

Abstract
A male Japanese domestic cat with retarded growth in Hokkaido, Japan, showed progressive motor dysfunction, such as ataxia starting at 3 months of age and tremors, visual disorder and seizure after 4 months of age. Finally, the cat died of neurological deterioration at 9 months of age. Approximately half of the peripheral blood lymphocytes had multiple abnormal vacuoles. Magnetic resonance imaging showed bisymmetrical hyperintensity in the white matter of the parietal and occipital lobes in the forebrain on T2-weighted and fluid-attenuated inversion recovery images, and mild encephalatrophy of the olfactory bulbs and temporal lobes. The activity of lysosomal acid β-galactosidase in leukocytes was negligible, resulting in the biochemical diagnosis of GM1 gangliosidosis. Histologically, swollen neurons characterized by accumulation of pale, slightly granular cytoplasmic materials were observed throughout the central nervous system. Dysmyelination or demyelination and gemistocytic astrocytosis were observed in the white matter. Ultrastructually, membranous cytoplasmic bodies were detected in the lysosomes of neurons. However, genetic analysis did not identify the c.1448G>C mutation, which is the single known mutation of feline GM1 gangliosidosis, suggesting that the cat was affected with a new variant of the feline disease.
Keywords: β-galactosidase deficiency, feline GM1 gangliosidosis, Japanese domestic cat, lysosomal storage disease
GM1 gangliosidosis is a lysosomal storage disease that affects the brain, resulting in progressive neurodegeneration and premature death in both humans and animals [16]. The disease is caused by an autosomal recessive deficiency in lysosomal acid β-galactosidase, which is encoded by the GLB1 gene.
Feline GM1 gangliosidosis has been reported in Siamese cats in Japan [7] and the United States [2] and in Korat cats in Italy [6]. The disease also has been reported in non-purebred domestic cats in the United Kingdom [1, 4, 5, 15], Japan [11, 14] and Bangladesh [18]. To date, only one pathogenic mutation has been identified as a single nucleotide substitution from guanine to cytosine in exon 14 at nucleotide position 1448 (c.1448G>C) in the coding region of the feline GLB1 gene, resulting in the substitution of arginine with proline at amino acid position 483 (p.R483P). This mutation has been found in Siamese cats in the United States [12] and Japan [19] and in Korat cats in the United States, Canada and several European countries [3, 20]. The same mutation was also found in non-purebred domestic cats [18]. In feline GM1 gangliosidosis caused by this mutation, affected cats generally manifest neurological signs of progressive motor dysfunctions starting from 4 to 6 months of age and die prematurely at approximately 1 year of age.
The present case report, taken from Hokkaido, Japan, describes the clinical, magnetic resonance imaging (MRI), biochemical and pathological features in a Japanese domestic cat that was diagnosed biochemically with GM1 gangliosidosis that was not caused by the known c.1448G>C mutation, suggesting a new variant of the feline disease.
A 4-month-old, 1.78-kg, male Japanese domestic cat was referred to the Rakuno Gakuen University Veterinary Teaching Hospital (RGUVTH) in Hokkaido, Japan, for a complaint of progressive motor dysfunctions. When a combination vaccine was inoculated at 2 months of age at a private animal hospital (Sugiura Pet Clinic), no neurological abnormality was recognized. Three weeks after the vaccination, the cat had slight hindlimb ataxia with retarded growth and a hoarse cry at 3 months of age. A veterinarian in the private animal hospital prescribed prednisolone at that time, which was ineffective. At the initial examination at the RGUVTH, the clinical signs included progressive ataxia with a 6-week history of progression, head tremor with a 1-week history of progression and visual disorder with bilateral loss of menace response at 4 months of age. The neurological examination also revealed slight loss of postural reactions, such as decreased level in wheel barrowing and extensor, postural thrust tests and hyperreflexia of the spinal nerves in all four limbs. However, the cranial nerve reactions; mental status; and the olfactory, auditory and tactile senses were intact.
At 6 months of age, the cat’s ataxia and tremors worsened and generalized, and the owner reported an episode of seizures. The postural reaction was decreased, and the cat was not able to walk. At 7 months of age, the cat became bedridden. Although the cat survived until 9 months of age by compulsory feeding and compressive urination and defecation, the cat finally died of status epilepticus.
The hematological, MRI and cerebrospinal fluid (CSF) examinations were carried out at the RGUVTH when the cat was 4 months of age. The blood cell counts and serum profiles appeared within normal ranges, except for an increased activity in alkaline phosphatase (ALP; 2,344 U/l). Peripheral blood lymphocytes showed the presence of multiple abnormal vacuoles (Fig. 1), and the frequency of vacuolated lymphocytes was 47% of whole lymphocytes. MRI showed bisymmetrical hyperintensity in the white matter of the forebrain, especially parietal and occipital lobes, on the T2-weighted (T2W) and fluid-attenuated inversion recovery (FLAIR) images. Mild encephalatrophy was detected in both olfactory bulbs (Fig. 2A and 2B) and temporal lobes (Fig. 2C and 2D). However, there was no such lesion of increased T2W and FLAIR intensity in the white matter of the frontal and temporal lobes, the interbrain, brain stem and cerebellum. The corpus callosum and the rostral commissure were difficult to recognize in the T2W sagittal image (Fig. 2E). Gadodiamide did not enhance any lesion on the T1-weighted image (data not shown). Results of CSF analyses including the cell number (0.3/µl), specific gravity (1.005), protein concentration (12.7 mg/dl) and glucose concentration (144 mg/dl) appeared to be within normal ranges, but some lymphocytes in the CSF had multiple vacuoles like those in the peripheral blood (data not shown).
Fig. 1.
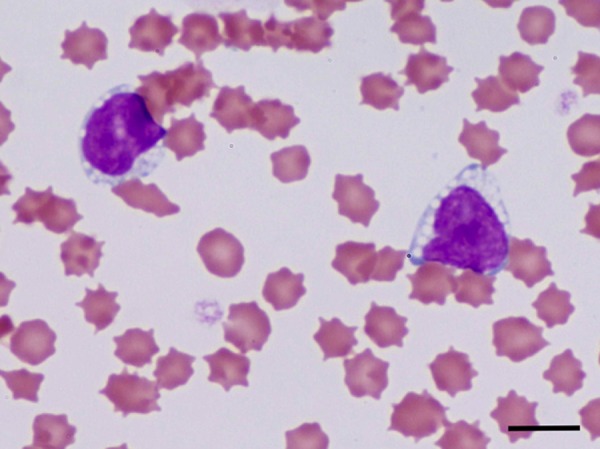
Vacuolated cytoplasm of peripheral blood lymphocytes from a cat with GM1 gangliosidosis. Giemsa stain. Bar=10 µm.
Fig. 2.

Magnetic resonance images from a cat with GM1 gangliosidosis. Transverse T2-weighted (A and C), fluid-attenuated inversion recovery (B and D) and midline sagittal T2-weighted (E) images. The mild encephalatrophy was detected in both olfactory bulbs (A and B) and temporal lobes (C and D). The corpus callosum and the rostral commissure were difficult to recognize (E).
On the basis of the clinical and MRI findings, a form of gangliosidosis was suspected in this cat. Therefore, when the cat was 6 months old, the activities of β-galactosidase and β-hexosaminidase A and B in leukocytes were measured spectrofluorometrically according to methods described previously [21, 22]. The activities were also measured in the cat’s clinically healthy dam and two littermates, which were owned by the same client. The β-galactosidase activity was very low (0.7 nmol/hr/mg protein) in the affected cat, compared to those in its dam and littermates and the reference data (Table 1). The activities of β-hexosaminidase A and B seemed higher in the affected cat than those in its pedigree members and the reference data.
Table 1. Activities of β-galactosidase and β-hexosaminidase in leukocytes of an affected cat and its relative cats.
| Animal | β-galactosidase* | β-hexosaminidase A* | β-hexosaminidase B* |
|---|---|---|---|
| Affected cat | 0.7 | 3,175 | 516 |
| Dam | 17.6 | 2,361 | 331 |
| Littermate 1 | 39.6 | 1,553 | 259 |
| Littermate 2 | 24.1 | 1,402 | 197 |
| Reference** | 51.0 ± 15.3 (n=19) | 1,135 ± 296 (n=16) | 294 ± 144 (n=16) |
*The activities are expressed as nmol/hr/mg protein. **Mean ± standard deviation. The reference data were collected at the Laboratory of Clinical Pathology, Joint Faculty of Veterinary Medicine, Kagoshima University.
At autopsy, no gross lesions were observed. Histologically, swollen neurons with accumulation of pale, slightly granular cytoplasmic materials were observed throughout the central nervous system (Fig. 3A and 3B). Dysmyelination or demyelination and gemistocytic astrocytosis were observed in the white matter. Ultrastructural analysis was carried out by a general method, and ultrathin sections were double-stained with uranyl acetate and lead citrate. Electron microscopy revealed membranous cytoplasmic bodies in the lysosomes of neurons (Fig. 3C).
Fig. 3.

Histopathology of a cat with GM1 gangliosidosis. Cerebral cortex (A). Hematoxylin and eosin stain. Bar=50 µm. Cerebral medulla (B). Hematoxylin and eosin stain. Bar=50 µm. Electron micrograph of a cerebral cortical neuron (C). Double-stained with uranyl acetate and lead citrate. Bar=5 µm.
Direct sequencing of a 326-base pair DNA fragment, including position 1448 in exon 14 of the feline GLB1 gene, was performed using forward (5ʹ-AGA GCA ATG TCT CCC GAG TCT G-3ʹ, c.1351–122 to c.1351–101) and reverse (5ʹ-GAG GAA GTC TTT GTA AAG CCA T-3ʹ, c.1482+51 to c.1482+72) primers designed based on the exonic and intronic sequences of the feline GLB1 gene (GenBank accession nos. AF006749 and ACBE01328632, respectively). The genotyping assay for the c.1448G>C mutation described previously [19] was also performed. However, the c.1448G>C mutation was not identified.
In this report, a juvenile domestic cat in Hokkaido, Japan, with retarded growth and progressive neurological signs was diagnosed with GM1 gangliosidosis by histopathological and ultrastructural examinations and by measuring the leukocyte acid β-galactosidase activity. The β-galactosidase activity of the affected cat was negligible, whereas the β-hexosaminidase activity was moderately increased, perhaps as a compensatory response (Table 1). The swollen neurons with ultrastructural membranous cytoplasmic bodies were observed morphologically in the central nervous system in the cat (Fig. 2). These biochemical and morphological changes showed that the cat was affected with GM1 gangliosidosis. However, the genetic analysis did not identify the c.1448G>C mutation, which is the single known mutation of feline GM1 gangliosidosis [12, 19], suggesting that the cat was affected with a new variant of the feline disease.
The cat had retarded growth and a hoarse cry and started to show neurological signs at 3 months of age and finally died of neurological deterioration at 9 months of age. The clinical signs included ataxia; tremor; visual disorder, which may have been due to cortical blindness; and seizures. These clinical signs seem more severe than those of feline GM1 gangliosidosis caused by the c.1448G>C mutation. Several researchers have reported that affected cats with the homozygous genotype for the c.1448G>C mutation start to show neurological signs at 4 to 6 months of age and die prematurely at approximately 1 year of age [2, 6, 7, 11, 18, 19]. The visual disorders, seizures, retarded growth and a hoarse cry have not been reported in cats homozygous for the c.1448G>C mutation. In addition, our patient had a relatively high frequency (47%) of vacuolated lymphocytes (Fig. 1) and high concentration (2,344 U/l) of serum ALP activity, perhaps due to the excretion of bone-specific ALP, findings that have not been reported previously in cats homozygous for the c.1448G>C mutation.
The difference of the clinical and clinico-pathologic features between these 2 variants of feline GM1 gangliosidosis may result from the difference of pathologic mutations in the feline GLB1 gene, suggesting our patient was affected with a different mutation(s) that are more deteriorative than the known c.1448G>C mutation. In the present report, the obligate carrier dam and one of the littermates of the affected cat showed slightly less than half of the reference activity in leukocyte β-galactosidase (Table 1), suggesting that they may be heterozygous for the mutation(s) [21]. Based on these data, further genetic studies are needed to identify the pathogenic mutation(s) of this new variant of feline GM1 gangliosidosis.
In the present report, the MRI examination revealed bisymmetrical T2W hyperintensity in the white matter of the parietal and occipital lobes (Fig. 2). In addition, the corpus callosum and the rostral commissure were difficult to recognize. The mild encephalatrophy was detected in both olfactory bulbs and temporal lobes. It is reported that the T2W hyperintensity in the white matter may result from the dysmyelinaton and/or demyelination, increased storage materials and astrocytosis in GM1 gangliosidosis in Shiba Inus [10]. In the present case, the abnormal T2W change was consistent with the histopathological abnormalities to some extent. The abnormal findings of the corpus callosum and the rostral commissure are common hypoplastic characteristics in juvenile-onset gangliosidoses in dogs and cats [8]. The encephalatrophy is observed in some canine and feline gangliosidoses, especially in the later and/or terminal stages of the disease [9, 10, 13, 17]. In the present case, the atrophy could be observed in an earlier stage (about 4 months of age) than in other animals with gangliosidoses that have been reported previously. This seems to be consistent with the severity of the new type’s clinical signs and may be attributed to the difference of a pathogenic mutation(s). In conclusion, the MRI findings of the present case were similar to those seen in previously reported animals affected with GM1 and GM2 gangliosidoses, although there were some distinctions; therefore, the findings in the present case can be useful for supporting the diagnosis of gangliosidosis before the definitive diagnosis is established by biochemical and/or genetic tests.
Acknowledgments
This study was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT grant nos. 25292181 and 26660242 OY).
REFERENCES
- 1.Barker C. G., Blakemore W. F., Dell A., Palmer A. C., Tiller P. R., Winchester B. G.1986. GM1 gangliosidosis (type 1) in a cat. Biochem. J. 235: 151–158. doi: 10.1042/bj2350151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker H. J., Lindsey J. R., McKhann G. M., Fallel A. F.1971. Neuronal GM1 gangliosidosis in a Siamese cat with β-galactosidase deficiency. Science 174: 838–839. doi: 10.1126/science.174.4011.838 [DOI] [PubMed] [Google Scholar]
- 3.Baker H. J., Smith B. F., Martin D. R., Foureman P.2001. Molecular diagnosis of gangliosidoses: a model for elimination of inherited diseases in pure breeds. pp. 615–620. In: Consultations in Feline Internal Medicine, 4th ed. (August, J. R. ed.) W. B. Saunders, Philadelphia. [Google Scholar]
- 4.Barnes I. C., Kelly D. F., Pennock C. A., Randell D. J.1981. Hepatic beta gangliosidase and feline GM1 gangliosidosis. Neuropathol. Appl. Neurobiol. 7: 463–476. doi: 10.1111/j.1365-2990.1981.tb00246.x [DOI] [PubMed] [Google Scholar]
- 5.Blakemore W. F.1972. GM1 gangliosidosis in a cat. J. Comp. Pathol. 82: 179–185. doi: 10.1016/0021-9975(72)90061-8 [DOI] [PubMed] [Google Scholar]
- 6.De Maria R., Divari S., Bo S., Sonnio S., Lotti D., Capuc- chino M., Castagnaro M.1998. β-glactosidase deficiency in a Korat cat: a new form of feline GM1 gangliodosis. Acta Neuropathol. 96: 307–314. doi: 10.1007/s004010050899 [DOI] [PubMed] [Google Scholar]
- 7.Handa S., Yamakawa T.1971. Biochemical studies in cat and human gangliosidosis. J. Neurochem. 18: 1275–1280. doi: 10.1111/j.1471-4159.1971.tb00226.x [DOI] [PubMed] [Google Scholar]
- 8.Hasegawa D., Tamura S., Nakamoto Y., Matsuki N., Takahashi K., Fujita M., Uchida K., Yamato O.2013. Magnetic resonance findings of the corpus callosum in canine and feline lysosomal storage diseases. PLoS ONE 8: e83455. doi: 10.1371/journal.pone.0083455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasegawa D., Yamato O., Kobayashi M., Fujita M., Nakamura S., Takahashi K., Satoh H., Shoda T., Hayashi D., Yamasaki M., Maede Y., Arai T., Orima H.2007. Clinical and molecular analysis of GM2 gangliosidosis in two apparent littermate kittens of the Japanese domestic cat. J. Feline Med. Surg. 9: 232–237. doi: 10.1016/j.jfms.2006.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hasegawa D., Yamato O., Nakamoto Y., Ozawa T., Yabuki A., Itamoto K., Kuwabara T., Fujita M., Takahashi K., Mizoguchi S., Orima H.2012. Serial MRI features of canine GM1 gangliosidosis: a possible imaging biomarker for diagnosis and progression of the disease. Scientific World Journal 2012: 250197. doi: 10.1100/2012/250197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawasaki Y., Nagatani I., Terashima Y., Koike H., Miyoshi N., Ohishi A.2009. Assessment of neurological function in three cats with homozygous or heterozygous GM1 gangliosidosis by brainstem auditory evoked potential. J. Jpn. Vet. Med. Assoc. 62: 148–154(in Japanese with English summary). [Google Scholar]
- 12.Martin D. R., Rigat B. A., Foureman P., Varadarajan G. S., Hwang M., Krum B. K., Smith B. F., Callahan J. W., Mahuran D. J., Baker H. J.2008. Molecular consequences of the pathogenic mutation in feline GM1 gangliosidosis. Mol. Genet. Metab. 94: 212–221. doi: 10.1016/j.ymgme.2008.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuki N., Yamato O., Kusuda M., Maede Y., Tsujimoto H., Ono K.2005. Magnetic resonance imaging of GM2-gangliosidosis in a golden retriever. Can. Vet. J. 46: 275–278. [PMC free article] [PubMed] [Google Scholar]
- 14.Mochizuki H., Kotani T.1977. GM1 gangliosidosis in a Japanese domestic cat. Exp. Anim. 26: 281–282(in Japanese). [PubMed] [Google Scholar]
- 15.Murray J. A., Blakemore W. F., Barnett K. C.1977. Ocular lesions in cats with GM1-gangliosidosis with visceral involvement. J. Small Anim. Pract. 18: 1–10. doi: 10.1111/j.1748-5827.1977.tb05818.x [DOI] [PubMed] [Google Scholar]
- 16.Suzuki Y., Oshima A., Nanba E.2001. β-Galactosidase deficiency (β-galactosidosis): GM1 gangliosidosis and Morquio B disease. pp. 3775–3809. In: The Metabolic and Molecular bases of Inherited Disease, 8th ed. (Scriver, C. R., Beaudet, A. L., Sly, W. S. and Valle, D. eds.), McGraw-Hill, New York. [Google Scholar]
- 17.Tamura S., Tamura Y., Uchida K., Nibe K., Nakaich M., Hossain M. A., Chang H.S., Rahman M. M., Yabuki A., Yamato O.2010. GM2 gangliosidosis variant 0 (Sandhoff-like disease) in a family of Toy Poodles. J. Vet. Intern. Med. 24: 1013–1019. doi: 10.1111/j.1939-1676.2010.0564.x [DOI] [PubMed] [Google Scholar]
- 18.Uddin M. M., Hossain M. A., Rahman M. M., Chowdhury M. A., Tanimoto T., Yabuki A., Mizukami K., Chang H.S., Yamato O.2013. Identification of Bangladeshi domestic cats with GM1 gangliosidosis caused by the c.1448G>C mutation of the feline GLB1 gene: case study. J. Vet. Med. Sci. 75: 395–397. doi: 10.1292/jvms.12-0307 [DOI] [PubMed] [Google Scholar]
- 19.Uddin M. M., Tanimoto T., Yabuki A., Kotani T., Kuwamura M., Chang H.S., Yamato O.2012. Mutation analysis of GM1 gangliosidosis in a Siamese cat from Japan in the 1960s. J. Feline Med. Surg. 14: 900–902. doi: 10.1177/1098612X12454120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C. Y., Smith B. F.2007. Development of quantitative polymerase chain reaction assays for allelic discrimination of gangliosidoses in cats. Am. J. Vet. Res. 68: 231–235. doi: 10.2460/ajvr.68.3.231 [DOI] [PubMed] [Google Scholar]
- 21.Yamato O., Kobayashi A., Satoh H., Endoh D., Shoda T., Masuoka Y., Hatakeyama A., Jo E. O., Asano T., Yonemura M., Yamasaki M., Maede Y.2004. Comparison of polymerase chain reaction-restriction fragment length polymorphism assay and enzyme assay for diagnosis of GM1-gangliosidosis in Shiba dogs. J. Vet. Diagn. Invest. 16: 299–304. doi: 10.1177/104063870401600407 [DOI] [PubMed] [Google Scholar]
- 22.Yamato O., Satoh H., Matsuki N., Ono K., Yamasaki M., Maede Y.2004. Laboratory diagnosis of canine GM2-gangliosidosis using blood and cerebrospinal fluid. J. Vet. Diagn. Invest. 16: 39–44. doi: 10.1177/104063870401600107 [DOI] [PubMed] [Google Scholar]