

Abstract
Behçet’s disease, also known as the Silk Road Disease, is a rare systemic vasculitis disorder of unknown etiology. Recurrent attacks of acute inflammation characterize Behçet’s disease. Frequent oral aphthous ulcers, genital ulcers, skin lesions and ocular lesions are the most common manifestations. Inflammation is typically self-limiting in time and relapsing episodes of clinical manifestations represent a hallmark of Behçet’s disease. Other less frequent yet severe manifestations that have a major prognostic impact involve the eyes, the central nervous system, the main large vessels and the gastrointestinal tract. Behçet’s disease has a heterogeneous onset and is associated with significant morbidity and premature mortality. This study presents a current immunological review of the disease and provides a synopsis of clinical aspects and treatment options.
Electronic supplementary material
The online version of this article (doi:10.1007/s13317-016-0074-1) contains supplementary material, which is available to authorized users.
Keywords: Behçet disease, Physiopathology, Autoimmunity, Inflammation
Description
Behçet’s Disease (BD) is a rare systemic vasculitis disorder of unknown etiology characterized by recurrent attacks of oral aphthous ulcers, genital sores, and ocular lesions (triple-symptom complex). Clinical manifestations of acute inflammation are typically self-limiting in time with relapsing episodes of varied intensity leading to sequelae. Frequency and duration of relapses are unpredictable and follow no discernible pattern of onset. The disease occasionally generates severe manifestations involving the cardiovascular system, the central nervous system, and the gastrointestinal tract. Behçet’s Disease is classified as a systemic vasculitis associated with significant morbidity and mortality, particularly in males with early age onset [1–3]. Currently, there exists no specific diagnostic tool or serum biomarker to identify and quantify the severity of BD; prognosis is difficult to ascertain. Behçet’s disease typically appears between the third and the fourth decades of age, infrequently developing before puberty or after the fifth decade of life. Earlier age onset is correlated with more severe clinical manifestations and mortality [2, 4, 5]. The disease appears to occur more frequently and is associated with more severe ocular, neurological, and cardiovascular clinical manifestations in males [1, 3]. Treatment options are unspecific and aim to relieve symptoms and control disease progression and severity. Disease management usually includes systemic anti-inflammatory and/or immune-modulating drugs [1, 3, 6].
Prevalence of BD worldwide ranges between 0.1/1000 and 1/10,000 with a significant presence in Asian countries 30°–40° north of the Equator from the Mediterranean to Japan, hence the term Silk Road Disease [1, 2]. Turkey has reported the highest prevalence ranging from 80 to 420/100,000 [7]. Disease occurrence in other Asian countries such as Saudi Arabia, Iran, Korea, China, and Japan, has been found to oscillate between 13.5 and 85/100,000 with latter possessing the highest reported incidence in East Asia [8]. Occurrence is far lower in Western countries ranging from 0.12 to 0.64/100,000 [1, 9]. Immigrants from countries with high prevalence tend to have inferior risks of developing the disease pursuant to emigration to low prevalence countries [2, 7]. Disease incidence amongst Turkish individuals in Germany was found to be 21/100,000, which, although significantly higher than that of the German population at large (0.42–0.55/100,000), remains inferior to that of their country of origin [5, 9]. Whereas BD primarily affects males in western Asia, it is more commonplace amongst females in Japan and Korea [5]. Familial aggregation of BD has been reported to occur in 1–18 % of the population, with a higher incidence of familial association in juvenile patients [10]. The pattern of disease inheritance does not follow Mendelian rules as narrated by multi-case family studies [10, 11]. A correlation with HLA class I antigens, notably HLA-B51, has been observed [2, 7].
Clinical features
As a systemic vasculitis disease, BD may affect almost all vascularized systems (Table 1) and is characterized by episodes of relapses and remissions with sequelae [1, 3, 6]. Although several clinical manifestations are associated with BD, the triple-system complex of oral and genital aphthae, and uveitis first described by Behçet in 1937 generally illustrates disease pattern. Children exhibit more frequent perianal aphthosis and arthralgia, less frequent genital ulcers and vascular involvement and a more severe course of uveitis [1, 10–12]. Over the past few years, modern treatment strategies, involving immunosuppressant therapy, and the use of aggressive approaches have led to improvements in the prognosis of severe forms of BD [13, 14]. Prognosis for the disease is usually reserved, especially when ocular, cardiovascular, neurological, and gastrointestinal manifestations appear [6, 15, 16].
Table 1.
Main manifestations of Behçet’s disease
| Manifestation | Prevalence (%) | Time after disease onset | Prognosis | Comments |
|---|---|---|---|---|
| Oral ulcers | 47–86 | – | Favorable | Appear in all patients during their clinical course |
| Genital ulcers | 57–93 | – | Favorable | Lesions of the scrotum will leave a scar |
| Ocular | 30–70 | 2–3 years | Poor | More frequent in males, high morbidity |
| Skin | 38–99 | – | Favorable | Erythema nodosum more frequent in females |
| Joints | 45–60 | – | Favorable | Non-erosive, non-deforming |
| Cardiovascular | 7–49 | 3–16 years | Poor | More frequent in males, high morbidity and mortality |
| Neurological | 5–10 | 5 years | Poor | More frequent in males, long-term morbidity and mortality |
| Gastrointestinal | 3–26 | – | Poor | More frequent in Japan, ileocecal region |
Oral ulcers
Recurrent oral ulcers represent the earliest disease manifestation in 47–86 % of patients [16]. It may take years for the other symptoms to appear afterwards, and oral ulcers are observed in all patients during their clinical course. Lesions have disciform appearance with round and sharp erythematous border, covered with a grayish-white pseudomembrane or a central yellowish fibrinous base and grow rapidly from a flat ulcer to a deep sore [1]. They may occur as single ulcers or in crops and heal with little scarring; prognosis is usually favorable [1, 5]. Oral ulcers most commonly affect the gingival and buccal mucosa, tongue and lips yet may also appear in the soft and hard palates, pharynx, and tonsils [1]. Minor ulcers (<1 cm in diameter) heal without scarring in 4–14 days whereas major ulcers (>1 cm in diameter) are more painful and heal within 2–6 weeks. Herpetiform ulcers occur in recurrent crops of small ulcers that are 0.2–0.3 cm in diameter are painful and may coalesce. Treatment is usually symptomatic and prognosis of oral ulcerations is favorable [5, 16].
Genital ulcers
Genital ulcers develop in 57–93 % of patients [17]. They are painful and morphologically resemble oral ulcers, but are larger, deeper, have more irregular margins and heal with white or pigmented scars [16]. Male genital lesions most commonly involve the scrotum and usually leave a scar that will help with the diagnosis retrospectively. They may also affect the epididymis; penile lesions are less frequent [1]. In females, vulvar, vaginal and cervical lesions are especially common [5]. Rarely, deep vaginal lesions may perforate the bladder resulting in fistulae [16, 17]. Both males and females may develop perineal, perianal and groin lesions [16]. In cases of fistulae and internal lesions, prognosis is unfavorable when the infectious risk is inadequately measured [1, 5].
Ocular manifestations
Ocular disease, involving the retina and the uvea, occurs in 30–70 % of BD patients and is associated with high morbidity [15]. It is the primary cause of blindness in approximately 25 % of patients despite aggressive corticosteroid treatment [15]. Ocular symptoms occur more frequently amongst males and are associated with disease severity, even though prognosis is improving with the use of aggressive immunosuppressant therapy [1, 5]. They usually occur 2–3 years after the onset of oral or genital ulcerations but remain the first disease manifestation in 10–20 % of patients [1]. Ocular disease is a chronic relapsing bilateral non-granulomatous uveitis that may involve the anterior segment, the posterior segment or both (panuveitis) [6, 13]. The latter is associated with worse prognosis and is more common among males [1]. Ocular disease is characterized by the formation of hypopyon—a visible layer of pus in the anterior chamber—observed in one-third of patients [5]. Episodes of anterior uveitis subside spontaneously; repeated attacks result in irreversible structural deformities [13, 17]. Ocular manifestations also include iridocyclitis, keratitis, episcleritis, scleritis, vitritis, vitreous hemorrhage, retinal vasculitis, retinal vein occlusion, retinal neovascularization and optic neuritis [15].
Ocular symptoms include blurred vision, photophobia, lacrimation, floaters, hyperemia and periorbital or global pain [1, 5]. Recurrent inflammatory attacks are associated with secondary complications such as posterior and peripheral anterior synechia, iris atrophy, cataract resulting from inflammation or treatment, secondary glaucoma (occasionally neovascular), atrophic retina, optic atrophy, macular edema, macular degeneration, retinal veins occlusion, sheathed vessels, chorioretinal scars, and proliferative vitreoretinopathy and phthisis bulbi [15]. Prognosis is correlated with frequency, severity of ocular inflammation, and extent of lesions, and remains in many cases unfavorable.
Cutaneous manifestations
Skin involvement affects 38–99 % of BD patients [1, 3]. Cutaneous manifestations commonly include papulopustular (28–96 %) and acne-like lesions [10, 17]. Wounds exhibit a wide distribution affecting the face, limbs, trunk, and buttocks [1]. Skin lesions are characterized by thrombosis and vasculitis [3]. Early lesions exhibit leukocytoclastic vasculitis or neutrophilic vascular reactions whereas mature lesions are characterized by lymphocytic vasculitis [17].
Erythema nodosum lesions occur in 15–78 % of patients, mainly in females, in the lower limbs [1, 5]. Erythema nodosum lesions do not ulcer and may heal spontaneously leaving residual pigmentation. Cutaneous ulcers are rare and only affect 3 % of BD patients [1]. They resemble aphthous ulcers, are recurrent and usually heal with scarring, in the neck, breast, axillae, inguinal region, legs and interdigital skin of the feet [16]. Prognosis for most cutaneous lesions in BD is usually favorable.
Neurological manifestations
Neurological involvement in BD (neuro-BD) occurs in 5–10 % of patients and is more frequent in males [18, 19]. It usually occurs around 5 years after the onset of the disease and is associated with long-term morbidity and mortality [1, 4, 20]. Neurological disease affects the central nervous system (CNS) more frequently than the peripheral nervous system [21, 22].
Neuro-BD may be parenchymal (80 % of patients), non-parenchymal or mixed brain disease [1, 18]. Parenchymal brain disease affects the brainstem and/or basal ganglia and is correlated with a poor prognosis [18, 23]. Non-parenchymal brain disease is characterized by subsets of cerebral venous thromboses, notably dural sinus thrombosis, arterial vasculitis and aseptic meningitis and comprises the most devastating symptom category of BD resulting in a range of neurological deficits that may lead to coma and death [18, 19].
Headache syndromes represent the most common neurological symptom and occur in 70 % of patients [18]. Most parenchymal neuro-BD cases present as subacute meningoencephalitis that exhibit subacute onset and is associated with exacerbation of systemic manifestations [21, 22]. Flare-ups reach a peak within few days and may last for periods of weeks. Brainstem involvement, including ophthalmoparesis, cranial neuropathy and cerebellar or pyramidal dysfunction, has additionally been reported [1, 18, 23, 24]. Cerebral or spinal cord involvement has been observed in association with subcortical dementia and accompanied by ataxia [21, 24]. Cerebral hemispheric involvement, including encephalopathy, hemiparesis, hemisensory loss, seizures and dysphasia, and mental changes, including cognitive dysfunction and psychosis, have been observed as well [18, 20, 24]. Spinal cord involvement, manifested by pyramidal signs in the limbs, sensory level dysfunction and sphincter dysfunction, has also been reported [18, 25]. Other less common clinical symptoms involving the CNS have been reported such as stroke, epilepsy, brain tumor-like neuro-BD, movement disorders, acute meningeal syndrome, optic neuropathy, spinal cord involvement and asymptomatic and subclinical neurological involvement [21, 24, 25].
Non-parenchymal neuro-BD, also referred to as vasculo-BD or angio-BD, involves the main vascular structures of the CNS [23]. Clinical syndromes include vascular disorders, intracranial hypertension and intracranial aneurysms [26]. More rarely, mixed parenchymal and non-parenchymal disease has also been reported [18].
Classically, meningitis or meningoencephalitis, neurological deficits including motor disturbances and brainstem symptoms and psychiatric symptoms including personality changes develop in patients with neuro-BD [5, 18]. These symptoms are associated with disease exacerbations and gradually cause irreversible disability [5]. At late stages, dementia develops in approximately one-third of patients. Cognitive impairment in neuro-BD patients includes poor memory, attention and motivation, in addition to personality changes [27]. Prognosis for neuro-BD, in all its forms, is unfavorable [17, 18].
Cardiovascular manifestations
Behçet’s disease may affect blood vessels of different sizes and types, including arteries and veins, as well as the heart organ [28]. Cardiovascular features have been reported to affect 7–49 % of patients, more frequently in males [1, 29]. They occur 3–16 years after the onset of BD [30].
Vascular BD commonly affects veins causing recurrent superficial thrombophlebitis and deep venous thrombosis in 30–40 % of patients [18, 31, 32]. Thromboses of the superior and inferior vena cava (0.2–9 % of patients), dural sinuses and supra-hepatic veins (2–3.2 % of patients) and pulmonary arterial aneurysms (1 % of patients) are associated with poor prognoses [1, 29, 34]. Occlusion and aneurysms of major arteries commonly lead to bleeding, infarction and organ failure, particularly in instances of pulmonary aneurysm [5, 33]. Rupture of aneurysms may be fatal. At the level of lungs, thrombosis, aneurysm and arteriobronchial fistula cause recurrent episodes of dyspnea, cough, chest pain, and hemoptysis [5].
Cardiac involvement includes pericarditis, myocarditis, endocarditis, mitral valve prolapse, valve lesions, intracardiac thrombosis, endomyocardial fibrosis, myocardiopathy, and coronary artery lesions [30, 34, 35]. The prognosis in these cases is unfavorable with frequent recurrences [1]. The highest direct mortality rate in cardiovascular involvement was attributed to large vessel vasculitis as a result of sudden death by aneurysm rupture or thrombosis (up to 9.8 % of patients in one Turkish study) [4, 12, 30].
Articular manifestations
Articular involvement occurs in 45–60 % of patients and includes arthralgia, monoarthritis or polyarthritis [36, 37]. Non-erosive, non-deforming oligoarthralgia commonly involving the knees, ankles, elbows, and wrists is the most frequent manifestation [1, 5]. Neutrophilic and mononuclear cell infiltrates in the synovium and small vessel lesions with thrombosis typically characterize articular disease [5]. Destructive changes rarely occur in patients with articular involvement [37]. As anti-inflammatory treatment is generally effective, prognosis is usually favorable [38].
Gastrointestinal manifestations
Gastrointestinal involvement occurs in 3–26 % of patients and varies among different populations [39, 40]. It is much more frequent in Japan than in the Middle East and the Mediterranean region [1–3]. Mucosal inflammation and ulceration occur throughout the gastrointestinal tract and are mostly common in the ileocecal region [40, 41]. The esophagus, ascending colon, and transverse colon are less frequently involved [5]. Clinical symptoms include anorexia, vomiting, dyspepsia, diarrhea, melena, abdominal pain and, less frequently, perforation requiring surgical intervention [39, 41]. Prognosis is unfavorable as gastrointestinal involvement is typically acute and chronic.
Diagnosis
A Behçet’s disease diagnosis is typically confirmed by elimination of other disease scenarios, even when a triple-symptom complex is evident [10]. As there exists no targeted diagnostic test for BD, diagnosis of clinical symptoms is challenging, especially when symptoms are non-concomitant. The International Chapel Hill guidelines of 1990 (revised in 2012) propose the presence of recurrent oral ulcerations (at least three times in 1 year) plus any two of the following: recurrent genital ulceration (aphthous ulceration with scarring); eye lesions (anterior uveitis, posterior uveitis, cells in vitreous on slit lamp examination, retinal vasculitis); skin lesions (erythema nodosum, pseudofolliculitis or papulopustular lesions, acneiform nodules in post-adolescent patients not on corticosteroid therapy); and/or a positive pathergy test (read by a physician at 24–48 h) (Supplementary Table 1) [42]. Nevertheless, given the difficulty of diagnosis, it is vital for clinicians to thoroughly investigate the clinical history of BD patients to exclude diseases with similar manifestations before providing confirmation (Table 2) [1].
Table 2.
Common erroneous diagnoses presented to Behçet’s disease patients
| Bullous skin disorders |
| Celiac disease |
| Eales disease |
| Erythema multiforme |
| Familial mediterranean fever |
| Herpes simplex virus infection |
| Inflammatory bowel disease (Crohn’s disease and ulcerative colitis) |
| Mixed connective tissue disease |
| Multiple sclerosis |
| Recurrent aphthous stomatitis |
| Reiter’s syndrome |
| Sarcoidosis |
| Seronegative arthropathies |
| Stevens–Johnson syndrome |
| Sweet’s syndrome |
| Syphilis |
| Systemic lupus erythematosus |
| Vogt–Koyanagi–Harada syndrome |
Behçet’s disease is characterized by episodes of relapses and remissions, and is associated with periods in which patients may exhibit few overt clinical symptoms [1].
Elevated levels of various inflammatory markers are sensitive (but not specific) to BD including C-reactive protein, erythrocyte sedimentation rate, peripheral leukocyte and platelet counts and serum cytokines (TNF-α, IFN-γ, IL-1β, IL-6, and IL-8) [35, 43]. Moderate anemia of chronic disease may also be present; however, autoantibodies, such as antinuclear, rheumatoid factor, cryoglobulinemia and antineutrophil cytoplasmic antibodies (ANCA) have not been detected in BD, hence the classification of the disease as a non-ANCA vasculitis [43]. Pathergy is a characteristic feature of BD and is characterized by a non-specific hyper-reactivity of the skin to minor trauma [1, 10]. The positivity of the test, reduced upon surgical pre-cleansing of the skin, also varies with geographical location (more than 60 % of Middle Eastern patients, 15 % of Korean patients and 5 % of Caucasian patients) [1].
Over the years, five major independent sets of criteria have been used for diagnosing BD, each characterized by its own clinical features and frequencies, as well as nature of criteria that should be met in order for the diagnosis to be positive [44, 45]. All required a confirmation of the three major symptoms initially described by Behçet as a separate clinical entity (oral ulceration, genital ulceration, and eye lesions). The International Study Group (ISG) for BD proposed the main international standards for diagnosing BD [1]. The study group compared the performance of the previously defined sets of criteria and established a new set of symptom manifestations for BD. These criteria, published in 1990 and last updated in 2013, provided simpler means for diagnosis of BD, excluded rare and subjective features, and showed more specificity with little or no loss of sensitivity to previously used tools [5, 48].
In 2004, Lawton et al. conducted a study in an attempt to define a set of clinical features that may be used as a standard index for measurement of BD activity [8]. Five countries participated in this study, namely China, Korea, Iraq, Turkey and UK. Using the Rasch method, the study analyzed 14 items that formed an index of disease activity (Supplementary Fig. 1) [8]. This BD Current Activity Form is used to develop an overall score for a disease activity index in clinical trials on BD involving therapeutic interventions [46].
Pathogenesis
Various hypotheses attempt to shed light on the etiology of BD including innate and adaptive immunity actors as well as environmental factors [47]. Epidemiological data suggest the involvement of both genetic and environmental factors in the development of BD [5, 10, 45]. The disease is assumed to stem from an autoimmune response, which, in turn may be triggered by infectious or other environmental agents in genetically susceptible individuals [1, 5] (Fig. 1).
Fig. 1.
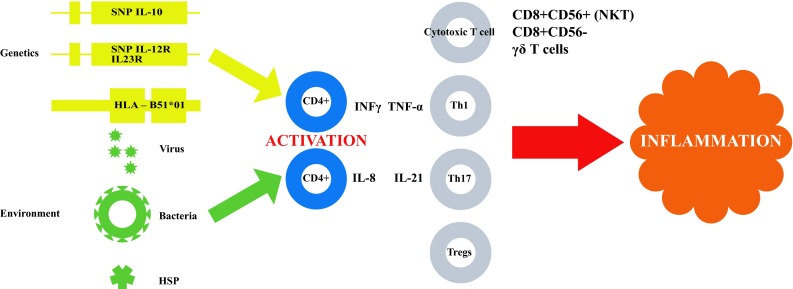
Summary of pathogenesis of Behçet disease. It is presumed that environmental factors trigger CD4+ T cells activation in genetically susceptible individuals, leading to the secretion of cytokines that stimulate other inflammation-inducing immune cells resulting in an uncontrolled autoimmune cascade in vascular tissue. SNP single nucleotide polymorphism, IL interleukin, HLA human Leukocyte Antigen, HSP heat shock proteins, INFγ interferon gamma, TNF-α tumor necrosis factor alpha, CD mature Th cells expressing a surface protein identified by a number, NKT natural killer T cells, Th T helper cells, ϒσ T cells T lymphocytes
Four criteria support evidence of genetic influence on susceptibility to BD: peculiar geographical distribution, occasions of familial aggregation, correlation with class I HLA antigens (HLA-B51) and polymorphisms in genes that control immune responses [2, 7, 48, 49]. The fact that Turkish immigrants in Germany had significantly higher disease prevalence than Turkish nationals who did not immigrate also corroborates the environment influence on susceptibility [7, 54]. Yazici et al. and Ohno et al. reported a close association between HLA-B5 and BD development [7, 54]. They demonstrate that HLA-B5 is heterogeneous in composition and includes at least HLA-B51 and HLA-B52. Within the MHC locus, HLA-B51 and HLA-B5701 were associated with disease pathogenesis, most notably in populations along the Old Silk Route. In Iranian Azari patients, HLA-B35 frequency was higher in the patient than control group, and the frequency of HLA-B51, HLA-B52, and HLA-BW4 was significantly elevated [50].
However, the reported risk increase varied between studies from 1.3 to 16 [7, 54]. Similarly, various independent associations of HLA-A and HLA-C regions have been described, with unspecific evidence that remains to be confirmed [51, 52]. Other genes from the MHC locus have additionally been investigated, including several TNF genes and the MHC class I-related gene (MICA), yet their exact involvement remains undetermined as they may conceivably be activated a posteriori by an HLA-B51-induced inflammatory cascade [55, 58]. Ombrello et al. depicted an association of the MHC-I loci and BD [53]. They propose that the peptide-binding specificity of MHC-I molecules by killer immunoglobulin-like receptors on natural killer cells and T lymphocytes plays a primary role in autoimmunity. Non-MHC genes, namely interleukin 1 (IL-1), coagulation factor V, intercellular adhesion molecule 1 (ICAM-1), and endothelial nitric oxide synthase (eNOS) have been also investigated in BD pathogenesis [56, 59]. Although several associations were determined, definitive conclusions have not been established as to their precise role. Similarly, association findings of genome-wide association studies have identified also established associations with IL10, IL23R, CCR1, STAT4, KLRC4, GIMAP2/GIMAP4, and UBAC2 genes in Behçet’s disease patients of different ethnicities. Deep resequencing of targeted genes identified additional associations with rare variants in TLR4, MEFV, and NOD2 genes as well as shared inflammatory pathways with spondyloarthropathies [51, 52].
Infectious agents such as bacteria and/or viruses have been implicated as possible instigators of a dysfunction in inflammatory response in BD patients [45, 54]. However, most viral, fungal, bacterial and parasitic causes have been eliminated as disease instigators; scans for microorganisms such as Streptococcus sanguis, Herpes simplex virus, Epstein–Barr virus and cytomegalovirus lead to negative results in BD patients during periods of both flare-up and remission [45, 55, 58]. Recent results indicate that both a peculiar dysbiosis of the gut microbiota and a significant decrease of butyrate production are present in patients with BD patients. As butyrate is able to promote differentiation of T regulatory cells, a defect of butyrate production might lead to both reduced Treg responses and activation of immuno-pathological T-effector responses [56, 59].
Matzinger proposed that immune responses in situations similar to BD develop from a continuous autoimmune cascade as a result of signals emitted by injured host cells [55, 58]. This ‘danger model’ reaction proposes that the immune reaction overreacts to external stimuli. T cells and other antigen-presenting cells (APC) take hold of a process that feeds on itself in cases of a favorable genetic terrain. The ‘pattern-recognition theory’ consigns the injury by a ‘non-self’ entity to a permanent aggression, activating an uncontrolled adaptive response. Heat-shock proteins (HSP60) given their resemblance with pathogenic proteins have been proposed in this context [61, 64]. Medzhitov proposes that autoimmune disease such as BD may be an autoantigen-derived autoinflammatory disease [56]. This adaptive reaction to external triggers persists in situations of permanent pathogenic presence via autoantigen-activated dendritic T or B cells. This explanation might help to explain why both anti-microbial agents and immunosuppressant therapies may come across as effective in autoimmune diseases in general and BD in particular [57].
The depiction of Toll-like receptors (TLRs) as innate structures that detect and react to permanent proteins in infectious agents additionally suggests mechanisms by which the body’s reaction to ‘non-self’ develops into a state of autoinflammation [50, 62]. This may play a role in BD for several reasons: either the loss of built-in regulation of TLRs, or the erroneous recognition of self-proteins by TLRs [62]. Cohen proposed that TLRs and the receptors IL-1, IL-18, and IL-33 play a crucial role in these circumstances: a state of hyperactivation becomes permanent, which brings about inflammatory tissue damage as observed in BD [58].
Histopathological observations in BD indicate diffuse necrosis, along with extensive intraluminal transudative fluid [59]. Extravasation of red blood cells has also been noted, suggesting occlusion with recanalization with rare areas of perforation [39, 42]. Diffuse venous thrombosis and focal myonecrosis are likewise frequent [18, 32]. Examined tissue exhibits necrotic and cystic parenchyma, along with high infiltration by mononuclear and polymorphonuclear leukocytes [60, 65]. Dense polymorphonuclear infiltration is commonly observed around arterioles, and is comprised mainly of eosinophils [19]. Macrophages are abundantly observed around venules, which, unlike arterioles, exhibit sparse acute inflammatory cells in infiltrates comprised mainly of lymphocytes [27, 32]. Areas surrounding lesions commonly exhibit edema, astrocytosis, and macrophage infiltrates with scattered microglial nodules [48]. Lymphocytic infiltrates are predominantly constituted of CD3+ T cells, of which CD4+ or CD8+ T cells can be (rarely) identified; CD20+ B cells are also present [61–64]. Some CD68+ macrophages and monocytes have been observed [65]. Consequently, a role for pro-inflammatory mediators secreted by infiltrating T cells and monocytes in inducing apoptosis of neurons has been suggested.
Several studies reported the involvement of specific cytokines as mediators in BD [66–70]. Peripheral blood of BD patients contains IFN-γ and IL-12, suggesting the involvement of Th1 cells [71–73]. Elevated IL-1, IL-6, IL-18, TNF-α and chemokine levels are thought to account for activation of innate and adaptive immune responses [73, 75]. Neutrophils of BD patients are hyperactivated, as shown by their increased phagocytosis and superoxide production, enhanced chemotaxis and elevated production of lysosomal enzymes [57, 73]. Accordingly, lymphocytes of BD patients exhibit abnormal functions as clonal expansion of autoreactive T cells specific for HSP60 peptides [59, 74]. Moreover, γδ T cells are more prevalent in blood and mucosal lesions of BD patients [42, 58]. It has consequently been suggested that interactions between T cells, neutrophils and APCs may contribute to the pathogenesis of BD (Fig. 1) [19, 57, 60, 77].
Saadoun et al. and Geri et al. additionally reported evidence for the involvement of IL-21 in BD [35, 75]. Both studies observed that Th1 cells and IL-21 were significantly increased in serum of active BD patients compared to patients in remission and healthy controls [41, 76, 81]. The addition of serum from active BD patients to that of healthy controls increased the frequency of Th17 and Th1 cells and decreased FoxP3 expression in CD4+ T cells. IL-21-producing CD4+ T cells were notably expanded in peripheral blood of patients with active disease compared to those in remission and healthy controls, and were positively correlated with Th17 cells, and negatively correlated with Tregs [77–79]. Levels of the other IL-17A-promoting cytokines (IL-1β, IL-6, TGF-β, and IL-23) did not show significant difference between the active, remission, and healthy groups [73, 78, 84]. The frequency of Tregs was significantly decreased in BD patients with active disease and remission compared to healthy controls; a similar pattern was noted for activated Tregs, notably in cases of CNS involvement [78]. These results suggest that IL-21 induces the differentiation of Th17 cells and the production of Th17 and polymorphonuclear leukocyte chemoattractants in CNS lesions of BD patients [80, 81]. IL-21 also demonstrated a role in T cell homeostasis of BD [81, 82]. IL-21 may contribute to the pathogenesis of BD by disrupting the equilibrium between Th17 cells and Tregs [77, 83]. This is consistent with the findings of Nurieva et al. who demonstrated the expression of IL-21 by mouse Th17 cells and its autocrine induction of Th17 cell differentiation while inhibiting the expression of FoxP3 [74]. The expression of IL-17 and IL-21 mRNA was significantly increased in Th17 cells compared to other effector cells [45, 84, 85]. IL-21 expression appeared to be induced by IL-6 alone, rather than by an adjoined effect of IL-6 and TGF-β [35, 82, 84]. IL-21 also upregulated its own expression, indicating an autocrine regulation [35, 50, 86]. IL-21 expression was also shown to be regulated by STAT3, unlike IL-17 expression, which was reported to be regulated by RORγ [74]. STAT3-deficient Th cells failed to produce IL-21 mRNA and protein, which were normally expressed in RORγ-deficient Th cells [90]. IL-21 thus appeared to play an important role in Th17 cell differentiation; activated naïve Th cells differentiated into Th17 cells in the presence of IL-21 [89]. This effect of IL-21 was synergized by the addition of TGF-β [82]. Combined treatment enhanced the generation of Th17 cells and inhibited FoxP3 expression [76, 84].
IL-23 was also found to potentiate the effect of IL-21/TGF-β co-treatment as IL-21 upregulated the expression of IL-23R, RORγ and Th17 cytokines and inhibited the expression of TGF-β and FoxP3 [75]. These results indicate that IL-21 selectively regulates Th17 differentiation [70, 79, 89].
Vollmer et al. reported differential effects for IL-21 during the initiation and progression of Experimental Allergic Encephalomyelitis (EAE) [83]. Treatment of mice with IL-21 prior to EAE induction significantly increased disease severity [67]. In contrast, administration of IL-21 during EAE progression had less effect [83]. IL-21 did not affect antigen-specific T cell proliferation, but rather increased autoreactive Th1 cells upon pretreatment [82]. IL-21 also showed differential effects on IFN-γ production by natural killer cells that was enhanced upon pretreatment [67, 70]. IL-21 pretreatment significantly increased the levels of circulating IgG and IgG2b antibodies, thus demonstrating an effect for IL-21 on natural killer cells and on autoantibody production [70, 74]. IL-21 seems to be induced in T cells by IL-6 and is necessary and sufficient for inducing Th17 cell differentiation via STAT3-dependent upregulation of RORγ [72, 74]. Thus, IL-21 acts in an autocrine manner, similar to the action of IFN-γ in Th1 cells and IL-4 in Th2 cells [46, 87]. IL-21 also appears to modulate the balance between Th17 cells and Tregs in an experimental model of colitis [86]. In this latter model, IL-21 treatment reduced the TGF-β-mediated expression of FoxP3 in CD4+ T cells and the suppressive function of Tregs, and preferentially induced the expansion of FoxP3− T cells [89]. Autocrine IL-21 expression suppresses the induction of Tregs and induces the generation of Th17 cells [70, 76, 84–86].
Furthermore, the effects of IL-21 on the differentiation of B cells and generation of plasma cells were reported by Ozaki et al. [67, 88]. IL-21 was found to enhance Ig production, isotype switching and production of plasma cells. Habib et al. reported the requirement of γc chain for IL-21R signaling and for IL-21-induced activation of the JAK/STAT pathway [96]. This study also demonstrated that γc-associated JAK3 tyrosine kinase is an important mediator of γc-dependent IL-21 signaling [89].
Reduced Tregs in BD patients have been shown to suppress antigen-specific T cell responses [71]. Immune tolerance towards self-antigens is initiated in the thymus and involves clonal deletion of autoreactive T lymphocytes. Peripheral exposure to extrathymic self-antigens as well as nonpathogenic foreign substances also plays a role in immune tolerance [60]. One proposed mechanism for peripheral tolerance involves active immune suppression mediated by T cells [71]. This study reported the generation of a unique CD4+ T cell subset that displayed immunoregulatory properties [71]. Mouse ovalbumin-specific naïve CD4+ T cells stimulated with splenic APCs and ovalbumin peptide in the presence of IL-10 displayed a cytokine profile (high levels of IL-10 and IL-5 and low levels of IL-2 and IL-4) that was distinct from that of Th0, Th1 or Th2 cells, and exhibited low proliferation in response to antigenic stimulation [46, 90, 94]. These IL-10-induced cells, now known as Tregs, were designated T regulatory cells 1 (Tr1) [11, 71]. It is worth mentioning that antigen-specific Tr1 cells could be generated in vitro from naïve CD4+ T cells of humans as well [80, 94, 97]. It was further demonstrated that antigen-specific activation of Tr1 inhibits antigen-specific proliferation of other T cells, and thus suppresses antigen-specific T cell responses, in both mice and humans [85, 86]. This may be explained by the cytokine profile of Tr1 cells that includes high levels of the immunosuppressive cytokine IL-10 and low levels of the T cell growth-promoting cytokines IL-2 and IL-4 [82, 93, 97]. Tr1 cells in essence prevented the development of T cell-mediated disease in mice.
Treatment
As no curative solution is currently available, treatment of BD attempts to relieve symptoms, resolve inflammation, limit tissue damage, reduce recurrence frequency and severity, and avoid life-threatening complications. Choice of treatment depends on the combinations of clinical symptoms and the severity of organ involvement, with priority given to treatment of ocular, gastrointestinal, CNS, and cardiovascular manifestations [6, 44, 45].
A variety of approaches have been proposed for BD including anti-inflammatory and immunosuppressive therapies [3, 14, 91]. Long-term treatment has been associated with significant adverse effects and comorbidity and no treatment protocol has demonstrated enduring healing outcomes [83, 92, 93]. Treatment in refractory disease usually involves a combination of therapies including corticosteroids in addition to chemical immunosuppressants and/or biologic monoclonal antibodies (Table 3) [17, 49, 50]. The European League Against Rheumatism (EULAR) proposed comprehensive treatment guidelines for the management of BD [5]. The multidisciplinary recommendations combined current evidence from clinical trials with input from an expert multinational committee (Table 4) [1, 44, 94].
Table 3.
Characteristic therapies for Behçet’s disease
| Treatment | Characteristic clinical manifestation focus | Immunosuppressive mechanisms of action |
|---|---|---|
| Apremilast | Mucocutaneous | Phosphodiesterase-4 inhibitor |
| Anti-Tumor Necrosis Factor-α | Ocular Neurological Cardiovascular Gastrointestinal |
Neutralize biological activity of TNF-α |
| Azathioprine | Articular Ocular Cardiovascular Mucocutaneous |
Inhibit synthesis of DNA and RNA and proliferation of T and B lymphocytes |
| Calcineurin inhibitors | Refractory | Inhibit activation and recruitment of T lymphocytes |
| Colchicine | Articular Mucocutaneous Ocular Neurological Cardiovascular Gastrointestinal |
Inhibit neutrophil function |
| Corticosteroids | Articular Mucocutaneous Ocular Neurological Cardiovascular Gastrointestinal |
Inhibit neutrophil function General immunosuppressive activity |
| Cyclosporine A (alkylating agents) | Neurological Ocular |
Inhibit lymphocyte function |
| Dapsone | Mucocutaneous | Antibacterial agent |
| Interferon-α | Ocular Neurological Cardiovascular Gastrointestinal |
Antiviral activity |
| Methotrexate | Mucocutaneous Neurological Ocular Articular |
Inhibit synthesis of DNA, RNA, and thymidylates |
| Pentoxifylline | Mucocutaneous | Inhibit synthesis of cytokines |
| Sulfasalazine | Articular Gastrointestinal |
General immunosuppressive activity |
| Thalidomide | Mucocutaneous Gastrointestinal |
Unidentified immunomodulatory activity |
Table 4.
EULAR general treatment recommendations for the treatment of Behçet’s disease
| Any patient with BD and inflammatory eye disease affecting the posterior segment should be on a treatment regime that includes azathioprine and systemic corticosteroids |
| If the patient has severe eye disease defined as >2 lines of drop in visual acuity on a 10/10 scale and/or retinal disease (retinal vasculitis or macular involvement), it is recommended that either cyclosporine A or infliximab be used in combination with azathioprine and corticosteroids. Alternatively IFN-α with or without corticosteroids could be used instead |
| There is no firm evidence to guide the management of major vessel disease in BD. For the management of acute deep vein thrombosis in BD, immunosuppressive agents such as corticosteroids, azathioprine, cyclophosphamide or cyclosporine A are recommended. For the management of pulmonary and peripheral arterial aneurysms, cyclophosphamide and corticosteroids are recommended |
| Similarly, there are no controlled data on, or evidence of benefit from uncontrolled experience with anticoagulants, antiplatelet or anti-fibrinolytic agents in the management of deep vein thrombosis or for the use of anticoagulation for the arterial lesions of BD |
| There is no evidence-based treatment that can be recommended for the management of gastrointestinal involvement of BD. Agents such as sulfasalazine, corticosteroids, azathioprine, TNF-α antagonists and thalidomide should be tried first before surgery, except in emergencies |
| In most patients with BD, arthritis can be managed with colchicine |
| There are no controlled data to guide the management of CNS involvement in BD. For parenchymal involvement, agents to be tried may include corticosteroids, IFN-α, azathioprine, cyclophosphamide, methotrexate and TNFα antagonists. For dural sinus thrombosis, corticosteroids are recommended |
| Cyclosporine A should not be used in BD patients with central nervous system involvement unless necessary for intraocular inflammation |
| The decision to treat skin and mucosa involvement will depend on the perceived severity by the doctor and the patient. Mucocutaneous involvement should be treated according to the dominant or codominant lesions present. Topical measures (i.e., local corticosteroids) should be the first line of treatment for isolated oral and genital ulcers. Acne-like lesions are usually of cosmetic concern only. Thus, topical measures as used in acne vulgaris are sufficient. Colchicine should be preferred when the dominant lesion is erythema nodosum. Leg ulcers in BD might have different causes. Treatment should be planned accordingly. Azathioprine, IFN-α and TNF-α antagonists may be considered in resistant cases |
As mentioned above, prior evidence suggests the involvement of IL-17, IL-21, and TGF-β in the pathogenesis of BD as the level of these markers was correlated with disease activity [73, 78, 82, 84]. Treatments that aim to target these cytokines may constitute a promising target for novel therapy [94, 95, 97, 98]. In this context, IL-21 blocking agents (neutralizing antibodies or receptors) have been suggested for patients with severe or refractory BD, given that IL-21 blockade restored the altered balance between Th17 cells and Tregs [35, 67, 70, 89]. Further studies are also warranted to elucidate signaling pathways associated with IL-17 in BD patients with the goal of developing targeted therapies [73, 75, 90]. Similarly, the regulation of anti-inflammatory cytokines such as TGF-β by current treatments such as Aprelimast show significant promise [95].
Conclusion
Irrespective of recent therapeutic advancements, the clinical prognosis of BD patients remains unfavorable. The primary goal is to focus on identifying the pathogenesis of the disease and providing therapies that inhibit the underlying autoimmune inflammatory cascade. Current research points towards targeted cytokines that play a role in the physiopathology of Behçet’s disease.
Highlights
Recurrent oral and genital ulcers and ocular lesions characterize Behçet’s disease (BD).
Severe manifestations involve the eyes, the CNS, the main large vessels and the gastrointestinal tract.
Innate and adaptive immunity responses result in an uncontrolled autoimmune cascade in vascular tissue.
Environmental factors trigger T cells activation in genetically susceptible individuals, resulting in secretion of cytokines that stimulate other inflammation-inducing immune cells.
New treatments point towards targeted cytokines such as TNF-alpha, IL-21 and IL-17.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors wish to thank the precious cooperation of the i2 lab—Integrative Immunology at Université Pierre et Marie Curie—The Sorbonne Universities, as well as the Department of Internal Medicine and Clinical Immunology at the La Pitié-Salpêtrière Hospital.
Abbreviations
- SNP
Single nucleotide polymorphism
- IL-
Interleukin
- HLA
Human leukocyte antigen
- HSP
Heat-shock proteins
- INFγ
Interferon gamma
- TNF-α
Tumor necrosis factor alpha
- CD
Mature Th cells expressing a surface protein identified by a number
- NKT
Natural killer T cells
- Th
T helper cells
- ϒσ T cells
T lymphocytes
Compliance with ethical standards
Funding
None.
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Mendes D, Correia M, Barbedo M, Vaio T, Mota M, Gonçalves O, Valente J. Behçet’s disease—a contemporary review. J Autoimmun. 2009;32(3–4):178–188. doi: 10.1016/j.jaut.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Verity DH, Marr JE, Ohno S, Wallace GR, Stanford MR. Behçet’s disease, the silk road and HLA-B51: historical and geographical perspectives. Tissue Antigens. 1999;54(3):213–220. doi: 10.1034/j.1399-0039.1999.540301.x. [DOI] [PubMed] [Google Scholar]
- 3.Sfikakis PP, Markomichelakis N, Alpsoy E, Assaad-Khalil S, Bodaghi B, Gul A, et al. Anti-TNF therapy in the management of behçet’s disease-review and basis for recommendations. Rheumatology (Oxford) 2007;46(5):736–741. doi: 10.1093/rheumatology/kem034. [DOI] [PubMed] [Google Scholar]
- 4.Saadoun D, Wechsler B, Desseaux K, Le Thi Huong D, Amoura Z, Resche-Rigon M, Cacoub P. Mortality in Behçet’s disease. Arthritis Rheum. 2010;62(9):2806–2812. doi: 10.1002/art.27568. [DOI] [PubMed] [Google Scholar]
- 5.Sakane T, Takeno M, Suzuki N. Behçet’s disease. N Engl J Med. 1999;341(17):1284–1291. doi: 10.1056/NEJM199910213411707. [DOI] [PubMed] [Google Scholar]
- 6.Sfikakis PP. Behçet’s disease: a new target for anti-tumor necrosis factor treatment. Ann Rheum Dis. 2002;61(Suppl 2):51–53. doi: 10.1136/ard.61.suppl_2.ii51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yazici H, Akokan G, Yalçin B, Müftüoğlu A. The high prevalence of HLA-B5 in Behçet’s disease. Clin Exp Immunol. 1977;30(2):259–261. [PMC free article] [PubMed] [Google Scholar]
- 8.Lawton G, Bhakta BB, Chamberlain MA, Tennant A. The Behçet’s disease activity index. Rheumatology (Oxford) 2004;43(1):73–78. doi: 10.1093/rheumatology/keg453. [DOI] [PubMed] [Google Scholar]
- 9.Savey L, Resche-Rigon M, Wechsler B, Comarmond C, Piette JC, Cacoub P, Saadoun D. Ethnicity and association with disease manifestations and mortality in Behçet’s disease. Orphanet J Rare Dis. 2014;27(9):42. doi: 10.1186/1750-1172-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gül A, Inanç M, Ocal L, Aral O, Koniçe M. Familial aggregation of Behçet’s disease in turkey. Ann Rheum Dis. 2000;59(8):622–625. doi: 10.1136/ard.59.8.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat Genet. 2010;42(8):698–702. doi: 10.1038/ng.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saadoun D, Wechsler B, Terrada C, Hajage D, Le Thi Huong D, Resche-Rigon M, et al. Azathioprine in severe uveitis of Behçet’s disease. Arthritis Care Res (Hoboken). 2010;62(12):1733–1738. doi: 10.1002/acr.20308. [DOI] [PubMed] [Google Scholar]
- 13.Desbois AC, Wechsler B, Resche-Rigon M, Piette JC, Huong D Le T, Amoura Z, et al. Immunosuppressants reduce venous thrombosis relapse in Behçet’s disease. Arthritis Rheum. 2012;64(8):2753–2760. doi: 10.1002/art.34450. [DOI] [PubMed] [Google Scholar]
- 14.Kump LI, Moeller KL, Reed GF, Kurup SK, Nussenblatt RB, Levy-Clarke GA. Behçet’s disease: comparing 3 decades of treatment response at the national eye institute. Can J Ophthalmol. 2008;43(4):468–472. doi: 10.3129/i08-080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alpsoy E, Zouboulis CC, Ehrlich GE. Mucocutaneous lesions of Behçet’s disease. Yonsei Med J. 2007;48(4):573–585. doi: 10.3349/ymj.2007.48.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alpsoy E, Donmez L, Bacanli A, Apaydin C, Butun B. Review of the chronology of clinical manifestations in 60 patients with Behçet’s disease. Dermatology. 2003;207(4):354–356. doi: 10.1159/000074113. [DOI] [PubMed] [Google Scholar]
- 17.Al-Araji A. Neuro-Behçet’s disease: epidemiology, clinical characteristics, and management. Lancet Neurol. 2009;8(2):192. doi: 10.1016/S1474-4422(09)70015-8. [DOI] [PubMed] [Google Scholar]
- 18.Arai Y, Kohno S, Takahashi Y, Miyajima Y, Tsutusi Y. Autopsy case of neuro-Behçet’s disease with multifocal neutrophilic perivascular inflammation. Neuropathology. 2006;26(6):579–585. doi: 10.1111/j.1440-1789.2006.00734.x. [DOI] [PubMed] [Google Scholar]
- 19.Hadfield MG, Aydin A, Lippman HR, Sanders KM. Neuro-Behçet’s disease. Clin Neuropathol. 1997;16(2):55–60. [PubMed] [Google Scholar]
- 20.Noel N, Bernard R, Wechsler B, Resche-Rigon M, Depaz R, Le Thi Huong Boutin D, et al. Long-term outcome of neuro-Behçet’s disease. Arthritis Rheum. 2014;66(5):1306–1314. doi: 10.1002/art.38351. [DOI] [PubMed] [Google Scholar]
- 21.Kidd D. Neurological complications of Behçet’s syndrome. Curr Neurol Neurosc Rep. 2012;12(6):675–679. doi: 10.1007/s11910-012-0316-1. [DOI] [PubMed] [Google Scholar]
- 22.Siva A, Kantarci OH, Saip S, Altintas A, Hamuryudan V, Islak C, et al. Behçet’s disease: diagnostic and prognostic aspects of neurological involvement. J Neurol. 2001;248(2):95–103. doi: 10.1007/s004150170242. [DOI] [PubMed] [Google Scholar]
- 23.Kikuchi H, Aramaki K, Hirohata S. Effect of infliximab in progressive neuro-Behçet’s syndrome. J Neurol Sci. 2008;272(1):99–105. doi: 10.1016/j.jns.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Saadoun D, Wechsler B, Resche-Rigon M, Trad S, Le Thi Huong D, Sbai A, et al. Cerebral venous thrombosis in Behçet’s disease. Arthritis Rheum. 2009;61(4):518–526. doi: 10.1002/art.24393. [DOI] [PubMed] [Google Scholar]
- 25.Akman-Demir G. Clinical patterns of neurological involvement in Behçet’s disease: evaluation of 200 patients. Brain. 1999;122(11):2171–2182. doi: 10.1093/brain/122.11.2171. [DOI] [PubMed] [Google Scholar]
- 26.Kizilkilic O, Albayram S, Adaletli I, Ak H, Islak C, Kocer N. Endovascular treatment of Behçet’s disease-associated intracranial aneurysms: report of two cases and review of the literature. Neuroradiology. 2003;45(5):328–334. doi: 10.1007/s00234-003-0952-x. [DOI] [PubMed] [Google Scholar]
- 27.Öktem-Tanör Ö, Baykan-Kurt B, Gürvit IH, Akman-Demir G, Serdaroğlu P. Neuropsychological follow-up of 12 patients with neuro-Behçet’s disease. J Neurol. 1999;246(2):113–119. doi: 10.1007/s004150050317. [DOI] [PubMed] [Google Scholar]
- 28.Saadoun D, Asli B, Wechsler B, Houman H, Geri G, Desseaux K, et al. Long-term outcome of arterial lesions in Behçet’s disease: a series of 101 patients. Medicine (Baltimore) 2012;91(1):18–24. doi: 10.1097/MD.0b013e3182428126. [DOI] [PubMed] [Google Scholar]
- 29.Desbois A, Wechsler B, Cluzel P, Helft G, Boutin D, Piette JC, et al. Cardiovascular involvement in Behçet’s disease. Rev Med Int. 2014;35(2):103–111. doi: 10.1016/j.revmed.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Tursen U, Gurler A, Boyvat A. Evaluation of clinical findings according to sex in 2313 turkish patients with Behçet’s disease. Int J Dermatol. 2003;42(5):346–351. doi: 10.1046/j.1365-4362.2003.01741.x. [DOI] [PubMed] [Google Scholar]
- 31.Kural-Seyahi E, Fresko I, Seyahi N, Ozyazgan Y, Mat C, Hamuryudan V, et al. The long-term mortality and morbidity of Behçet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine. 2003;82(1):60–76. doi: 10.1097/00005792-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Desbois A, Rautou P, Biard L, Belmatoug N, Wechsler B, Resche-Rigon M, et al. Behçet’s disease in Budd-Chiari syndrome. Orphanet J Rare Dis. 2014;9(1):104. doi: 10.1186/s13023-014-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamuryudan V, Yurdakul S, Moral F, et al. Pulmonary arterial aneurysms in behçet’s syndrome: a report of 24 cases. Br J Rheumatol. 1994;33(1):48–51. doi: 10.1093/rheumatology/33.1.48. [DOI] [PubMed] [Google Scholar]
- 34.Geri G, Terrier B, Rosenzwajg M, Wechsler B, Touzot M, Seilhean D, et al. Critical role of IL-21 in modulating Th17 and regulatory T cells in Behçet’s disease. J Allergy Clin Immunol. 2011;128(3):655. doi: 10.1016/j.jaci.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 35.Atzeni F, Sarzi-Puttini P, Doria A, Boiardi L, Pipitone N, Salvarani C. Behçet’s disease and cardiovascular involvement. Lupus. 2005;14(9):723–726. doi: 10.1191/0961203305lu2208oa. [DOI] [PubMed] [Google Scholar]
- 36.Ait Badi MA, Zyani M, Kaddouri S, Niamane R, Hda A, Algayres J. Les manifestations articulaires de la maladie de Behçet. À propos de 79 cas. Rev Med Int. 2008;29(4):277–282. doi: 10.1016/j.revmed.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 37.Kaklamani VG, Vaiopoulos G, Kaklamanis PG. Behçet’s disease. Sem Arthritis Rheum. 1998;27(4):197–217. doi: 10.1016/S0049-0172(98)80001-2. [DOI] [PubMed] [Google Scholar]
- 38.Kastner DL. Intermittent and periodic arthritic syndromes. In: Koopman WJ (ed.) Arthritis and allied conditions: a textbook of rheumatology. Vol. 1. 13th ed. Baltimore: Williams and Wilkins; 1997:1279–1306
- 39.Beales IL. Gastrointestinal involvement in Behçet’s syndrome. Am J Gastroenterol. 1998;93(12):2633. doi: 10.1111/j.1572-0241.1998.02633.x. [DOI] [PubMed] [Google Scholar]
- 40.Marshall SE. Behçet’s disease. Best Pract Res Clin Rheumatol. 2004;18(3):291–311. doi: 10.1016/j.berh.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 41.Lee KS, Kim SJ, Lee BC, Yoon DS, Lee WJ, Chi HS. Surgical treatment of intestinal Behçet’s disease. Yonsei Med J. 1997;38(6):455–460. doi: 10.3349/ymj.1997.38.6.455. [DOI] [PubMed] [Google Scholar]
- 42.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65:1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 43.Link J, Söderström M, Olsson T, Höjeberg B, Ljungdahl A, Link H. Increased transforming growth factor-beta, interleukin-4, and interferon-gamma in multiple sclerosis. Ann Neurology. 1994;36(3):379–386. doi: 10.1002/ana.410360309. [DOI] [PubMed] [Google Scholar]
- 44.Cho SB, Cho S, Bang D. New insights in the clinical understanding of Behçet’s disease. Yonsei Med J. 2012;53(1):35–42. doi: 10.3349/ymj.2012.53.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.International study group for Behçet’s disease Criteria for diagnosis of Behçet’s disease. Lancet. 1990;335(8697):1078–1080. [PubMed] [Google Scholar]
- 46.Türsen Ü. Activation markers in Behçet disease. Turkderm. 2009;43:74–86. [Google Scholar]
- 47.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 48.de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behçet’s disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 2009;61(10):1287–1296. doi: 10.1002/art.24642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohno S, Ohguchi M, Hirose S, Matsuda H, Wakisaka A, Aizawa M. Close association of HLA-Bw51 with Behçet’s disease. Arch Ophthalmol. 1982;100(9):1455–1458. doi: 10.1001/archopht.1982.01030040433013. [DOI] [PubMed] [Google Scholar]
- 50.Shahneh FZ, Hamzavi F, Bayazi B, Bandehagh A, Baradaran B. New insights into HLA class I association to Behçet’s syndrome in Iranian Azari patients. Auto Immun Highlights. 2013;4(3):101–102. doi: 10.1007/s13317-013-0047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gül A. Genetics of Behçetʼs disease: lessons learned from genomewide association studies. Curr Opin Rheumatol. 2014;26:56–63. doi: 10.1097/BOR.0000000000000003. [DOI] [PubMed] [Google Scholar]
- 52.Takeuchi M, Kastner DL, Remmers EF. The immunogenetics of Behçet’s disease: a comprehensive review. J Autoimmun. 2015;64:137–148. doi: 10.1016/j.jaut.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ombrello MJ, Kirino Y, de Paul Bakker IW, Gül A, Kastner DL, Remmers EF. Behçet disease-associated MHC class I residues implicate antigen binding and regulation of cell-mediated cytotoxicity. Proc Natl Acad Sci USA. 2014;111:8867–8872. doi: 10.1073/pnas.1406575111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kobayashi M, Ito M, Nakagawa A, Matsushita M, Nishikimi N, Sakurai T, Nimura Y. Neutrophil and endothelial cell activation in the vasa vasorum in vasculo-Behçet disease. Histopathology. 2000;36(4):362–371. doi: 10.1046/j.1365-2559.2000.00859.x. [DOI] [PubMed] [Google Scholar]
- 55.Lehner T. The role of heat shock protein, microbial and autoimmune agents in the aetiology of Behçet’s disease. Int Rev Immunol. 1997;14(1):21–32. doi: 10.3109/08830189709116842. [DOI] [PubMed] [Google Scholar]
- 56.Consolandi C, Turroni S, Emmi G, Severgnini M, Fiori J, Peano C, et al. Behçet’s syndrome patients exhibit specific microbiome signature. Autoimmun Rev. 2015;14(4):269–276. doi: 10.1016/j.autrev.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Hirohata S, Oka H, Mizushima Y. Streptococcal-related antigens stimulate production of IL6 and interferon-γ by T cells from patients with Behçet’s disease. Cell Immunol. 1992;140(2):410–419. doi: 10.1016/0008-8749(92)90207-6. [DOI] [PubMed] [Google Scholar]
- 58.The Matzinger P, Model Danger. A Renewed Sense of Self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 59.Medzhitov R, Janeway CA. Decoding the patterns of self and non self by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 60.Direskeneli H. Autoimmunity vs autoinflammation in Behcet’s disease: do we oversimplify a complex disorder? Rheumatology (Oxford) 2006;45:1461–1465. doi: 10.1093/rheumatology/kel329. [DOI] [PubMed] [Google Scholar]
- 61.Cohen P. The TLR and IL-1 signalling network at a glance. J Cell Sci. 2014;127:2383–2390. doi: 10.1242/jcs.149831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirohata S. Histopathology of central nervous system lesions in Behçet’s disease. J Neurol Sci. 2008;267(1):41–47. doi: 10.1016/j.jns.2007.09.041. [DOI] [PubMed] [Google Scholar]
- 63.Kidd G, Kivisäkk P, Ransohoff RM. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3(7):569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 64.Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of blimp-1 and bcl-6. J Immunol. 2004;173(9):5361. doi: 10.4049/jimmunol.173.9.5361. [DOI] [PubMed] [Google Scholar]
- 65.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172(1):146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23(1):447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 67.Bucher C, Koch L, Vogtenhuber C, Goren E, Munger M, Panoskaltsis-Mortari A, et al. IL-21 blockade reduces graft-versus-host-disease mortality by supporting inducible T regulatory cell generation. Blood. 2009;114(26):5375–5384. doi: 10.1182/blood-2009-05-221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Garra A, Rouleau M, Antonenko S, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389(6652):737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 69.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26(3):371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 70.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25(1):821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 71.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 72.Lee JW, Wang P, Kattah MG, Youssef S, Steinman L, DeFea K, Straus DS. Differential regulation of chemokines by IL-17 in colonic epithelial cells. J Immunol. 2008;181(9):6536. doi: 10.4049/jimmunol.181.9.6536. [DOI] [PubMed] [Google Scholar]
- 73.Eastaff-Leung N, Mabarrack N, Barbour A, Cummins A, Barry S. Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease. J Clin Immunol. 2010; 2009;30(1):80–9 [DOI] [PubMed]
- 74.de Pineton Chambrun M, Wechsler B, Geri G, Cacoub P, Saadoun D. New insights into the pathogenesis of Behçet’s disease. Autoimmun Rev. 2012;11(10):687–698. doi: 10.1016/j.autrev.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 75.Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121(7):906–915. doi: 10.1161/CIRCULATIONAHA.109.872903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakane T. New perspective on Behçet’s disease. Int Rev Immunol. 1997;14(1):89–96. doi: 10.3109/08830189709116847. [DOI] [PubMed] [Google Scholar]
- 77.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human Th17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13(10):1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365:2067–2077. doi: 10.1056/NEJMoa1105143. [DOI] [PubMed] [Google Scholar]
- 79.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448(7152):480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 80.Vollmer TL, Liu R, Price M, Rhodes S, La Cava A, Shi F. Differential effects of IL-21 during initiation and progression of autoimmunity against neuroantigen. J Immunol. 2005;174(5):2696–2701. doi: 10.4049/jimmunol.174.5.2696. [DOI] [PubMed] [Google Scholar]
- 81.Brustle A, Heink S, Huber M, Rosenplänter C, Stadelmann C, Yu P, et al. The development of inflammatory Th-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8(9):958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- 82.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6):899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 83.Bottinelli D, Uccelli A, Engelhardt B, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of T H-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10(5):514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 84.Engelhardt B, Wolburg-Buchholz K, Wolburg H. Involvement of the choroid plexus in central nervous system inflammation. Micros Res Tech. 2001;52(1):112–129. doi: 10.1002/1097-0029(20010101)52:1<112::AID-JEMT13>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 85.Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, Pernis AB. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity. 2008;29(6):899–911. doi: 10.1016/j.immuni.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fantini MC, Rizzo A, Fina D, Caruso R, Becker C, Neurath MF, et al. IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur J Immunol. 2007;37(11):3155–3163. doi: 10.1002/eji.200737766. [DOI] [PubMed] [Google Scholar]
- 87.Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5(2):101–104. doi: 10.1191/135245899678847275. [DOI] [PubMed] [Google Scholar]
- 88.Dagur PK, Biancotto A, Stansky E, Sen HN, Nussenblatt RB, McCoy JP. Secretion of interleukin-17 by CD8+ T cells expressing CD146 (MCAM) Clin Immunol. 2014;152:36–47. doi: 10.1016/j.clim.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of pro-inflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 90.Seder RA, Paul WE, Davis MM, de Fazekas St Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med. 1992;176(4):1091–1098. doi: 10.1084/jem.176.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu X, Tan Y, Xing Q, Wang S. IL-21 accelerates xenogeneic graft-versus-host disease correlated with increased B-cell proliferation. Protein Cell. 2013;4(11):863–871. doi: 10.1007/s13238-013-3088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Habib T, Senadheera S, Weinberg K, Kaushansky K. The common gamma chain (gamma c) is a required signaling component of the IL-21 receptor and supports IL-21-induced cell proliferation via JAK3. Biochemistry. 2002;41(27):8725. doi: 10.1021/bi0202023. [DOI] [PubMed] [Google Scholar]
- 93.Touzot M, Cacoub P, Bodaghi B, Soumelis V, Saadoun D. IFN-α induces IL-10 production and tilt the balance between Th1 and Th17 in Behçet disease. Autoimmun Rev. 2015;14(5):370–375. doi: 10.1016/j.autrev.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 94.Comarmond C, Wechsler B, Bodaghi B, Cacoub P, Saadoun D. Biotherapies in Behçet’s disease. Autoimmun Rev. 2014;13(7):762–769. doi: 10.1016/j.autrev.2014.01.056. [DOI] [PubMed] [Google Scholar]
- 95.Takamoto M, Kaburaki T, Numaga J, Fujino Y, Kawashima H. Long-term infliximab treatment for behçet’s disease. Jpn J Ophthalmol. 2007;51(3):239–240. doi: 10.1007/s10384-006-0424-z. [DOI] [PubMed] [Google Scholar]
- 96.Sbaï A, Wechsler B, Duhaut P, Du-Boutin LT, Amoura Z, Cacoub P, et al. Neuro-Behçet’s disease (isolated cerebral thrombophlebitis excluded). Clinical pattern, prognostic factors, treatment and long term follow-up. Adv Exp Med Biol. 2003;528:371–376. doi: 10.1007/0-306-48382-3_75. [DOI] [PubMed] [Google Scholar]
- 97.Wechsler B, Lê Thi Huong DB, Saadoun D. EULAR recommendations for the management of Behçet’s disease: evidence-based or experience-based medicine. Rev Med Interne. 2009;30(11):939–941. doi: 10.1016/j.revmed.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 98.Hatemi G, Melikoglu M, Tunc R, et al. Apremilast for Behçet’s syndrome–a phase 2, placebo-controlled study. N Engl J Med. 2015;372:1510. doi: 10.1056/NEJMoa1408684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.