

Oxidative stress is considered a risk factor of atherosclerosis. Nevertheless, antioxidant therapy had failed to limit vascular disease development in human. Here we demonstrate that the NADPH oxidase Nox4, an important source of reactive oxygen species in the vascular system, limits arteriosclerosis development in two different mouse models. Deletion or inhibition of Nox4 induced a vascular inflammatory phenotype, which probably promoted atherosclerosis development. As Nox inhibitors are currently in clinical testing for the treatment of fibrosis, these drugs should be carefully evaluated for cardiovascular side-effects.
Keywords: NADPH oxidase, Lipids, Inflammation, Remodelling, Reactive oxygen species, ApoE, Arteriosclerosis
Abstract
Aims
Oxidative stress is thought to be a risk for cardiovascular disease and NADPH oxidases of the Nox family are important producers of reactive oxygen species. Within the Nox family, the NADPH oxidase Nox4 has a unique position as it is constitutively active and produces H2O2 rather than . Nox4 is therefore incapable of scavenging NO and its low constitutive H2O2 production might even be beneficial. We hypothesized that Nox4 acts as an endogenous anti-atherosclerotic enzyme.
Methods and results
Tamoxifen-induced Nox4-knockout mice were crossed with ApoE−/− mice and spontaneous atherosclerosis under regular chow as well as accelerated atherosclerosis in response to partial carotid artery ligation under high-fat diet were determined. Deletion of Nox4 resulted in increased atherosclerosis formation in both models. Mechanistically, pro-atherosclerotic and pro-inflammatory changes in gene expression were observed prior to plaque development. Moreover, inhibition of Nox4 or deletion of the enzyme in the endothelium but not in macrophages resulted in increased adhesion of macrophages to the endothelial surface.
Conclusions
The H2O2-producing NADPH oxidase Nox4 is an endogenous anti-atherosclerotic enzyme. Nox4 inhibitors, currently under clinical evaluation, should be carefully monitored for cardiovascular side-effects.
See page 3457 for the editorial comment on this article (doi:10.1093/eurheartj/ehv518)
Translational perspective.
Oxidative stress is considered a risk factor of atherosclerosis. Nevertheless, antioxidant therapy had failed to limit vascular disease development in human. Here we demonstrate that the NADPH oxidase Nox4, an important source of reactive oxygen species in the vascular system, limits arteriosclerosis development in two different mouse models. Deletion or inhibition of Nox4 induced a vascular inflammatory phenotype, which probably promoted atherosclerosis development. As Nox inhibitors are currently in clinical testing for the treatment of fibrosis, these drugs should be carefully evaluated for cardiovascular side-effects.
Introduction
Limited NO availability, vascular inflammation, and noxious environmental conditions promote atherosclerosis development1 and increase the level of reactive oxygen species (ROS). Although there is little evidence in humans that changing ROS formation alters the course of arteriosclerosis development, positive results have been published in animal studies.2 The failure of antioxidants to delay arteriosclerosis development in man is usually explained by insufficient dosing, unfavourable reaction kinetics, or counter-regulatory responses of the body. As a consequence, a concept emerged that rather than scavenging ROS, blocking their enzymatic production might be a better way to prevent vascular oxidative stress.3
Numerous enzymes are capable of increasing vascular ROS formation.1 In particular, the members of the Nox family NADPH oxidases, however, gained considerable interest in recent years.4 Nox2 is the source of superoxide () formation in leucocytes. Patients carrying mutations of Nox2 are immunocompromised but present with increased endothelium-dependent relaxation and enhanced NO availability.5 Nox1 is another NADPH oxidase, which is induced by cytokines and together with Nox2 contributes to vascular formation. Genetic deletion of Nox1 or pharmacological inhibition reduces atherosclerosis development in diabetic ApoE−/− mice6 but in the absence of diabetes this effect was not observed.6,7
For the Nox2 system, deletion of the cytosolic p47phox subunit reduced atherosclerosis development in mice8 but reducing Nox2 activity had little effect on disease progression. Although vascular ROS formation was impaired and endothelium-dependent relaxation was improved,9 plaque load between wild-type (WT) and Nox2-knockout mice was similar.9,10 In human subjects deficient of Nox2 activity, carotid artery intima volume was reduced when compared with healthy subjects, but coronary artery calcification was similar.11 Moreover, in a case–control study of 31 subjects carrying mutations of Nox2, loss of the function of the enzyme results in greater flow-mediated relaxation and reduced intima-media thickness when compared with healthy controls which was associated with increased NO availability.12
Another Nox enzyme expressed in the vascular system is Nox4. Nox4 is different to Nox1 and Nox2 as it produces H2O2 rather than . Moreover, Nox4 is constitutively active and induced in the course of differentiation rather than by inflammatory cytokines.13 Whether Nox4 impacts on arteriosclerosis is unclear. Cell culture experiments with siRNA support Nox4 as a potential target to prevent vascular inflammation. Knockout mouse data, however, suggest that Nox4 might be protective in the vascular system.14,15 The present study was therefore conducted to determine whether genetic deletion of Nox4 alters the course of atherosclerosis development in mice. To address this, we generated tamoxifen-inducible Nox4-ApoE double-knockout mice and subjected them to two models of atherosclerosis development.
Materials and methods
For an extended material and methods section, see Supplementary material online.
Animal experiments
All animal experiments were performed in accordance with the National Institutes of Health Guidelines on the Use of Laboratory Animals. The University Animal Care Committee and the Federal Authorities for Animal Research (Darmstadt, Germany) approved the study protocol.
Global tamoxifen-inducible Nox4-ApoE−/− double-knockout (Nox4flox/flox-Cre-ERT2+/0ApoE−/−) mice were generated by crossing Nox4flox/flox-C57/Bl6J14 with Cre-ERT2 transgenics16 and ApoE−/− mice (Taconics). Activation of Cre-ERT2 was achieved by oral tamoxifen administration with the chow ad libitum for 10 days. In all experiments only male animals were used. Cre-positive and Cre-negative litter mates received tamoxifen to exclude direct effects of the anti-oestrogen. According to the expression of Cre-ERT2, Nox4 carrying WT equivalent to Nox4-flox/flox-ApoE−/− Cre-ERT20/0 mice are denoted as WT, whereas Nox4-deleted Nox4-flox/flox-ApoE−/− Cre-ERT2+/0 are denoted as Nox4*/* throughout the study.
Atherosclerosis model
Atherosclerosis formation was analysed at 44–49 weeks of age in mice fed standard rodent chow or in animals fed a western-type diet with subsequent partial left common carotid artery ligation17 at an age of 10–14 weeks.
Results
Nox4-ApoE double-knockout mice exhibit altered reactive oxygen species production
Tamoxifen resulted in an efficient loss of vascular Nox4 expression: by qRT-PCR, mRNA expression was >100 times lower in the carotid artery of Nox4*/* when compared with that of WT mice. Nox4 protein was readily detectable in aortic extracts of WT animals, whereas only a very faint band was observed in samples from Nox4*/* aortas (Figure 1A). The strains did not differ in plasma glucose, body weight, plasma triglycerides or cholesterol, and triglyceride fractions in serum electrophoresis (Figure 1Band C). Amplex red assays from aortic rings demonstrated that the H2O2 production of vessel from Nox4*/* was lower than that of WT animals (Figure 1D), which is compatible with the notion that Nox4 is an important source for vascular H2O2 production. Interestingly, in the dihydroethidium assay (DHE), some evidence for increased superoxide () formation after deletion of Nox4 was found: whereas ethidium formation, as a measure for unspecific oxidation of DHE, tended to be attenuated, the formation of the adduct 2-hydroxyethidium was significantly increased in the aorta of Nox4*/* mice when compared with WT controls. This might indicate an increased leucocyte adhesion after deletion of Nox4 or compensatory induction of Nox2 (Figure 1E).
Figure 1.

Characterization of inducible Nox4-ApoE double-knockout mice. (A) Nox4 RT-PCR from Nox4*/* and WT carotid arteries, n > 6 (left). Western blot for Nox4 and β-actin in aortae from Nox4*/* and WT mice (right). (B) Morning plasma glucose, body weight, plasma triglycerides, and cholesterol, n = 6. (C) Representative plasma lipoprotein FPLC for cholesterol and triglyceride. (D) Aortic H2O2 determined by Amplex Red/HRP from the sites indicated, n = 4. #P < 0.05 with vs. without catalase. (E) Dihydroethidium assay in arbitrary units (aU) for the adduct oxyethidium (EOH) and the oxidation product ethidium (E) from aortic rings, n = 4. *P < 0.05 WT vs. Nox4*/*.
Deletion of Nox4 results in increased atherosclerosis
In order to determine the role of Nox4 for spontaneous atherosclerosis development in the ApoE−/− background, animals were aged for 9 months after tamoxifen treatment without dietary intervention. Genetic deletion of Nox4 approximately doubled plaque load in the aortic sinus, the thoracic, and the abdominal aorta (Figure 2A and B). Deletion of Nox4 resulted in plaques with a higher amount of collagens as determined by Sirius red morphometry and macrophages but with a trend towards reduced neutral lipid content (Figure 2C). Thus, deletion of Nox4 results in increased atherosclerosis with enhanced fibrotic and inflammatory characteristics. Deletion of Nox4 led to a trend towards an increased expression of the fibroblast marker S100a4, whereas the smooth muscle marker smooth muscle actin was identical between both groups. Importantly, the macrophage marker Emr1 was double as high in vessels of Nox4*/* when compared with WT aorta (see Supplementary material online, Figure S1). Although it is usually difficult to derive mechanistic insights for disease development from late stage atherosclerosis, qRT-PCR experiments were performed for some redox-regulating genes. Expression of Nox1 in the aorta was low and variable. Nox2 expression, in contrast, was two-fold higher in the aorta of Nox4*/* when compared with WT mice, which is probably a reflection of the increased macrophage content in these vessels. Indeed, p67phox expression which is most highly expressed in leucocytes was similarly changed, whereas no differences were found for the more widely expressed p47phox and p22phox subunit of the NADPH oxidase. Endothelial eNOS expression was measured relatively to the endothelial housekeeping marker PECAM-1 and was similar between the groups and also no differences were obtained for SOD1 and SOD2. SOD3 expression was significantly increased after deletion of Nox4 but the underlying mechanism of this observation is unknown (see Supplementary material online, Figure S1).
Figure 2.
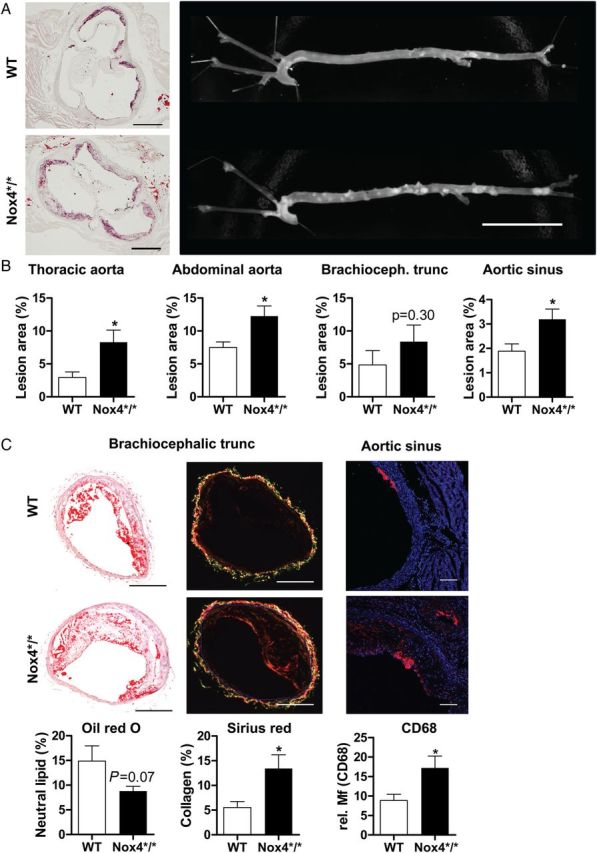
Role of Nox4 for spontaneous atherosclerosis development. (A) Photographs of oil red O staining of the aortic sinus and of the naive aorta. Scale bars are 0.5 and 20 mm, respectively. (B) Statistics of the planimetry of the vessels indicated, n ≥ 9. (C) Oil red O, Sirius red or CD68 (CD68, red and DAPI, blue) stained cross sections of brachiocephalic arteries or aortic sinus and statistics, n = 12–15 and n = 6 for CD68, Scale bars are 200 and 20 µm, respectively. *P < 0.05 WT vs. Nox4*/*. Tissue was collected at 44–49 weeks of age.
Nox4 knockout increases plaque development in the partial carotid artery ligation model
In order to support the observation of an endogenous anti-atherosclerotic function of Nox4, a second, more inflammation-driven model of accelerated atherosclerosis was performed—partial carotid artery ligation in ApoE−/− mice subjected to western-type diet. Morphometric analyses were performed 2 and 4 weeks after operation by in vivo micro CT, and quantitative histology and gene expression were determined 28 and 7 days after the operation, respectively. Lumen loss within the first 2 weeks after flow reduction was similar between both strains. Subsequently, however, lumen loss was more rapid in Nox4-deficient animals, so that after 28 days, the lumen of the carotid artery of Nox4*/* was much smaller than that of WT vessels. This was also reflected by the plaque burden: plaque load was similar 2 weeks after the operation, whereas after 28 days, the plaque load of Nox4*/* animals was double as high as that of WT mice (Figure 3). The number of haemorrhages as a marker for plaque angiogenesis and plaque cholesterol content were similar between both groups (data not shown). Remodelling of the contra-lateral control side was also similar between both strains (see Supplementary material online, Figure S2).
Figure 3.
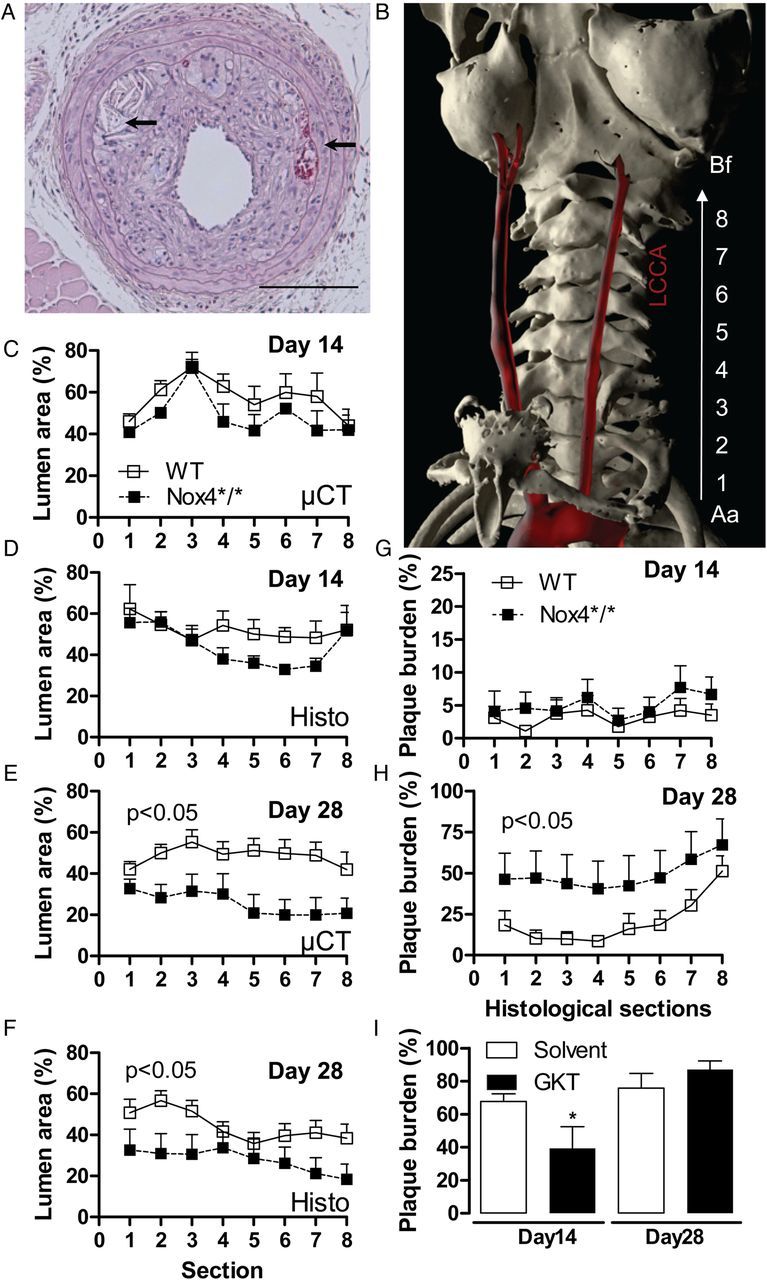
Role of Nox4 for accelerated plaque formation after partial carotid artery ligation and high-fat diet. (A) H&E cross sections of the common carotid artery of Nox4*/* mouse 28 days after partial carotid artery ligation. Arrows point to cholesterol clefts and plaque haemorrhages. (B) Volume rendering of in vivo micro-CT angiography of an Nox4*/* mouse day 14 after partial ligation of the left common carotid artery. The common carotid artery is shown and was segmented into eight equidistant parts from aorta (Aa) to bifurcation (Bf). (C–F) Side-dependent vascular lumen profile of the operated normalized to the non-operative vessels according to the segmentation denoted in (B), n ≥ 7. (G–I) Plaque volume determined by serial histological sections and time points in Nox4*/* and WT mice (G–H) and ApoE−/− mice treated with and without GKT137928 (20 mg/kg/day) (I). *P < 0.05 with vs. without GKT.
Given that Nox enzymes are drug targets,3 the impact of the combined low-selectivity Nox1/Nox4 GKT137928 inhibitor on plaque development in this model was determined, too. Whereas the compound significantly inhibited atherosclerosis development during the first 2 weeks after operation, the protective effect was rapidly lost and after 4 weeks, animals receiving the drug showed a trend towards more atherosclerosis development (Figure 3I).
Nox4 knockout increases pro-atherosclerosclerotic pathways
To identify the molecular basis of increased atherosclerosis after deletion of Nox4, cDNA microarrays were performed 7 days after ligation. This early time-point was chosen as plaques had not yet developed.
Comparison of the ligated vessels of WT and Nox4*/* animals applying a P < 0.1 without Bonferroni correction and a threshold of 1.5-fold, recovered 51 up-regulated genes and 16 down-regulated genes after deletion of Nox4. Differences in the non-operated carotid artery were more discrete with 10 genes up- and 16 genes down-regulated >1.3-fold in Nox4*/* (Figure 4). Interestingly, KEGG pathway analysis provided signs of increased immuno- and inflammatory signalling in Nox4*/* mice like enrichment of genes for matrix-receptor interaction, lysosomal signal but also allograft rejection and diabetes (see Supplementary material online, Table S2). A more detailed analysis focusing on redox-signalling, inflammation, and matrix indicate that ligation results in more substantial expression changes in Nox4*/* than WT mice (see Supplementary material online, Table S3). Similar to in the chronic atherosclerosis model, Nox4-knockout results in an increase in the NADPH oxidase Nox2 and some of its cytosolic subunits, whereas expression of the other Nox genes decreased or remained unchanged. Interestingly, carotid artery ligation decreased vascular Nox4 expression, compatible with the view that the enzyme is involve predominantly in differentiation rather than proliferation (see Supplementary material online, Table S3). These data suggest that deletion of Nox4 results in an inflammatory state which may subsequently favour atherosclerosis development. Obviously, these data should be handled with caution as a P threshold of 0.1 without Bonferroni correction was applied and thus the differences are not statistically significant for the individual gene. To obtain confirmation for a more pro-inflammatory situation, protein arrays were performed from the aorta of the spontaneous atherosclerosis model (see Supplementary material online, Figure S3) and from the secretome of the carotid artery 7 days after partial ligation (see Supplementary material online, Figure S4). Although changes in protein expression were low and Bonferroni correction was not applied, they also suggest a situation of increased inflammation and adhesion molecule expression in response to the knockout of Nox4.
Figure 4.

Carotid gene expression of ApoE−/− mice 7 days after ligation. Illumina MouseWG-6 v2 BeadChip array. L indicates operated left side, R indicates non-operated control side, n = 5. (A) Log2 normalized heat map of selected genes (ligated, >1.5-fold, P < 0.1) in Nox4*/* compared with WT. (B) Log2 normalized heat map of selected genes (non-ligated, >1.3-fold, P < 0.1) in Nox4*/* compared with WT. (C) Dot plot displaying relative expression level of genes in operated left (L) and control right (R) carotids of Nox4*/* and WT mice. Values are the ratios of Log10 normalized fold change.
Endothelial deletion of Nox4 increases leucocyte adhesion
The combination of increased macrophage content together with different array data is indicative towards increased inflammatory activation. Thus, deletion of Nox4 may promote trapping of inflammatory cells at the vascular wall. Indeed, already 7 days after the operation leucocyte makers were increase in the operated vessel of Nox4*/* animas but not WT mice (see Supplementary material online, Table S3). To address this possibility, adhesion experiments were performed in flow chambers. Macrophages from Nox4*/* and WT mice exhibited a similar adhesion to cytokine-stimulated endothelial cells. In contrast, adhesion of the monocytic cell line THP-1 was higher to endothelial cells obtained from the lung or the carotid artery of Nox4*/* than to cells from WT mice (Figure 5A–D). Moreover, pre-treatment of human umbilical vein endothelial cells with the low-selective Nox1/Nox4 inhibitor GKT137928 in the absence of cytokine stimulation increased the surface E-selectin expression and the adhesion of monocytes (Figure 5E and F).
Figure 5.

Leucocyte adhesion on endothelial cells in vitro. (A) Adhesion of peritoneal macrophages from WT and Nox4*/* to TNFα-stimulated (10 ng/mL, 25 h) lung endothelial cells from WT mice in response to flow, n = 5–6. (B and C) Statistics and exemplary pictures of THP1 adhesion to lung endothelial cells from WT and Nox4*/* during exposure to laminar flow, n = 3. (D) Statistics of THP1 adhesion to carotid artery endothelial cells from WT and Nox4*/* during exposure laminar flow, n = 4. (E) Normalized human peripheral blood monocyte adhesion under static conditions to HUVECs pre-treated with GKT137831 (20 µM, 2 h) or solvent, n = 6. (F) HUVEC surface to total E-selectin (selE) expression determined by FACS pre-treated with GKT137831 (20 µM, 2 h) or solvent, n = 6.
As these data suggest that deletion of Nox4 in the vessel rather than in macrophages is responsible for the atherosclerosis prone phenotype, Nox4 expression in myeloid cells was further characterized. Whereas the mRNA of the oxidase was readily detected in endothelial cells, no specific signal was obtained in macrophages (see Supplementary material online, Figure S5A). Also the search for splice variants did not yield protein coding transcripts (see Supplementary material online, Figure S5B). Similarly, Nox4 protein was undetectable in monocytes by western blot (data not shown). Thus, Nox4 expression in endothelial cells is tremendously higher than that in macrophages, and it appears that the latter cells may not even express a functional version of the enzyme under the present conditions.
Discussion
In the present study, we observed that acute genetic deletion of Nox4 accelerates atherosclerosis development in ApoE−/− mice and results in greater disease development after partial carotid artery ligation in mice fed a high-fat diet. We observed that deletion of Nox4 in the endothelium increases leucocyte adhesion, which might be a consequence of increased inflammatory activation of the endothelium. Thus, Nox4 by preventing vascular inflammation could in part limit vascular influx of leucocytes which eventually should result in attenuated atherosclerosis development (Figure 6).
Figure 6.

Schematic on the pro-atherosclerotic effect of loss of Nox4.
Over the past years, evidence is accumulating that supports the view that Nox4 is an endogenous protective system that by maintaining antioxidant defence prevents oxidative injury.14,15 In part, this is mediated by controlling NO levels14,15 and in part by maintaining Nrf2 signalling.14,18 This view is contrasted by reports in epithelial cells where Nox4 mediates TGFβ-induced apoptosis. The potential negative function of Nox4, however, might be specific for the lung as in the kidney, at least in response to ureter ligation, loss of Nox4 also resulted in a greater inflammatory response.19 It was also observed that inflammation-induced kidney destruction leads to loss of Nox4 expression,19 which is compatible with the view that Nox4 is induced during differentiation and orchestrates the differentiation process.20,21 Interestingly, also in the present work, carotid artery ligation, a pro-inflammatory, pro-proliferative setting was associated with a marked down-regulation of Nox4.
A main limitation of the present work is that we failed to identify a single specific, ideally redox-controlled, mechanism by which deletion of Nox4 promoted atherosclerosis development. We, however, doubt that such a mechanism exists as Nox4, being a redox-modulator impacts on numerous signalling pathways like those involving the important transcription factors HIF1α, NFκB, and Nrf2.18,22 In addition to these pathways, the enzyme has been shown to contribute to senescence development, and thus its deletion would favour proliferation.23 Nox4 contributes to endoplasmic reticulum stress and autophagy, other growth-inhibiting situations24 and subsequent apoptosis induction. The mechanisms underlying this effect are, however, not well understood but may involve oxidative modifications of Ras or of phosphatases.24
Also functional, some data may suggest that Nox4 limits atherosclerosis development: the enzyme is induced in the consolidation phase after vascular injury when proliferation decreases25 and in smooth muscle cells grown from ApoE-LDLR double-knockout mice, Nox4 had direct anti-proliferative and pro-senescent effects compatible with a direct anti-atherosclerotic function.26
In the present study, carotid artery ligation and Nox4 deletion were associated with an increase in Nox2 expression. Importantly, increased Nox2 expression has also been observed in human atherosclerosis,27 but it appears that this might be a reflection of leucocyte content, which increases in atherosclerosis in general. Not much is known concerning Nox4 under this situation. In a monkey atherosclerosis model, expression of Nox2 and Nox1 increased while that of Nox4 and Nox5 remained unchanged28 but data from humans are lacking. These observations are in line with the failure to observe cross talk between the different Nox homologues. Given that Nox4 is induced by TGFβ and hypoxia, whereas Nox1 is increased by cytokines but suppressed by nitric oxide,29 this aspect is well understandable.
The data on cell adhesion appear to suggest that Nox4 in the vessel wall rather than in myeloid cells is important for the atherosclerosis modifying effect. Indeed, Nox4 expression was basically undetectable in macrophages in the present study. This, however, does not exclude a contribution of Nox4 in these cells to disease development. We and others have shown that myeloid cell differentiation may involve Nox4 induction20 and in response to combined hyperglycaemia and hyperlipidaemia Nox4 expression increases in macrophages and contributes to macrophage apoptosis.30 At least for spontaneous atherosclerosis, thus without fat feeding, the present study, however, favours an importance of endothelial rather than myeloid Nox4.
A recent study revealed that Nox1 contributes to accelerated atherosclerosis of ApoE−/− mice with streptozotocin-induced diabetes.6 This study also included ApoE-Nox4 double-knockout mice and treatment with the low-selective Nox1/Nox4 inhibitor GKT137928 in non-diabetic animals. Although this scenario is similar to the present study, no role of Nox4 in atherosclerosis development was observed.6 This result, however, was expected as the animals were studied at an age of 26 weeks, which is too early for spontaneous atherosclerosis development in ApoE−/− mice not treated with high-fat diet. Detailed analysis of this particular study, however, yielded data in support to the present work as plaque area was slightly higher in non-diabetic Nox4-ApoE double-knockout mice than ApoE−/− mice (although not significant). Moreover, GKT137931, which was used as a low-selectivity Nox1/Nox4 inhibitor, resulted by tendency in an increase aortic arch plaque load in non-diabetic mice.6 The complexity of the action of GKT137931 and the lack of specific information about the extent of inhibition of individual Nox enzymes, however, should limit any conclusions drawn from effects of this compound.
In summary, with the present study Nox4 is established as an endogenous protective system which limits atherosclerosis development in mice. Nox4 inhibitors, which are currently in clinical testing for fibrotic diseases, should be carefully monitored for a potential to increase cardiovascular disease rates.
Supplementary material
Supplementary material is available at European Heart Journal online.
Author's contributions
C.S., F.R., C.K.,O.L., C.F., B.v.d.S., R.B., and K.S. performed statistical analysis; K.S. and R.B. handled funding and supervision; C.S., F.R., C.K., Y.Y., O.L., C.F., B.v.d.S. and R.B. acquired the data; C.S., N.W., A.S., R.B., and K.S. conceived and designed the research; C.S., R.B. and K.S. drafted the manuscript; and N.W., A.S., H.J., R.B. and K.S. made critical revision of the manuscript for key intellectual content.
Funding
The study was supported by the DFG Excellence Cluster ‘Cardiopulmonary System – ECCPS’, SFB 815 (TP A1 to K.S.) and SFB 834 (TPA2 to R.P.B.), the Faculty of Medicine, Goethe-Universität, Frankfurt am Main, Germany and the Heinrich und Fritz-Riese-Stiftung to F.R. A.M.S. is supported by the British Heart Foundation.
Conflict of interest: The authors declare that they have no relevant financial, personal, or professional relationships to disclose which could be perceived as a conflict of interest or as potentially influencing or biasing the authors' work.
Acknowledgement
We are grateful for excellent technical assistance of Susanne Schütz, Katalin Palfi, and Daphne Dekker.
References
- 1.Li H, Horke S, Forstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014;237:208–219. [DOI] [PubMed] [Google Scholar]
- 2.Lonn ME, Dennis JM, Stocker R. Actions of “antioxidants” in the protection against atherosclerosis. Free Radic Biol Med 2012;53:863–884. [DOI] [PubMed] [Google Scholar]
- 3.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 2011;10:453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med 2010;49:687–706. [DOI] [PubMed] [Google Scholar]
- 5.Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De MD, Martire B, Pietrogrande MC, Martino S, Gambineri E, Soresina AR, Pignatelli P, Martino F, Basili S, Loffredo L. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation 2009;120:1616–1622. [DOI] [PubMed] [Google Scholar]
- 6.Gray SP, Di ME, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013;127:1888–1902. [DOI] [PubMed] [Google Scholar]
- 7.Sobey CG, Judkins CP, Rivera J, Lewis CV, Diep H, Lee HW, Kemp-Harper BK, Broughton BR, Selemidis S, Gaspari TA, Samuel CS, Drummond GR. NOX1 deficiency in apolipoprotein E-knockout mice is associated with elevated plasma lipids and enhanced atherosclerosis. Free Radic Res 2015;49:186–198. [DOI] [PubMed] [Google Scholar]
- 8.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE(–/–) mice. J Clin Invest 2001;108:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Judkins CP, Diep H, Broughton BR, Mast AE, Hooker EU, Miller AA, Selemidis S, Dusting GJ, Sobey CG, Drummond GR. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE–/– mice. Am J Physiol Heart Circ Physiol 2010;298:H24–H32. [DOI] [PubMed] [Google Scholar]
- 10.Kirk EA, Dinauer MC, Rosen H, Chait A, Heinecke JW, LeBoeuf RC. Impaired superoxide production due to a deficiency in phagocyte NADPH oxidase fails to inhibit atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2000;20:1529–1535. [DOI] [PubMed] [Google Scholar]
- 11.Sibley CT, Estwick T, Zavodni A, Huang CY, Kwan AC, Soule BP, Long Priel DA, Remaley AT, Rudman Spergel AK, Turkbey EB, Kuhns DB, Holland SM, Malech HL, Zarember KA, Bluemke DA, Gallin JI. Assessment of atherosclerosis in chronic granulomatous disease. Circulation 2014;130:2031–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Violi F, Pignatelli P, Pignata C, Plebani A, Rossi P, Sanguigni V, Carnevale R, Soresina A, Finocchi A, Cirillo E, Catasca E, Angelico F, Loffredo L. Reduced atherosclerotic burden in subjects with genetically determined low oxidative stress. Arterioscler Thromb Vasc Biol 2013;33:406–412. [DOI] [PubMed] [Google Scholar]
- 13.Montezano AC, Burger D, Ceravolo GS, Yusuf H, Montero M, Touyz RM. Novel Nox homologues in the vasculature: focusing on Nox4 and Nox5. Clin Sci (Lond) 2011;120:131–141. [DOI] [PubMed] [Google Scholar]
- 14.Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 2012;110:1217–1225. [DOI] [PubMed] [Google Scholar]
- 15.Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF., Jr NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011;124:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res 1999;27:4324–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol 2009;297:H1535–H1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brewer AC, Murray TV, Arno M, Zhang M, Anilkumar NP, Mann GE, Shah AM. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic Biol Med 2011;51:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schroder K, Brandes RP. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 2012;53:842–853. [DOI] [PubMed] [Google Scholar]
- 20.Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, Weissmann N, Weinberger V, Benkhoff S, Kampschulte M, Obermayer-Pietsch B, Hofbauer LC, Brandes RP, Schroder K. NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Invest 2013;123:4731–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 2007;27:42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA 2010;107:18121–18126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lener B, Koziel R, Pircher H, Hutter E, Greussing R, Herndler-Brandstetter D, Hermann M, Unterluggauer H, Jansen-Durr P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem J 2009;423:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol 2010;30:3553–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szocs K, Lassegue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, Wilcox JN, Quinn MT, Lambeth JD, Griendling KK. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol 2002;22:21–27. [DOI] [PubMed] [Google Scholar]
- 26.Xu S, Chamseddine AH, Carrell S, Miller FJ., Jr Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol 2014;2:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002;105:1429–1435. [DOI] [PubMed] [Google Scholar]
- 28.Stanic B, Pandey D, Fulton DJ, Miller FJ., Jr Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arterioscler Thromb Vasc Biol 2012;32:2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pleskova M, Beck KF, Behrens MH, Huwiler A, Fichtlscherer B, Wingerter O, Brandes RP, Mulsch A, Pfeilschifter J. Nitric oxide down-regulates the expression of the catalytic NADPH oxidase subunit Nox1 in rat renal mesangial cells. FASEB J 2006;20:139–141. [DOI] [PubMed] [Google Scholar]
- 30.Lee CF, Qiao M, Schroder K, Zhao Q, Asmis R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ Res 2010;106:1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]