

Abstract
The unscheduled DNA synthesis (UDS) assay measures the ability of a cell to perform global genomic nucleotide excision repair (NER). This chapter provides instructions for the application of this technique by creating 6-4 photoproducts and pyrimidine dimers using UV-C irradiation. This procedure is designed specifically for quantification of the 6-4 photoproducts. Repair is quantified by the amount of radioactive thymidine incorporated during repair synthesis after this insult, and radioactivity is evaluated by grain counting after autoradiography. The results are used to clinically diagnose human DNA repair deficiency disorders and provide a basis for investigation of repair deficiency in human tissues or tumors. No other functional assay is available that directly measures the capacity to perform NER on the entire genome without the use of specific antibodies. Since live cells are required for this assay, explant culture techniques must be previously established. Host cell reactivation (HCR), as discussed in Chapter 37, is not an equivalent technique, as it measures only transcription-coupled repair (TCR) at active genes, a small subset of total NER.
Keywords: Unscheduled DNA synthesis (UDS), Nucleotide excision repair (NER), DNA repair, DNA damage, UV light, Pyrimidine dimers, 6-4 Photoproducts, Global genomic repair (GGR), Transcription-coupled repair (TCR)
1 Introduction
Long patch, or Nucleotide Excision Repair (NER), is the primary process by which cyclobutane pyrimidine dimers, 6-4 pyrimidine-pyrimidone photoproducts, and DNA cross-links are removed from the DNA [1, 2]. Ultraviolet (UV)254nm light (UV-C), as well as UV-mimetic drugs, induces DNA lesions that are corrected by the NER pathway. Damage lesions caused by agents that act as interstrand and intrastrand cross-linkers, such as cisplatin [3], covalently bind to DNA creating “bulky” adducts, such as N-acetoxyaminoacetylfluorene (AAAF) [4] and perhaps alkylating agents, such as cyclophosphamide [5, 6] are also remediated by this pathway. NER is a complicated process requiring the protein products of 25–30 genes [7]. NER involves the recognition of a damage lesion causing distortion of the DNA helix, incisions flanking the lesion on the damaged strand, excision of 27–29 bases including the damage lesion, and replication and ligation to replace the excised information and seal the strand breaks at each end of the newly synthesized region [8–11]. This pathway can also be recruited for other types of DNA damage lesions that have not been corrected by base excision and other single stranded DNA repair mechanisms [12, 13]. In effect, the NER pathway provides redundancy for these other repair systems should they be overwhelmed by a genotoxic exposure [1]. Since the environment generally contains mixtures of chemicals, a significant genotoxic exposure is not an unlikely event. In the clinical setting, cancer chemotherapy is generally a cocktail of several genotoxic agents that together can overwhelm the repair capacities of the tumor cells.
Human cells perform NER at two levels: the rapid and efficient removal of lesions that block ongoing transcription and thus need to be eliminated quickly, also known as transcription-coupled repair (TCR) [14, 15]; see Chapter 37, and the slower, less efficient repair of bulk DNA, including the nontranscribed strand of active genes, also referred to as global genomic repair (GGR). In addition, the sense strand of the actively transcribed gene is repaired before the antisense strand. Rodent cells apparently do not perform repair of the bulk DNA and therefore cannot serve as fully accurate model systems for NER. This underscores the need for primary cultures to evaluate tissue-specific repair differences in the human system.
NER of the overall genome can be measured quantitatively using the unscheduled DNA synthesis (UDS) assay. The UDS assay involves the measurement of labeled base incorporation into the DNA after in vitro exposure to UV light or certain chemicals (Fig. 1). The UDS assay is a cell-autonomous, functional assay, in that it allows one to look at the complex process of NER as a whole, at least as it is expressed in a particular cell type [16–18]. As applied in our laboratory, this assay predominantly quantifies the repair of UV-induced DNA 6-4 photoproducts, and elements of both the “global genomic” as well as the “transcription-coupled” components of NER contribute to the results (see Note 1). The commercially available “Click-iT” cell proliferation assay can also quantify NER by such repair synthesis, using a new fluorescently labeled nucleoside, 5-ethynyl-2′-deoxyuridine (EdU) [19]. Other existing functional assays for NER include an ELISA-based method that uses specific antibodies against damage lesions (thymine dimers or 6-4 photoproducts (see Chapter 37), which can also be performed with standardized antibodies [20] or even with a kit (CycLex Cellular UV DNA-Damage Detection Kit, MBL International, Woburn, MA). More recently, there are also PCR-based assays (see Chapter 31), and flow cytometry-based assays [21, 22]. Recent studies using the UDS protocol in this chapter include functional analyses of primary cultures of human lymphocytes, breast and ovarian tissue, and early-stage breast tumors [23–26].
Fig. 1.
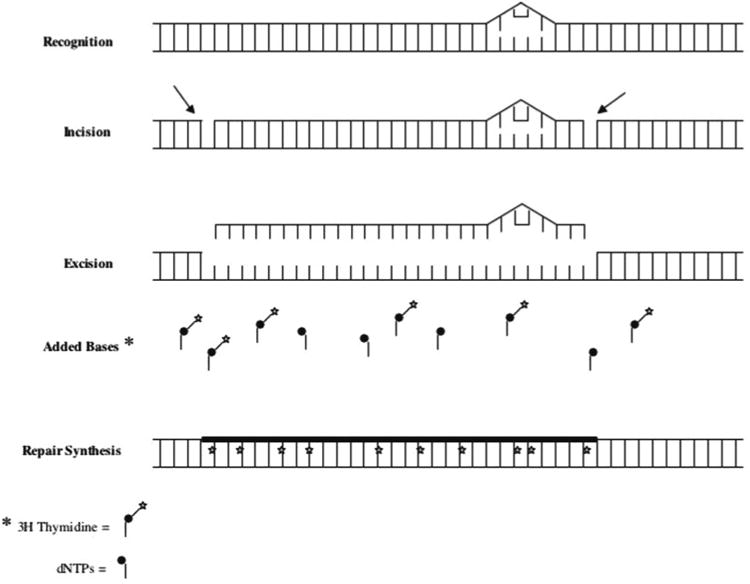
NER schematic as measured by the UDS assay. The assay involves damaging cells with UV-C light, then allowing the cells to repair themselves over a designated period of time. The UV light causes the formation of pyrimidine dimers and 6-4 photoproducts. Over time, the cells utilize the NER pathway to remove a single stranded DNA molecule containing 23–27 bases including the damage lesion. Resynthesis of the removed strand is then performed in the presence of radioactive thymidine. The amount of repair is therefore correlated with the total incorporation of radioactive thymidine
The autoradiographic UDS assay requires the analysis of living cells. It has previously been applied primarily to skin fibroblasts and peripheral blood lymphocytes (PBLs) for diagnosis of xeroderma pigmentosum (XP) and other DNA repair diseases impacting specifically on the NER pathway. Classical NER deficiency disorders are characterized by UV sensitivity manifesting mainly in the skin and cornea [1].
Studies involving functional assays in general, and specifically functional assays of DNA repair capacity, have been hampered by a technical lack of ability to perform primary explant culture on all cell types. The one notable exception is that of rat hepatocyte primary cultures, which have been used extensively in UDS assays for evaluation of the carcinogenic potential of chemicals [27, 28]. Although repair assays can be performed on established, transformed cell lines, the generation of cell lines from normal adult tissue has proven to be a technical challenge. In addition, during the process of passaging, established cell lines undergo clonal evolution that may alter or extinguish many of the original characteristics of the cells, including their intrinsic repair capacity [29, 30]. Techniques that use Epstein-Barr virus [31], SV40 [32], or papilloma virus [33] to immortalize cells actually reduce NER, and are not recommended for determining the baseline repair in a cell lineage or tumor cells. The potential impact of hTERT (human telomerase) that has been more recently used to immortalize cells has not yet been determined [34]. Until recently, cell culture techniques did not exist to support primary culture explants of most human tissues. These tissues require attachment to a substratum of some sort of extracellular matrix (ECM). Our laboratory has developed methods for primary culture of various cell types [23, 35].
The UDS assay was first described by Rassmussen and Painter [36]. Its name stems from the fact that the assay looks at the DNA repair mechanisms of cells in all stages of replication except synthesis or S phase (we know of no technique that will measure NER during S phase). There are two methods of quantifying data from the assay: autoradiography or scintillation counting. Autoradiography is the preferred method and is described in this Chapter. Although it is labor intensive, software packages have been designed in an attempt to remove the human subjectivity of this aspect of the data analysis [37–39]. However, our laboratory still performs grain counting with 2–3 independent counters on each slide, due to the fact that the current software is still inadequate for appreciating the differences between silver grains and darkly stained nucleoli, for example, in some cases. Finding rare cells can also be difficult without an automated stage for a grain counting software program. The greatest strength of software-based grain counting is the evaluation of background, as this is the area where individual reader subjectivity is greatest.
Scintillation counting is a simplified form of the autoradio-graphic assay that was popularized in industry for the hepatocyte evaluation of mutagenic chemicals. In order for it to be accurate, all of the S phase cells would have to be eliminated. In an attempt to achieve this, the use of hydroxyurea was incorporated into the original UDS protocol. However, we have found that up to 40 % of the cells can still be in S phase even in the presence of hydroxyurea, which would significantly affect the results and render them inaccurate. Indeed, since S phase nuclei incorporate hundreds of times more radioactive label than non-S phase cells, only a few cells that happen to be in synthesis mode would render these experiments quite inaccurate. We therefore recommend the auto-radiographic form of this assay more than the scintillation counting form, which probably should only be used to determine major trends.
Controls for use in combination with this assay can include commercially available, repair deficient XP cell lines. Our laboratory standardly utilizes foreskin fibroblasts as a positive standard for comparison, grown on tissue culture plastic in MEM medium. These cells can reliably be used as extended explants up to passage 13. Explants beyond 13 passages show a decreasing repair capacity relative to the earliest passages. We have recently demonstrated considerable tissue variation in NER capacity, suggesting that a tissue matched control should be used [23–26, 35]. For controls in lymphocyte studies, we recommend the transformed lymphoblastoid cell line TK6 [23].
Published doses of UV irradiation utilized for UDS experiments range from 5 to 50 J/m3. It is recommended that a dose– response curve be performed in order to determine the optimal dose of UV-C for a given cell type.
2 Materials
2.1 Cell Irradiation, Labeling and Fixation
Viable experimental samples (see Note 2).
Positive (and, if appropriate, negative) controls, including “tester” slides (see Notes 3–5).
2-chamber chamber slides (made by Nunc, ordered from Fisher Scientific, Pittsburgh, PA), 2–4 slides per sample, controls, 4 slides minimum.
10-cm2 round cell culture dish (one per chamber slide) (made by Corning, ordered from Fisher).
Whatman filter paper (3 mm CHR in 35 cm × 45 cm sheets, Thomas Scientific, Swedeboro, NJ) cut into 2.5 cm × 5 cm strips, wrapped in tin foil and autoclaved (one strip per chamber slide).
Appropriate growth medium for each type of cell that will be analyzed.
Fetal bovine serum (FBS, Hyclone, Logan, UT).
Sterile tissue culture hood (Class IIA/B3 Biological Safety Cabinet, ThermoForma Scientific, Mariette, OH).
Tissue culture incubator with 5 % CO2 (ThermoForma Series II Water Jacketed CO2 Incubator, Forma Scientific).
Vortex mixer.
Specialized UV delivery device (Design Specialties, Bethel Park, PA) (Fig. 2) [40].
Short wave UV meter (e.g., Spectroline model DM-254XA, Spectronics, Westbury, NY).
80 Ci/mmol 3H thymidine (NET 027Z, Perkin Elmer, Boston, MA). Thaw and allow to warm to room temperature before use (see Note 6).
Radiolabeled thymidine incubation medium: 10 μCi 3H thymidine label (80 Ci/mmol) per mL of appropriate medium for each cell type, including serum and 1× Penicillin– Streptomycin (10,000 U/mL penicillin G sodium, 100,000 μg/mL streptomycin sulfate in 85 % saline, Invitrogen [Gibco], Carlsbad, CA). For example, for 20 slides, in a total volume of 20 mL of medium with serum, 200 μL of label are added. Add 3H thymidine to the medium immediately before use.
Nonradioactive thymidine cold chase medium: medium of choice supplemented with 10-3 M thymidine nucleoside (Sigma, St. Louis, MO). Add 10 % serum just prior to use and filter through a 0.45 μm filter (Nalgene, through Fisher) (see Note 7).
1× Salt Sodium Citrate (SSC) (Sigma).
100 mL 70 % ethanol (made fresh to prevent evaporation) (Fisher).
33 % acetic acid (Fisher) in ethanol (made fresh), total 100 mL.
Scalpel.
Hemostats (2).
Vertical glass slide “Copeland” jars (Fisher). Each holds five slides, so the number of jars needed depends on the size of the experiment.
4 % perchloric acid (diluted from stock 60 % perchloric acid [Fisher]) (store in a fume hood, can be explosive). Need about 60 mL per Copeland jar.
Fig. 2.

Specialized UV delivery device created to deliver a specific damaging dosage of UV-C light. The machine consists of three appropriate UV bulbs, a turntable and timed shutter, all enclosed by plexiglass. The distance from the bulbs to the sample (3 ft) determines the amount of damage the bulbs can give over a set time
2.2 Slide Processing I: Dipping in Emulsion
Rectangular glass slide jars with glass slide holders (Fisher). Each holds ten slides.
Distilled water.
Paper towels.
Sealed darkroom (see Note 8).
Carestream Autoradiography Emulsion (catalog no. 889 566, Type NTB) 118 mL/vial (stored at 4 °C) (see Note 9).
48 °C water bath in dark room.
Lab tape.
Small 3–4 cm diameter funnel (Fisher).
50, 400, 1,000 mL beakers.
Amersham dipping chamber (Piscataway, NJ).
Darkroom drying box (see Fig. 3).
Light tight slide boxes containing desiccant at one end behind a glass slide.
Regular and heavy duty tin foil.
Refrigerator.
Fig. 3.

An example of a drying box for slides just dipped in emulsion. It is best to have a box with a removable lid (in this example, a copier paper box). Four pieces of string are strung across the length of the box like clothes lines, with nine small bulldog clips evenly spaced on the knotted string, to hang the slides
2.3 Slide Processing II: Developing Emulsion
Four rectangular glass slide dishes (Fisher).
Three lab timers (VWR, Pittsburgh, PA).
Tap water, ice.
Medium sized plastic developing tray (Gage Industrial, Lake Oswego, OR).
Thermometer.
Kodak D-19 Developer (stored at 4 °C) (Eastman Kodak, Rochester, NY).
Kodak fixer (stored at 4 °C) (Eastman Kodak).
Two glass staining dishes for slides (Copeland jars).
0.5 g Giemsa powder (Fisher).
Glycerol.
60 °C water bath.
Methanol.
Stirring plate.
Parafilm.
Whatman filter paper (3 mm CHR) and funnel.
100 mL brown glass bottle.
Giemsa stock solution (see Note 10).
0.1 M citric acid.
0.2 M Na2HPO4.
-
Staining solution final concentration: 0.015 % (w/v) Giemsa, 0.6 M methanol, 7 mM citric acid, 0.018 M Na2 HPO4, pH 5.75. This pH can be achieved by mixing sodium phosphate dibasic buffer with sodium phosphate monobasic buffer, both at 0.2 M. Use this phosphate solution in both the stain and the rinse buffer. For 100 mL: 2 mL Giemsa stock solution, 2.4 mL methanol, 6.8 mL 0.1 M citric acid, 9.2 mL 0.2 M Na2 HPO4, and 80 mL distilled H2O.
For 100 mL:- 2 mL Giemsa stock solution.
- 2.4 mL methanol.
- 6.8 mL 0.1 M citric acid.
- 9.2 mL 0.2 M Na2HPO4.
- 80 mL distilled H2O.
-
Rinse buffer final concentration: 7 m M citric acid, 0.018 M Na2 HPO4.
For 100 mL:- 6.8 mL 0.1 M citric acid.
- 9.2 mL 0.2 M Na2HPO4.
- Bring up to 100 mL with distilled H2O.
Upright microscope with oil immersion 100× objective (e.g., Zeiss Axioskop, Oberkochen, Germany).
2.4 Grain Counting and Normalization
Create a standardized counting sheet, with columns for grains over nuclei and background (grains over a nucleus-sized area of the acellular field). Several other pieces of data that we suggest should be recorded include: total number of cells in the microscope field (countable nuclei and S phase cells), S phase cells and morphology of the cells. These parameters may be correlated with NER, or define differential cell populations.
Grain counts can be processed with any suitable statistical software, such as SAS (SAS Institute, Cary, NC) or the statistical package included in the Excel spreadsheet program (Microsoft, Redmond, WA).
3 Methods
3.1 Cell Irradiation, Labeling, and Fixation
All cells should be placed into culture at least 2 days before the performance of the UDS assay. Ideally, the cells on the final slides should be easy to find, but not identifiable as clumps or clones (i.e., the slides should be less than semi-confluent on the day of the assay). Therefore, seed appropriate numbers of each experimental and control cell populations (see Notes 2–4) into both chambers of four 2-chamber slides (total volume of 1 mL, free of DMSO, trypsin, etc.).
Place each chamber slide into a 10-cm2 round cell culture dish with a piece of dampened sterilized filter (see Note 11).
Incubate at 37 °C in a standard tissue culture incubator with 5 % CO 2 for 2 days.
Turn on UV bulbs on the UV delivery device (Fig. 2) and allow them to warm up for at least 1 h. Test the dose delivery rate under experimental conditions with a short wave (254 nm) UV Spectroline meter and adjust if necessary (time of exposure and/or distance from bulbs; see Note 12).
Thaw radioactive label and allow to warm to room temperature.
Feed all cells (replace with fresh medium) 1 h before UV exposure. Label all slides in pencil.
Thoroughly mix the 3H-thymidine label by vigorous vortexing. For up to 20 slides, add 200 μL label to 20 mL medium with serum to create “hot incubation media” with a final concentration of 10 μCi/mL. Vortex until foam appears.
Remove regular medium from all chamber slides. Divide experimental and control chamber slides into groups of four, ensuring there is a positive control slide (and a negative controls slide, if necessary) in each group.
Leave the chamber of each slide closest to the ground class label covered with the plastic lid and uncover the other chambers.
Start the turntable (set at “mid” speed) to equalize exposure. Expose slides in sets of four, as determined in Subheading 3.1, step 7.
Immediately after irradiation, add 0.5 mL of “hot incubation medium” to each chamber of each chamber slide, for a total of 1 mL of media per slide. 10–12 slides should be handled at a time to prevent the cells from drying out, with a maximum of 40 slides per experiment.
Incubate the slides for 2 h in tissue culture incubator (may wish to designate a “radioactive” incubator and/or “radioactive” shelf; see Note 13).
Add FCS to the cold chase medium.
In the radioactive hood, remove the “hot incubation medium” from all chamber slides by pipetting and replace with 0.5 mL of cold chase media.
Incubate the slides for 2 h in tissue culture incubator.
Prepare fresh 70 % ethanol and fixative solution (33 % acetic acid in 100 % ethanol).
In the radioactive hood, remove the cold chase medium by aspiration and gently rinse each side of the chamber slides with 1 mL of 1× SSC.
Remove the 1× SSC immediately by aspiration, and replace it with 0.5 mL fixative solution in each chamber. Leave at room temperature for 15 min.
Remove fixative by aspiration and leave slides to partially dry for 5 min. Add 0.5 mL of 70 % ethanol to each chamber and leave at room temperature for 15 min.
Remove the ethanol by aspiration. Remove the chambers and rubber gaskets from the slides, using a sharp scalpel to loosen one corner of the rubber gasket. Once the corner is free, use a pair of hemostats to pull the remaining gasket away from the slide. Any remaining gasket can be lightly scraped off with the scalpel. All traces of the gasket must be removed or it will cause the emulsion to be too thick on the edges of the slide.
Place the slides in radioactively labeled Copeland jars with 4 % perchloric acid. The jars are then placed in a refrigerator overnight.
3.2 Slide Processing I: Dipping in Emulsion
Caution: All steps in the dipping process must be done in complete darkness (with no safe lights) until the slides are dry, packaged and wrapped in foil (see Note 8).
Remove the slides from Copeland jars and rinse by letting them sit in distilled H2O for 3–4 min. Allow the slides to dry in the hood for 24 h in glass slide holders sitting on paper towels.
Melt emulsion and heat to 48 °C in a water bath in dark room. First, place the emulsion in a 1,000 mL beaker with 400 mL of water and then place the beaker into the water bath for 1.5 h. To keep the emulsion from floating, fill a 50 mL beaker and place it on top of the emulsion container. Stir the emulsion every 30 min, to ensure complete thawing.
Prepare a “drying box” in which to hang and dry the slides (Fig. 3).
Before taking the slides into the darkroom, place a triangular piece of tape on the upper right corner of each slide on the ground glass. This is to allow the slides to be oriented by touch in the dark while dipping. At this time, select one slide of each type of control to be “tester” slides. These slides will be placed in a separate drying box, and will be developed first in order to determine when the slides from the entire experiment have developed sufficiently for scoring.
Place an Amersham dipping container in a beaker half full of 48 °C water. In the darkroom, with all of the lights off including the red light, carefully pour the emulsion into the dipping container with a small funnel. Use your ungloved little finger to ensure the emulsion has filled the dipping container (the emulsion is nontoxic).
Orienting them by the piece of tape from Subheading 3.2, step 4, take each slide and dip it into the dipping container (lightly tap the slide on both sides of the container to ensure it is fully in the dipping container).
Allow each slide to drip a few seconds after removing it from the dipping container, and then hang the slide in the box with the bulldog clips with the ground glass at the top.
The “tester” slides should be hung exclusively on the first string and all subsequent slides hung on the other three pieces of string. The emulsion will have to be refilled every 15–25 slides.
After all the slides have been dipped the lid should be placed over the box. Allow the slides to dry in the box in the dark for 1 h in complete darkness.
Prepare plastic slide boxes by taping a clean glass slide toward one end of the box. In the space behind the slide, add desiccant. Place a piece of tape on the lid of the slide box to orient the front of the box in the dark. For each box, have three layers of foil ready to wrap the boxes in the dark after the slides have been placed in them (two layers regular, one heavy duty).
Place the tester slides only in the first slide box, close the lid, wrap it in foil, and mark it with a “T” (for testers) on the foil, with the date.
Remove the remaining slides from the drying box and place them in the other slide boxes, wrapping them in foil and marking them “E” (for experimental) and dating them.
Place the emulsion back in its original box and wrap it with foil as well. The emulsion is very light sensitive so any measures that are taken to lessen the exposure to light will prolong the useful lifetime of the emulsion and maintain low background on the slides.
Place the slide boxes in a refrigerator until developing.
3.3 Slide Processing II: Developing Emulsion
The tester slides are developed 12 days after dipping (day 1 being the day after dipping).
Take all slide boxes out of the refrigerator and allow to warm to room temperature for a minimum of 5 h.
In the darkroom, create a 15 °C water bath using water and ice in a medium sized developing tray.
Place four slide dishes into the 15 °C water bath. In the first dish, place D19 developer (1:1 dilution with dH2O), place undiluted fixer in the third, and water only in the second and fourth (each slide dish holds 250 mL of liquid). The red light may be on while these solutions come down to temperature.
Set three timers (one for 4 min and two for 5 min) before you enter the darkroom. It helps to use timers that beep when the start button is pressed, as well as when time has expired.
In complete darkness, place the tester slides in a glass slide holder and attach the wire holder. Lower the slide holder into the first dish (developer) and start the timer set for 4 min.
Move the slides into the second dish (water) for a count of 10 s, then place them into the third dish (fixer). Start a timer set for 5 min.
Move the slides into the fourth dish (water again) and start the second timer set for 5 min. The lights can then be turned back on, as long as there are no other open slide boxes or unexposed slides.
Dry the tester slides for at least 1 h.
Giemsa stain the slides for 7 min in Copeland jars (see Note 14).
Rinse the slides in the rinse buffer for 3 min and allow to air dry in a dust free environment for 2–3 h.
Score the tester slides according to Subheading 3.4. If the slides are determined to have developed long enough (see Note 15), develop the experimental slides in batches of 5–10 according to Subheadings 3.3, steps 1–9. The experimental slides can dry overnight in a dust free environment, to be stained the following day. If development is not sufficient, return the experimental slides to the refrigerator and determine when they should be developed (see Note 13).
3.4 Grain Counting and Normalization
- Once all of the slides have been stained, the nuclei of the cells can be counted (Fig. 4). There should be at least two slides per type of cell. Two criteria must be met for both the controls and the experimental slides for a successful experiment:
- there must be a reasonable number of grains per nucleus, particularly with regard to the background (25–50 for the irradiated chamber, 1–5 in the unirradiated chamber).
- there must be sufficient cells (or nuclei) on at least two slides (100 scorable nuclei is ideal, less than 25 is insufficient).
Orient the slide to be counted on the microscope stand so that the ground glass is to the left (the unirradiated side of the slide will now always be to the left and the irradiated to the right).
Scan both sides of the slide with a low power objective before counting, to ensure that there are at least 20 cells on both sides. If less than 20 cells are visible in each chamber then it will be necessary to pool multiple slides of the same time to obtain 20 unirradiated nuclei and 20 irradiated nuclei. If multiple slides do not exist, then this specimen cannot be evaluated for repair capacity. It is best to count a minimum of 100 cells on both sides of a slide. There should be some nuclei that are black with grains indicating cells that are in S phase (see Fig. 4). These cells can be marked in a separate column on the counting sheets.
If the slide is scorable, view it under oil immersion at 1,000× magnification (10× eyepiece × 100× objective) to resolve individual silver grains.
The Giemsa stain should provide three shades of purple color at high magnification. The lightest purple is the cytoplasm of the cells. There should be few to no grains in this area (see Note 16). The nuclei are the second darkest purple. These will contain the majority of silver grains representing 3H thymidine incorporated into the cell's DNA. The number of grains over the nuclei is proportional to the amount of repair. These are the grains that are counted (see Note 17). For both the positive control and the experimental slides, if the positive control is appropriate, the average number of grains per nuclei should be ∼50 on the irradiated side of the slide. The third deeper purple is the nucleoli of the cell.
To determine the “background” (grain count outside of the nuclei), the counter should define an acellular area that is about the same size as the nuclei in that field and count the number of grains. Background counts should be usually 5 or less. For evaluation of tester slides, see Note 15.
For each experiment two slides should be counted for each type of cell, and two individuals should count each slide. Slides of the same cell type analyzed in the same experiment and developed on the same day should have a coefficient of variance of 10–15 %. Counters on the same slide should have results that are within 10–15 % as well.
-
After the unirradiated and irradiated side of a slide is counted, statistical analysis may be completed. To arrive at the corrected value for the individual nuclei, take the counts per nuclei and subtract the background for that field for both the unirradiated and irradiated counts. To find the mean number of grains for each slide take the corrected value for the unirradiated cells and divide by the total number of nuclei counted (not including S phase cells). Do the same for the irradiated cells, and then take the average number of grains per nuclei for the unirradiated nuclei and subtract it from the average number of grains for the irradiated nuclei.
(Corrected irradiated grains per nucleus/total number nuclei counted - corrected unirradiated grains per nucleus/total nuclei counted) = average number of grains per nuclei.
-
To compare the experimental slides to the control, take the average of grains per nuclei for the experimental slide and divide by the average number of grains per nuclei for the control. Multiply this number by 100 to arrive at the percent repair for the experimental slide as compared to the control.
(Average number of grains per nuclei of experimental/average number grains per nuclei of control × 100) = % repair as compared to control.
-
To compare the experimental slides to a population of controls, take the % repair as determined relative to the concurrent control and multiply this by the ratio of the concurrent control divided by the average repair for the population. Multiply this number by 100 to arrive at the percent repair for the experimental slide as compared to the average of the control population.
(% repair as compared to concurrent control × average number grains per nuclei of concurrent control/average number grains per nuclei of average of control population × 100) = % repair as compared to average of control population.
Fig. 4.
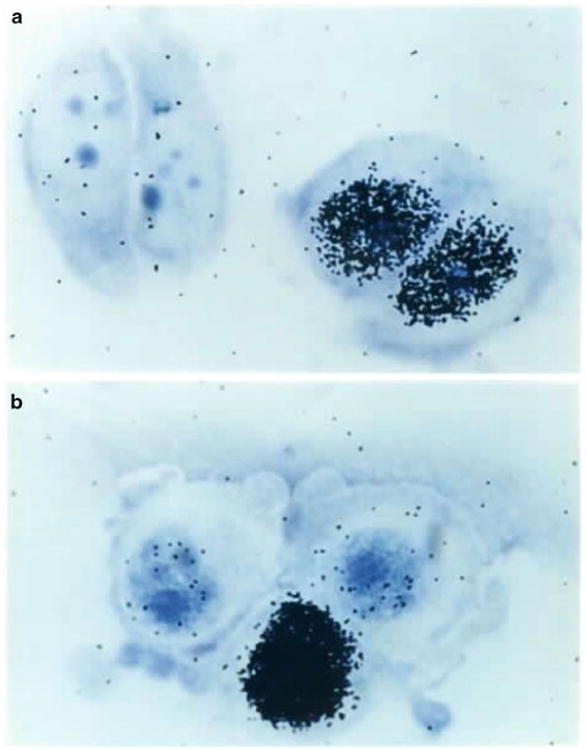
100× Micrograph of Giemsa-stained MCF-7 cells after UDS: (a) unirradiated cells, (b) irradiated cells
4 Notes
There is a perception that the HCR assay is specific for the TCR component of NER, while the UDS assay is specific for GGR. The specificity of the HCR assay (see Chapter 37) comes from the fact that repair is detected through repair of a reporter gene (although it is a foreign gene in an unnatural context). However, for UDS to be totally exclusive of TCR, the GGR mechanism would have to specifically avoid or exclude repairing damage in transcribed sequences, and there is no evidence that GGR has such specificity. Indeed, it has been estimated that ∼10 % of the repair measured in the HCR assay occurs through the incidental activity of GGR on the reporter gene [41–43]. The specificity of the UDS assay, if it has any, comes simply from the fact that the vast majority of the genome is either noncoding, or non-transcribed in any particular cell type. Indeed, given the faster kinetics of TCR, the UDS assay probably quantitates a greater proportion of this type of repair than GGR, however, how much this contributes to the final result is unknown.
The UDS assay has traditionally been performed on monolayer cell cultures, such as skin fibroblasts, transformed fibroblastic cell lines or hepatocytes. The stable attachment of such cells to glass slides allows the entire assay to be performed as described, on cells cultured on chamber slides. However, there is no reason that irradiation and labeling cannot be done prior to the attachment of the cells to a slide for quantification. Indeed, with cells, such as lymphocytes, that have a weaker attachment to the slides themselves, we see a significant attrition in scorable cells through slide processing. Thus, we have coated the slides to enhance their attachment [23, 44]. Similarly, we have attached single layer sheets of cells to slides for processing, or even isolated labeled nuclei and attached them to the slide [35]. We have also found that 3-dimensional cellular structures can be analyzed, as long as only the outer layer of cells is scored, since they are the only ones who receive an unattenuated dose of UV, and are directly exposed to the emulsion [23].
-
Since the UDS assay is affected by the strength of the radiolabel as well as the emulsion, it cannot be considered an absolute assay of DNA repair, and raw grain counts in and of themselves, are relatively meaningless (although they are often reported). Instead, repair capacity should be reported relative to a standardized positive control, analyzed concurrently in each experiment. Foreskin fibroblasts (FF) have traditionally been used as the positive control for the UDS assay (see Note 18 for a protocol for establishing FF cultures). Besides being relatively easy to acquire, FF are described as having relatively high levels of repair [18], suggesting that NER declines with age, which some studies have observed [45], but not our own [23]. The positive control should ideally provide two different, but related references: first, it should provide a baseline measure of the “normal” level of NER for the experimental samples to be compared against, and second, it should provide a guide as to when the experiment, specifically the exposure of the emulsion, can be successfully concluded (see Notes 4 and 15). FF have long been considered to provide both references, both for analyses of cell lines and for fibroblast samples taken from patients for diagnosis. However, it is significant that diagnostic studies of PBLs have used normal PBLs as controls; we have recently shown that there is a 20-fold difference in the baseline NER capacity of the two cell types [23]. In addition, a single “normal” sample, often randomly acquired, usually serves as the control, whereas we have also shown that there is considerable inter-individual variability in NER capacity in the “normal” population. Mixtures of FF from several babies have sometimes been used to attempt to account for possible inter-individual differences. Instead, we suggest that a population of normal samples should first be analyzed, with the “concurrent” control included. The final grain counts can then be normalized not just against the concurrent normal sample, but, through it, to the normal population.
If lymphocytes are being run in the experiment, an appropriate control appears to be the lymphoblastoid cell line TK6 (although transformed cells, in general, cannot be assumed to have “normal” repair). Lymphocytes need to be developed for a longer period of time because of their low repair, usually 14–16 days instead of 12. The accuracy of the final determination is based on the number of grains scored, not the number of cells. Thus, if the slides are developed too early (which would be the case if lymphocyte slides were developed on a timetable based on FF controls) they have very few grains, making it difficult to unambiguously establish a difference between the irradiated and unirradiated populations. In general, grain counts of 50–100 should be projected prior to emulsion development, and we regularly count 200–300 nuclei per slide.
Tester slides are extra positive control slides that must be designed into an experiment in order to determine the optimal exposure time for the emulsion. The freshness of the label and of the emulsion (background grains increase with each thawing of the emulsion), as well as the changing conditions of the darkroom can all be variables in terms of the length of time autoradiographic slides should be developed for maximal signal to noise ratio. Generally two extra FF (or other control) slides are made for this purpose and are packaged separately for development at 12 days, to be grain counted before the slides of the rest of the experiment are developed. If the rest of the experimental slides are developed on the same day as the tester slides, then the tester slides can be used as additional positive control slides in the experiment. If the tester slides are developed on a day other than the experimental slides, they may not be included (as positive standards of comparison) in the analysis of the experimental slides.
If the experiment is designed to document repair deficiency, “negative” controls, i.e., repair-deficient cells, may be included, in addition to the standardized positive controls. Indeed, a second set of slides may be prepared, and developed using the negative controls slides as guides although these will not in any way replace the unirradiated chamber controls. Three human diseases have been found to be associated with defects in the NER genes: xeroderma pigmentosum (XP) with seven complementation groups, Cockayne syndrome, with five complementation groups, three of which overlap with those of XP, and trichothiodystrophy, with three complementation groups, two of which overlap with XP [1]. UDS has historically been used to diagnose XP in skin fibroblasts or in peripheral blood lymphocytes, since these patients are more or less deficient in NER depending on their complementation group. Cockayne syndrome affects primarily transcription-coupled repair (TCR). There is a considerable range of residual activity amongst patients in these various XP complementation groups (<10 % in groups A, B, and G up to 50 % in groups D and E), with none exhibiting complete deficiency [46]. Complete NER deficiency may be a prenatal lethal condition; a mutation in ERCC1, the first NER gene cloned, has only recently been identified in a single patient [47], perhaps because they are usually inviable at the organismal level [48]. XP cell lines (immortalized both with and without the use of exogenous agents) can be purchased from the American Type Culture Collection (ATCC, Manassas, VA) or Coriell Cell Repositories (Camden, NJ) for negative control use.
Date the label when it arrives and discard 1 month from arrival date due to chemical deterioration. Label should be stored at −20 °C.
Cold thymidine media should be stored without the presence of serum, which can bind the thymidine and effectively lower the bioavailable thymidine. For 100 mL of medium, add 0.0242 g of thymidine (this solution can be stored at 4 °C for a period of 3 months as long as it lacks serum).
This portion of the assay is extremely light sensitive. All light sources must be covered or removed, including lights on water baths, temperature control for the room, light leaks from doorways and light fixtures and any other sources in the dark room including fluorescent watches. Spend 20 min in the room with all the lights off to identify light sources and cover them before the procedure is begun if you are uncertain of the integrity of your darkroom. The assay can also be affected by static electricity during the winter months.
Reuse of previously melted emulsion can cause higher background in sequential experiments, due to factors like static electricity that expose it with each use. Use the same emulsion at most three times to avoid increasing spurious background grains.
Giemsa stock solution: 0.8 % (w/v) Giemsa in 1:1 glycerol: methanol. Warm glycerol in a 60 °C water bath, add 0.5 g Giemsa powder to 33 mL glycerol, and place on stir plate overnight without heat. The following morning, add 33 mL methanol and cover with Parafilm and Saran wrap. The next day, filter the solution using Whatman filter paper and store in a dark bottle at room temperature. The filtration step takes several hours.
The petri dish also provides an extra layer of protection against air-born contamination. The moistened filter paper creates an additional humidity chamber for the cells.
The required dosage of UV-C light at 254 nm is 14 J/m2 to produce the desired amount of DNA damage. For a desired dose of 14 J/m2, with a mean fluence of 1.2 J/m2/s from the UV bulbs, a 12 s exposure of the cells is generally used by our laboratory (dose in J/m2 = fluence [read from the meter] × number of seconds).
This allows sufficient time for the cells to repair the 6-4 photo-products that were created by the UV-C light but not the pyrimidine dimers (to quantitate the repair of pyrimidine dimers would require an 8 h incubation) [49, 50].
Slides may be stained as many times as needed in order to clearly view the nuclei. Alternatively, methanol can be used to lighten the stain if it is too heavy. Some cells such as CHO cells only need to be stained for 2 min instead of 7 min.
For the tester slides, once the slides have dried, view under an oil immersion lens at a total magnification of 1,000× and count 25 nuclei on both the irradiated and unirradiated sides of the slide. This will allow a decision to be made as to whether or not the experimental slides should be developed that day. If no grains are observed, then the label was either not radioactive or was not added to the incubation, and the experiment is unusable. If an average of ∼50 grains/nucleus or more above background are observed in the irradiated chamber, the experimental slides are ready to be developed (unless you know the experimental cells have lower NER activity than the controls, in which case, you must compensate for their relatively longer development time). However, both control and experimental slides must be counted accurately under the same conditions in order to perform the normalization. S phase cells should be visible on both sides of the slide (in approximately equal numbers). If the controls do not exhibit the required number of grains/nucleus the remaining control and experimental slides should be developed 24–72 h later. If longer development seems necessary, it is likely that the signal to noise (background) ratio will be too high to accurately determine NER capacity. If background grains appear to be unevenly distributed, or distributed in a pattern, there is likely a light leak in the slide boxes used to store the dipped slides.
If the cytoplasm is covered with grains and the nucleus is relatively uncovered, it is most likely a result of mycoplasma contamination or some other bacterial contamination. These slides are not scorable and the cell population will have to be reacquired or rendered free of contamination in order to be assayed.
If the tester slides have been used correctly and the experimental slides average about 50 grains/nucleus, nuclei that have more the ∼100 grains when counted should be considered in S phase.
FF can be generated in large quantities from a single foreskin by finely mincing the tissue and stirring it in trypsin for 5 h at room temperature, followed by repeated tituration with a 25 mL pipet and subsequent plating in tissue culture dishes. The first few passages should be performed to remove cell clumps and create even monolayers of fibroblasts. Then these early explant passages can be frozen in 10 % DMSO for use in many UDS experiments. FF repair capacity is stable for about 18 passages, so we recommend the use of FF up to passage 13 to retain consistency [27].
References
- 1.Thompson LH. Nucleotide excision repair: its relation to human disease. In: Nickoloff JA, Hoekstra MF, editors. DNA damage and repair, vol 2: DNA repair in higher eukaryotes. Humana; Totowa, NJ: 1998. pp. 335–393. [Google Scholar]
- 2.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 3.Reed E. Platinum-DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat Rev. 1998;24:331–344. doi: 10.1016/s0305-7372(98)90056-1. [DOI] [PubMed] [Google Scholar]
- 4.Kaneko M, Cerutti PA. Excision of N-acetoxy-2-acetylaminofluorene-induced DNA adducts from chromatin fractions of human fibroblasts. Cancer Res. 1980;40:4313–4319. [PubMed] [Google Scholar]
- 5.Andersson BS, Sadeghi T, Siciliano MJ, Legerski R, Murray D. Nucleotide excision repair genes as determinants of cellular sensitivity to cyclophosphamide analogs. Cancer Chemother Pharmacol. 1996;38:406–416. doi: 10.1007/s002800050504. [DOI] [PubMed] [Google Scholar]
- 6.Gamesik MP, Dolan ME, Andersson BS, Murray D. Mechanisms of resistance to the toxicity of cyclophosphamide. Curr Pharm Des. 1999;5:587–605. [PubMed] [Google Scholar]
- 7.Mullenders LHF, Berneberg M. Photoimmunology and nucleotide excision repair: impact of transcription coupled and global genome excision repair. J Photochem Photobiol B. 2001;56:97–100. doi: 10.1016/s1011-1344(01)00244-5. [DOI] [PubMed] [Google Scholar]
- 8.Covertey D, Kenney MK, Rupp WD, Lane DP, Wood RD. Requirement of the replication protein SSB in human DNA excision repair. Nature. 1991;347:538–541. doi: 10.1038/349538a0. [DOI] [PubMed] [Google Scholar]
- 9.Huang JC, Svoboda DL, Reardon JT, Sancar A. Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc Natl Acad Sci USA. 1992;89:3664–3668. doi: 10.1073/pnas.89.8.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shivji KK, Kenney MP, Wood RD. Proliferating cell nuclear antigen is required for DNA excision repair. Cell. 1992;69:367–374. doi: 10.1016/0092-8674(92)90416-a. [DOI] [PubMed] [Google Scholar]
- 11.Grossman L, Thiagalingam S. Nucleotide excision repair, a tracking mechanism in search of damage. J Biol Chem. 1993;268:16871–16874. [PubMed] [Google Scholar]
- 12.Satoh MS, Jones CJ, Wood RD, Lindahl T. DNA excision-repair defect of xeroderma pigmentosum prevents removal of a class of oxygen free radical-induced base lesions. Proc Natl Acad Sci USA. 1993;90:6335–6339. doi: 10.1073/pnas.90.13.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang JC, Hsu DS, Kazantsev A, Sancar A. Substrate specificity of human exinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc Natl Acad Sci USA. 1994;91:12213–12217. doi: 10.1073/pnas.91.25.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- 15.Bootsma D, Hoeijmakers JHJ. The molecular basis of nucleotide excision repair syndromes. Mutat Res. 1994;307:15–23. doi: 10.1016/0027-5107(94)90273-9. [DOI] [PubMed] [Google Scholar]
- 16.Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218:652–656. doi: 10.1038/218652a0. [DOI] [PubMed] [Google Scholar]
- 17.Painter RB, Cleaver JE. Repair replication, unscheduled DNA synthesis and the repair of mammalian DNA. Radiat Res. 1969;37:451–466. [PubMed] [Google Scholar]
- 18.Cleaver JE, Thomas GH. Measurement of unscheduled synthesis by autoradiography. In: Friedberg EC, Hanawalt PC, editors. DNA repair: a laboratory manual of research procedures, vol I. Marcel Dekker; New York: 1981. pp. 277–287. [Google Scholar]
- 19.Limsirichaikul S, Niimi A, Fawcett H, Lehmann A, Yamashita S, Ogi T. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU) Nucleic Acids Res. 2009;37:e31. doi: 10.1093/nar/gkp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsunaga T. In vitro assays for evaluating the cellular responses to DNA damage induced by solar UV. AATEX. 2007;14:637–640. [Google Scholar]
- 21.Thyagarajan B, Anderson KE, Lessard CJ, et al. Alkaline unwinding flow cytometry assay to measure nucleotide excision repair. Mutagenesis. 2007;22:147–153. doi: 10.1093/mutage/gel071. [DOI] [PubMed] [Google Scholar]
- 22.Rouget R, Auclair Y, Loignon M, Affar EB, Drobetsky EA. A sensitive flow cytometry- based nucleotide excision repair assay unexpectedly reveals that mitogen-activated protein kinase signaling does not regulate the removal of UV-induced DNA damage in human cells. Biol Chem. 2008;283:5533–5541. doi: 10.1074/jbc.M706257200. [DOI] [PubMed] [Google Scholar]
- 23.Latimer JJ, Nazir T, Flowers LC, et al. Unique tissue-specific level of DNA nucleotide excision repair in primary human mammary epithelial cultures. Exp Cell Res. 2003;291:111–121. doi: 10.1016/s0014-4827(03)00368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Latimer JJ, Rubinstein WS, Johnson JM, et al. Haploinsufficiency for BRCA1 is associated with normal levels of DNA nucleotide excision repair in breast tissue and blood lymphocytes. BMC Med Genet. 2005;6:26. doi: 10.1186/1471-2350-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latimer JJ, Johnson JM, Miles TD, et al. Cell-type-specific level of DNA nucleotide excision repair in primary human mammary and ovarian epithelial cell cultures. Cell Tissue Res. 2008;333:461–467. doi: 10.1007/s00441-008-0645-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latimer JJ, Johnson JM, Kelly CM, et al. Nucleotide excision repair deficiency is intrinsic in sporadic stage I breast cancer. Proc Natl Acad Sci USA. 2010;50:21725–21730. doi: 10.1073/pnas.0914772107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalopoulos G, Sattler GL, O'Connor L, Pitot HC. Unscheduled DNA synthesis induced by procarcinogens in suspensions and primary cultures of hepatocytes on collagen membranes. Cancer Res. 1978;38:1866–1871. [PubMed] [Google Scholar]
- 28.Williams GM, Mori H, McQueen CA. Structure-activity relationships in the rat hepatocyte DNA-repair test for 300 chemicals. Mutat Res. 1989;221:263–286. doi: 10.1016/0165-1110(89)90039-0. [DOI] [PubMed] [Google Scholar]
- 29.Killary AM, Fournier REK. A genetic analysis of extinction: trans-dominant loci regulate expression of liver-specific traits in hepatoma hybrid cells. Cell. 1984;38:523–534. doi: 10.1016/0092-8674(84)90507-5. [DOI] [PubMed] [Google Scholar]
- 30.Clarke R, Leonessa F, Brunner WN, Thompson EW. In vitro models. In: Harris JR, Lippman ME, Morrow M, Osborne CK, editors. Diseases of the breast. Lippincott Williams and Wilkins; Philadelphia, PA: 2000. pp. 347–348. [Google Scholar]
- 3.Liu MT, Chen YR, Chen SC, et al. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene. 2004;23:2531–2539. doi: 10.1038/sj.onc.1207375. [DOI] [PubMed] [Google Scholar]
- 32.Bowman KK, Sicard DM, Ford JM, Hanawalt PC. Reduced global genomic repair of ultraviolet light-induced cyclobutane pyrimidine dimers in simian virus 40-transformed human cells. Mol Carcinogen. 2000;29:17–24. [PubMed] [Google Scholar]
- 33.Ford JM, Baron EL, Hanawalt PC. Human fibroblasts expressing the human papillomavirus E6 gene are deficient in global genomic nucleotide excision repair and sensitive to ultraviolet irradiation. Cancer Res. 1998;58:599–603. [PubMed] [Google Scholar]
- 34.Elenbaas B, Spirio L, Koemer F, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Latimer JJ, Hultner ML, Cleaver JE, Pederson RA. Elevated DNA excision repair capacity in the extra embryonic mesoderm of the midgestation mouse embryo. Exp Cell Res. 1996;228:19–28. doi: 10.1006/excr.1996.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasmussen RE, Painter RB. Evidence for repair of UV damaged deoxyribonucleic acid in cultured mammalian cells. Nature. 1964;203:1360–1362. doi: 10.1038/2031360a0. [DOI] [PubMed] [Google Scholar]
- 37.Kam EY, Pitts JD. Computer-assisted grain counting for autoradiography. Comput Programs Biomed. 1984;19:81–83. doi: 10.1016/0010-468x(84)90042-4. [DOI] [PubMed] [Google Scholar]
- 38.Schellart NA, Zweijpfenning RC, van Marle J, Huijsmans DP. Computerized pattern recognition used for grain counting in high resolution autoradiography with low grain densities. Comput Meth Programs Biomed. 1986;23:103–109. doi: 10.1016/0169-2607(86)90105-7. [DOI] [PubMed] [Google Scholar]
- 39.Mize RR, Thouron C, Lucas L, Harlan R. Semiautomatic image analysis for grain counting in in situ hybridization experiments. Neuroimage. 1994;1:163–172. doi: 10.1006/nimg.1994.1001. [DOI] [PubMed] [Google Scholar]
- 40.Steier H, Cleaver JE. Exposure chamber for quantitative ultraviolet photobiology. Lab Prac. 1969;18:1295. [PubMed] [Google Scholar]
- 41.Carreau M, Eveno E, Quilliet X, et al. Development of a new easy complementation assay for DNA repair deficient human syndromes using cloned repair genes. Carcinogenesis. 1995;16:1003–1009. doi: 10.1093/carcin/16.5.1003. [DOI] [PubMed] [Google Scholar]
- 42.Qiao Y, Spitz MR, Shen H, et al. Modulation of repair of ultraviolet damage in the host-cell reactivation assay by polymorphic XPC and XPD/ERCC2 genotypes. Carcino-genesis. 2002;23:295–299. doi: 10.1093/carcin/23.2.295. [DOI] [PubMed] [Google Scholar]
- 43.Svetlova M, Solovjeva L, Pleskach N, et al. Clustered sites of DNA repair synthesis during early nucleotide excision repair in ultraviolet light-irradiated quiescent human fibroblasts. Exp Cell Res. 2002;276:284–295. doi: 10.1006/excr.2002.5519. [DOI] [PubMed] [Google Scholar]
- 44.Forlenza M, Latimer J, Baum A. The effects of stress on DNA repair capacity. Psychol Health. 2000;15:881–891. doi: 10.1080/08870440008405589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moriwaki S, Ray S, Tarone RE, Kraemer KH, Grossman L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: reduced DNA repair capacity and increased DNA mutability. Mutat Res. 1996;364:117–123. doi: 10.1016/0921-8777(96)00029-8. [DOI] [PubMed] [Google Scholar]
- 46.Kraemer KH, Levy DD, Parris CN, et al. Xeroderma pigmentosum and related disorders: examining the linkage between defective DNA repair and cancer. J Invest Dermatol. 1994;103(suppl 5):96S–101S. doi: 10.1111/1523-1747.ep12399329. [DOI] [PubMed] [Google Scholar]
- 47.Kashiyama K, Nakazawa Y, Pilz DT, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013;92:807–819. doi: 10.1016/j.ajhg.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsia KT, Millar MR, King S, et al. DNA repair gene Ercc1 is essential for normal spermatogenesis and oogenesis and for functional integrity of germ cell DNA in the mouse. Development. 2003;130:369–378. doi: 10.1242/dev.00221. [DOI] [PubMed] [Google Scholar]
- 49.Roza L, Vermeulen W, Bergen Henegouwen JB, et al. Effects of microinjected photo-reactivating enzyme on thymine dimer removal and DNA repair synthesis in normal human and xeroderma pigmentosum fibroblasts. Cancer Res. 1990;15:1905–1910. [PubMed] [Google Scholar]
- 50.Ye N, Bianchi MS, Bianchi NO, Holmquist GP. Adaptive enhancement and kinetics of nucleotide excision repair in humans. Mutat Res. 1999;435:43–61. doi: 10.1016/s0921-8777(99)00022-1. [DOI] [PubMed] [Google Scholar]