuoles increased very early following drug exposure particularly for MonoD, and decreased much more rapidly for MonoD as compared to Digitxoin (Fig. 1D). Maximal autophagic flux (measured using Cyto-ID®) was observed with 1 h MonoD treatment, which decreased at 8 h and disappeared at 24 h. Early induction of autophagy leading to apoptosis observed with Digitoxin and MonoD treatments is in agreement with other findings where gefitinib treated cells induced an autophagic flux in MCF7 cells in as little as 45 min, and a natural triterpenoid GA-DM increased Beclin-1 and LC3 expression in 3 h to induce autophagy in melanoma cells eventually leading to apoptosis (Hossain et al., 2012; Dragowska et al., 2013). Contrastingly, rottlerin induced autophagy in prostate cancer stem cells at 24 h, and metformin increased the autophagic flux at 48 h in esophageal squamous cell carcinoma eventually inducing the cells to undergo apoptosis (Feng et al., 2014; Kumar et al., 2014). We attribute temporal kinetics associated with increased accelerated autophagic flux with MonoD to its higher efficacy and potential differences in global signaling mechanisms as compared to Digitoxin.
Autophagic progression was verified by comparing the LC3-II levels between different treatments, as increase in LC3-II intensity corresponds to increased autophagy (Mizushima and Yoshimori, 2007). In several reports, the ratios of LC3-II/LC3-I and LC3-II/Total LC3 (LC3II + LC3-I) have been reported as a quantitative index for autophagy (Karim et al., 2007; Kadowaki and Karim, 2009; Eisenberg-Lerner and Kimchi, 2012). However, difference in the immunoreactivity of LC3-I and LC3-II may result in such ratio-based interpretations regarding the autophagic flux inappropriate. Comparing the amount of LC3-II alone between samples has been determined to be a more accurate method of interpreting autophagy, and we have adopted this approach to interpret our data (Mizushima and Yoshimori, 2007).
Beclin-1, one of the first autophagy effectors to be identified, is recruited early in the formation of pre-autophagosomal structures where it forms a complex with vacuolar protein sorting family members Vps15 and Vps34, which are responsible for vesicle nucleation (Liang et al., 1999). Moreover, the induction of autophagy mediated by p38 MAPK is often accompanied by increased expression of Beclin-1 (Cui et al., 2007). Beclin-1 and LC3-II are well established and widely used markers of induction of autophagy (Liang et al., 1999; Tanida et al., 2008; Cherra et al., 2010; Maejima et al., 2013). Therefore, we probed for LC3-II and Beclin-1 as key markers of autophagy in response to MonoD.
Digitoxin and MonoD treatment caused an increase in oxidative stress leading to excess production of ROS and NO in H460 cells. This finding is in agreement with earlier reports that demonstrate increased production of ROS in response to cardiac glycosides (Tian et al., 2003; Pasdois et al., 2007). Assessment of autophagy under various experimental conditions such as starvation, mitochondrial electron transport chain inhibitors and exogenous H2O2 concluded that increased autophagy levels correlated with increased •O2- production (Chen et al., 2009). This is consistent with previous reports showing a role for NO production in mediating autophagic flux (Tripathi et al., 2013). However, immunoblotting showed that inhibition of NO had no effect on regulation of LC3-II, Beclin-1, NF-κB, and p38 MAPK, suggesting that although NO was being produced, it did not directly drive Digitoxin- and MonoD-dependent autophagy in our cellular model at least for short-term exposure with the drugs (Fig. 3D and E). On the other hand, MnTBAP treatment not only inhibited the production of Digitoxin- and MonoD-induced •O2-, it was also accompanied by inhibition of MonoD induced upregulation of Beclin-1, NF-κB, p38 MAPK, and LC3-II levels (Fig. 4C). Previous studies have also demonstrated that overexpression of SOD2 has been shown to inhibit autophagy, supporting a role for •O2- in autophagy induction (Chen et al., 2007; Chen et al., 2008). These data prove that Digitoxin- and MonoD-induced autophagy is mediated by superoxide production in H460 cells.
The oncogenic potential of Bcl-2 is well established, with its overexpression reported in various cancers, making Bcl-2 an attractive chemotherapeutic target (Yip and Reed, 2008; Kang and Reynolds, 2009; Thomas et al., 2013). Additionally, Bcl-2 functions as an anti-autophagic protein by binding Beclin-1 via its BH3 (Bcl-2 homology 3) domain, thereby preventing Beclin-1 from forming a pre-autophagosome complex with Vps15 and Vps34 (Pattingre et al., 2005). To evaluate the function of Bcl-2 in Digitoxin- and MonoD-induced autophagic cell death both in terms of protein expression as well as activity, we used a H460 model overexpressing Bcl-2 as well as a small molecule inhibitor of Bcl-2 (ABT-737). Our results show that both Digitoxin and MonoD failed to induce expression of Beclin-1 and NF-κB in Bcl-2-overexpressing H460 cells (Fig. 5C). Furthermore, inhibition of Bcl-2 activity by co-treatment with ABT-737 led to an increase in autophagic vacuoles (Fig. 5G). These results clearly validate the role of Bcl-2 as an important inhibitor of MonoD-driven autophagy in our system.
In addition to the well-documented role of Akt in tumorigenesis (Testa and Bellacosa, 2001), there is emerging evidence of Akt inhibiting autophagy by targeting Beclin-1 (Wang et al., 2012). In keeping with such observations, we find that the reversible Akt-inhibitor LY294002 had an augmentative effect on autophagy. Both Akt and Bcl-2 are inhibitors of autophagy, and therefore inhibiting their activity resulted in an increased basal autophagy in H460 cells indicated by increased LC3-II and Beclin-1 expression. Concomitant treatment of these inhibitors with MonoD resulted in further elevation in levels of LC3-II and Beclin-1 expression, with increased punctae and vacuole formation. Concomitant treatment modicums such as that involving MonoD and inhibitors for Bcl-2 and Akt that targets distinct intracellular cell-death/cell-survival pathways in addition to the autophagic pathway may represent a novel paradigm for triggering effective and accelerated cancer cell death.
Autophagy is by definition a self-eating catabolic process that recycles misfolded and aggregated cytoplasmic proteins and organelles, and maintains cellular homeostasis by protecting cells from unfavorable conditions such as nutrient starvation by acting as an energy source. This confers upon cancer cells the ability to tolerate stressful conditions, eventually leading to a drug-resistant phenotype (Degenhardt et al., 2006), a concept that has encouraged strategies that inhibit autophagy as a viable therapeutic approach in cancer treatment (Townsend et al., 2012). There is plenty of recent evidence to suggest that autophagy acts as a double-edged sword, where short term autophagy may be thought of as cell survival mechanism while sustained autophagic flux causes progressive cellular consumption eventually leading to cell death (Mathew et al., 2007). We posit this latter role for autophagy with MonoD and Digitoxin, where short term autophagic signaling contributes to eventual apoptotic death. Co-treatment of MonoD with autophagic inducers and inhibitors has a direct effect on overall apoptotic death (Fig. 7A and B). It would be interesting to assess whether a complete loss of autophagic machinery in our cell model at the genetic level might recapitulate some of the effects we observe using pharmacological inhibitors. Also, while we have shown that inhibition of pro-survival proteins increases autophagy and eventually potentiates apoptosis in the context of our study, we have not identified all the factors that trigger the lateral cross talk between these two pathways of cell death in the context of MonoD action, which will be the focus of future evaluation of these compounds.
We have summarized our findings in the Schematic provided (Fig. 7C). Digitoxin and MonoD treatment leads to the production of superoxide, which drives early autophagy via the up-regulation of Beclin-1. MnTBAP inhibits the superoxide production, leading to down-regulation of Beclin-1 thereby inhibiting autophagy. Pro-survival factors Bcl-2 and Akt inhibit autophagy via Beclin-1, which is reversible in the presence of Bcl-2 and Akt inhibitors, leading to an increase in Beclin-1 expression and autophagic flux. Longer treatment with Digitoxin and MonoD leads to sustained autophagy, which eventually sensitizes cells to apoptosis, with a possible cross talk between autophagic and apoptotic mediators. We believe that we have presented an elegant mode of action for a hitherto uncharacterized drug, MonoD, which directs its action through both autophagic and apoptotic signaling. We propose that such multifarious effects of MonoD may be exploited not only for targeting resistant cancers of the lung such as NSCLC, but other cancers that show limited sensitivity to apoptotic stimuli.
Supplementary Material
Acknowledgments
Contract grant sponsor: NCI;
Contract grant number: CA173069.
Contract grant sponsor: NHLBI;
Contract grant number: HL112630.
Footnotes
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
Conflicts of interest: The authors disclose no potential conflicts of interest.
Literature Cited
- Altman BJ, Rathmell JC. Autophagy: Not good OR bad, but good AND bad. Autophagy. 2009;5:569–570. doi: 10.4161/auto.5.4.8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad N, Iyer AK, Manosroi A, Wang L, Rojanasakul Y. Superoxide-mediated proteasomal degradation of Bcl-2 determines cell susceptibility to Cr(VI)-induced apoptosis. Carcinogenesis. 2008;29:1538–1545. doi: 10.1093/carcin/bgn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad N, Iyer AK, Wang L, Liu Y, Lu Y, Rojanasakul Y. Reactive oxygen species-mediated p38 MAPK regulates carbon nanotube-induced fibrogenic and angiogenic responses. Nanotoxicology. 2013;7:157–168. doi: 10.3109/17435390.2011.647929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J Biol Chem. 2006;281:34124–34134. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- Bhutia SK, Mukhopadhyay S, Sinha N, Das DN, Panda PK, Patra SK, Maiti TK, Mandal M, Dent P, Wang XY, Das SK, Sarkar D, Fisher PB. Autophagy: Cancer’s friend or foe? Adv Cancer Res. 2013;118:61–95. doi: 10.1016/B978-0-12-407173-5.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Facts & Figures. American Cancer Society; 2013. [Google Scholar]
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–1052. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15:171–182. doi: 10.1038/sj.cdd.4402233. [DOI] [PubMed] [Google Scholar]
- Cherra SJ, 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190:533–539. doi: 10.1083/jcb.201002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Tashiro S, Onodera S, Minami M, Ikejima T. Oridonin induced autophagy in human cervical carcinoma HeLa cells through Ras, JNK, and P38 regulation. J Pharmacol Sci. 2007;105:317–325. doi: 10.1254/jphs.fp0070336. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragowska WH, Weppler SA, Wang JC, Wong LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge M, Gorski SM, Gelmon KA, Bally MB. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS ONE. 2013;8:e76503. doi: 10.1371/journal.pone.0076503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012;19:788–797. doi: 10.1038/cdd.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Ke C, Tang Q, Dong H, Zheng X, Lin W, Ke J, Huang J, Yeung SC, Zhang H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014;5:e1088. doi: 10.1038/cddis.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti L, Cossa G, Tinelli S, Carenini N, Arrighetti N, Pennati M, Cominetti D, De Cesare M, Zunino F, Zaffaroni N, Perego P. Improved apoptotic cell death in drug-resistant non small cell lung cancer cells by TRAIL-based treatment. J Pharmacol Exp Ther. 2013 doi: 10.1124/jpet.113.210054. [DOI] [PubMed] [Google Scholar]
- Hippert MM, O’Toole PS, Thorburn A. Autophagy in cancer: Good, bad, or both? Cancer Res. 2006;66:9349–9351. doi: 10.1158/0008-5472.CAN-06-1597. [DOI] [PubMed] [Google Scholar]
- Hossain A, Radwan FF, Doonan BP, God JM, Zhang L, Bell PD, Haque A. A possible cross-talk between autophagy and apoptosis in generating an immune response in melanoma. Apoptosis. 2012;17:1066–1078. doi: 10.1007/s10495-012-0745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer AK, Zhou M, Azad N, Elbaz H, Wang L, Rogalsky DK, Rojanasakul Y, O’Doherty GA, Langenhan JM. A direct comparison of the anticancer activities of digitoxin meon-neoglycosides and o-glycosides: Oligosaccharide chain length-dependent induction of caspase-9-mediated apoptosis. ACS Med Chem Lett. 2010;1:326–330. doi: 10.1021/ml1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: A link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki M, Karim MR. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 2009;452:199–213. doi: 10.1016/S0076-6879(08)03613-6. [DOI] [PubMed] [Google Scholar]
- Kang MH, Reynolds CP. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim MR, Kanazawa T, Daigaku Y, Fujimura S, Miotto G, Kadowaki M. Cytosolic LC3 ratio as a sensitive index of macroautophagy in isolated rat hepatocytes and H4-II-E cells. Autophagy. 2007;3:553–560. doi: 10.4161/auto.4615. [DOI] [PubMed] [Google Scholar]
- Kim DS, Kim JH, Lee GH, Kim HT, Lim JM, Chae SW, Chae HJ, Kim HR. P38 Mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol Pharm Bull. 2010;33:545–549. doi: 10.1248/bpb.33.545. [DOI] [PubMed] [Google Scholar]
- Kumar D, Shankar S, Srivastava RK. Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett. 2014;343:179–189. doi: 10.1016/j.canlet.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Lerena C, Calligaris SD, Colombo MI. Autophagy: For better or for worse, in good times or in bad times. Curr Mol Med. 2008;8:92–101. doi: 10.2174/156652408783769634. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M, Sadoshima J. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manish Shanker DW, Jack Roth A, Rajagopal Ramesh. Drug resistance in lung cancer. Lung Cancer Targets Ther. 2010;1:23–36. [PMC free article] [PubMed] [Google Scholar]
- Marx J. Autophagy: Is it cancer’s friend or foe? Science. 2006;312:1160–1161. doi: 10.1126/science.312.5777.1160. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medan D, Luanpitpong S, Azad N, Wang L, Jiang BH, Davis ME, Barnett JB, Guo L, Rojanasakul Y. Multifunctional role of Bcl-2 in malignant transformation and tumorigenesis of Cr(VI)-transformed lung cells. PLoS ONE. 2012;7:e37045. doi: 10.1371/journal.pone.0037045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Quinlan CL, Rissa A, Tariosse L, Vinassa B, Costa AD, Pierre SV, Dos Santos P, Garlid KD. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am J Physiol Heart Circ Physiol. 2007;292:H1470–H1478. doi: 10.1152/ajpheart.00877.2006. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Pongrakhananon V, Stueckle TA, Wang HY, O’Doherty GA, Dinu CZ, Chanvorachote P, Rojanasakul Y. Monosaccharide digitoxin derivative sensitize human non-small cell lung cancer cells to anoikis through Mcl-1 proteasomal degradation. Biochem Pharmacol. 2013 doi: 10.1016/j.bcp.2013.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA. 2001;98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Quinn BA, Das SK, Dash R, Emdad L, Dasgupta S, Wang XY, Dent P, Reed JC, Pellecchia M, Sarkar D, Fisher PB. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets. 2013;17:61–75. doi: 10.1517/14728222.2013.733001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Liu J, Garlid KD, Shapiro JI, Xie Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol Cell Biochem. 2003;242:181–187. [PubMed] [Google Scholar]
- Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ. Autophagy inhibition in cancer therapy: Metabolic considerations for antitumor immunity. Immunol Rev. 2012;249:176–194. doi: 10.1111/j.1600-065X.2012.01141.x. [DOI] [PubMed] [Google Scholar]
- Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, Wogan GN. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci USA. 2013;110:E2950–E2957. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–959. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.