

Abstract
Our previously published data show that As+3 in vivo and in vitro, at very low concentrations, inhibits lymphoid, but not myeloid stem cell development in mouse bone marrow. We also showed that the As+3 metabolite, monomethylarsonous acid (MMA+3), was responsible for the observed pre-B cell toxicity caused by As+3. Interleukin-7 (IL-7) is the primary growth factor responsible for pre-lymphoid development in mouse and human bone marrow, and Signal Transducer and Activator of Transcription 5 (STAT5) is a transcriptional factor in the IL-7 signaling pathway. We found that MMA+3 inhibited STAT5 phosphorylation at a concentration as low as 50 nM in mouse bone marrow pre-B cells. Inhibition of STAT5 phosphorylation by As+3 occurred only at a concentration of 500 nM. In the IL-7 dependent mouse pre-B 2E8 cell line, we also found selective inhibition of STAT5 phosphorylation by MMA+3, and this inhibition was dependent on effects on JAK3 phosphorylation. IL-7 receptor expression on 2E8 cell surface was also suppressed by 50 nM MMA+3 at 18 h. As further evidence for the inhibition of STAT5, we found that the induction of several genes required in B cell development, cyclin D1, E2A, EBF1, and PAX5, were selectively inhibited by MMA+3. Since 2E8 cells lack the enzymes responsible for the conversion of As+3 to MMA+3 in vitro, the results of these studies suggest that As+3 induced inhibition of pre-B cell formation in vivo is likely dependent on the formation of MMA+3 which in turn inhibits IL-7 signaling at several steps in mouse pre-B cells.
Keywords: Arsenite, monomethylarsonous acid, lymphoid progenitors, selective toxicity, STAT5, mouse pre-B cells
Arsenic exposure has been associated with many diseases including immunosuppression, hematotoxicity, and carcinogenesis (Kozul et al., 2009; Li et al., 2010). Trivalent inorganic arsenic (As+3) and its metabolite, monomethylarsonous acid (MMA+3), have also been associated with many toxicities in mammalian systems (Styblo et al., 2000). Several studies have examined mechanisms associated with high dose acute exposures to As+3 with resultant DNA damage and alterations of DNA repair systems (Qin et al., 2012; Wang et al., 2013; Wei et al., 2009). In general, several factors including oxidative stress (Flora, 2011), chromosomal aberration (Oya-Ohta et al., 1996), inhibition of DNA repair and PARP activity (Qin et al., 2012), and altered gene expression could collectively contribute to As+3 toxicity.
Only a few studies have attempted to address mechanisms associated with As+3 toxicity from physiological, long term, low dose, and environmentally relevant exposures. Our lab previously observed that low doses of As+3 inhibited mouse bone marrow (BM) lymphoid but not myeloid progenitor cell development in vivo (Ezeh et al., 2014). We attributed the in vivo toxicity of As+3 to one of its primary organic metabolites, MMA+3, which is formed by the liver and kidneys (Aposhian et al., 2000). We showed that MMA+3 inhibited pre-B development from hematopoietic stem cells at concentrations that can be achieved with environmental exposures.
For the selective targeting of mouse pre-lymphoid stem cells found in BM, we hypothesized that As+3 and MMA+3 could modulate the BM microenvironment, leading to alteration of IL-7 signaling which is critical for lymphoid cell formation and expansion. The cytokine, IL-7 has been shown to specify B-cell fate from the common lymphoid to the pre-pro B stage (Kikuchi et al., 2005). It is also required for further differentiation of B and T lineage cells. We utilized both primary mouse BM cells and 2E8 cells, an IL-7-dependent mouse pre-B cell line, to elucidate the mechanism(s) by which As+3 and MMA+3 could target the lymphoid progenitors in the BM. Using similar concentration ranges of As+3 and MMA+3 that were previously studied in BM primary cells, we conducted studies to determine whether As+3 and/or MMA+3 alter STAT5 activation and IL-7 signaling in mouse pre-B cells.
MATERIALS AND METHODS
Reagents and chemicals
Hanks Balanced Salt Solution from Lonza (Walkersville, Maryland), Dulbecco’s phosphate buffered saline w/o Ca+2 or Mg+2 (DPBS−), Iscoves Modified Dulbecco’s Medium (IMDM) purchased from American Type Culture Collection (ATCC, Manassas, Virginia), Penicillin/Streptomycin 10 000 (mg/ml)/10 000 (U/ml) (Pen/Strep) Life Technologies (Grand Island, New York), β-Mecaptoethanol, Sigma Aldrich (St Louis, Missouri), 1 mg/ml mouse IL-7 (mIL-7) purchased from Peprotech, Fetal Bovine Serum (FBS) Atlanta Biologicals (Flowery Branch, Georgia), sodium arsenite (CAS 774-46-5, NaAsO2) purchased from Sigma-Aldrich (St Louis, Missouri); MMA+3 was the generous gift of Dr Terry Monks and Dr. Todd Camenisch at the University of Arizona Superfund Center and the Southwest Environmental Health Science Center. Alexa Fluor 488 Rat Anti-Mouse CD45R (Cat. No. 560733) and PE-Cy 7 Rat Anti-Mouse CD127 (Cat. No. 560733), Alexa Fluor 488 Rat Anti-Mouse CD127 Clone SB/199 (Cat. No. 561533), and Alexa Fluor 488 Rat IgG2b Isotope Control (Cat. No. 4003598) were purchased from BD Biosciences (San Jose, California). Anti-Hu/Mo pSTAT5 (Y694) PE (Cat. No. 12-9010-42) and Mouse IgG1 Iso Conttrol (Cat. No. 12-4714-42) were purchased from eBioscience (San Diego, California). STAT5 (clone 3H7, Cat. No. 9358). Phospho-STAT5 (Tyr694) (clone C11C5, Cat No. 9359), phospho-JAK1 (Tyr1022/1023) (Cat. No.3331), phosphor-JAK3 (Tyr 980/981) (Cat. No. 5031), JAK3 (Cat. No. 3775), and anti-rabbit IgG –HRP secondary antibodies (Cat. No. 7074) were purchased from Cell Signaling Technology (Danvers, Massachusetts). Purified mouse anti-JAK1 (Cat. No. 610231) monoclonal antibody was obtained from BD Transduction Laboratories (Franklin Lakes, New Jersey).
BM cell in vitro treatments and flow cytometry assay
BM cells were isolated according to the procedure described by Ezeh et al. (2014) from 3 C57BL/6 J male mice purchased at 8 weeks of age from Jackson Laboratory (Bar Harbor, Maine) and pooled. All animals were handled in accordance to procedures and protocols approved by the Institutional Animal Use and Care Committee at the University of New Mexico Health Sciences Center in our AAALAC-approved animal facility. Cells were treated with As+3 or MMA+3 in medium consisting of IMDM, 10% FBS, 1 % Pen/Strep, and 0.05 µM β-mecaptoethanol. Briefly, 1 × 106 BM cells were suspended in 200 µl of DPBS− and incubated with 1 µg of CD45R and CD127 antibodies for 30 min at RT in dark. Cells were fixed by adding 200 µl of IC fixation buffer (eBioscience) and incubated for 30 min at RT in dark. After washing with DPBS−, cells were incubated in 200 µl of 90% methanol for 30 min at 4°C in dark. After another wash with DPBS−, cells were resuspended in 100 µl of DPBS- and stained with 5 µl of pSTAT5 (Y694) antibody or isotope control and incubated for 30 min at RT in dark. Cells were washed twice with DPBS- and analyzed using an AccuriC6 Flow Analyzer (BD Biosciences).
Cell culture and treatments
The 2E8 cell line was purchased from ATCC and was first described by Ishihara et al. (1991). 2E8 cells were grown and maintained in our laboratory following ATCC’s protocol using complete medium consisting of IMDM 1 ng/ml mIL-7, 20% FBS, 1% Pen/Strep, and 0.05 µM β-mecaptoethanol. As+3 and MMA+3 were prepared in sterile water, as 1000× stock solutions and used to treat 2E8 cell preparations at final concentration ranges of 5–500 nM. mIL-7 was prepared as 10 µg/ml stock solution and used to treat 2E8 cell preparations at a final concentration of 10 ng/ml in treatment medium or 1 ng/ml in culture medium. Cells were first seeded at 5 × 105 cells/ml, in T75 flasks using complete 2E8 medium with 1 ng/ml mIL-7 for 3 days, then washed twice in DPBS- to remove all mIL-7. Cells were re-suspended in 2E8 medium without mIL-7 and treated with As+3 or MMA+3 overnight (approximately 24 h.). mIL-7 was re-introduced to treated cells at 10 ng/ml and cells were incubated for an additional 30 min to evaluate signaling. Post incubation, cells were harvested into 50 ml centrifuge tubes containing equal volume of ice cold DPBS− and washed twice by centrifugation at 400 × g, 4°C for 10 min. Washed cells were used for Total RNA extraction or stored at −80°C for protein extraction and immunoblotting.
Cell proliferation and viability
To determine cell proliferation and IL-7 dependence, 2E8 cells were grown at starting concentrations of 2.5 × 105 cells/ml in 2E8 medium with or without mIL-7 for 5 days. IL-7 was not replenished, and medium was not changed during the 5-day period. Cell counts were obtained in triplicate for each sample each day for 5 days, using the Nexcelom Cellometer Auto 2000 cell counter. Viability counts were also obtained using the same instrument with Acridine Orange/Propidium Iodide (AO/PI) staining. To test the sensitivity of 2E8 cells to treatment, proliferation, and viability were also determined daily during the 5-day period for cells exposed to different concentrations of As+3 or MMA+3 in complete medium.
2E8 protein extraction and immunoblotting analyses
Treated and washed cell pellets were re-suspended at 2 × 105 cells/µl in radioimmune precipitation assay buffer (50 mM Tris, 150 mM sodium chloride, 0.5 % sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulphate, pH 8.0) with added protease inhibitors including one 25 mg protease inhibitor mini tablet (Roche Diagnostics), 1% sodium orthovanadate (Na3VO4), and 200 mM phenylmethylsufonyl fluoride. Samples were sonicated for 10 s on ice and centrifuged at 20 000 × g. Total protein (the supernatant) from each sample was quantified using the Bicinchoninic Acid protein assay procedure (Pierce, Thermo Scientific). The protein extracts were resolved by electrophoresis on Criterion TGX precast gels (Bio-Rad) and transferred to nitrocellulose membranes. Antibodies against STAT5, pSTAT5, JAK1, pJAK1, JAK3, pJAK3 were used to identify these proteins by immunoblotting. Using the Super Signal West Femto Maximum Sensitivity Substrate kit (Thermo Scientific), protein bands were resolved on the FLuorChem R ProteinSimple imager and analyzed with Image J (1.48v) software. Protein molecular weights were determined using the Precision Plus Protein Pre-stained Standards (Bio-Rad).
CD127 expression by flow
2E8 cells were treated with As+3 or MMA+3 for 4 and 18 h 1 × 106 cells from each treatments were washed and stained with 0.5 µg of Rat Anti-Mouse CD127 antibody or Rat IgG2b Isotope Control for 30 min in dark. Cells were washed and with DPBS- and analyzed using an AccuriC6 Flow Analyzer.
Total RNA isolation
Total RNA was isolated from treated and washed triplicate samples using RNeasy Mini Kit and Shredder (Qiagen, Valencia, California) following manufacturer’s instructions. Briefly, treated and washed cells were pelleted in 15-ml centrifuge tubes, resuspended in 600 µl RLT cell lysis buffer (contained in kit) with 10 µl/ml ß-mercaptoethanol and mixed by vortex. Each sample was transferred into a labeled QIA Shredder column and centrifuged at 20 000 × g for 2 min. The isolation process was completed by following subsequent steps in the kit instructions, with final elution of total RNA using 50 µl, nuclease-free water. Total RNA concentration was calculated using the Nanodrop procedure for RNA quantitation. Results ranged from 3.7 to 7.5 µg/sample.
Synthesis of cDNA from total RNA
cDNA samples were synthesized from the isolated total RNA by reverse transcriptase reaction using High Capacity cDNA Archive Kit (Applied Biosystems) according to kit instructions. A 2 × Master mix (MM) was prepared with 10 × buffer, 25 × dNTP, 10 Random primers, nuclease-free water and multiscribe and was placed into each polymerase chain reaction (PCR) tube. The samples were placed in PCR tubes containing equal volumes of the 2 × MM and centrifuged for approximately 10 s before incubating on to the Thermal Cycler DNA Machine set at 25°C for 10 min, 37°C for 2 h. The cDNA samples were stored at −80°C for later use.
Quantitative real-time PCR
Synthesized cDNA was used as template to amplify gene and measure the fold change in induction of PAX5 (Applied Biosystems, Assay ID: Mm00435501_m1), E2A (Applied Biosystems, Assay ID: Mm01175588_m1), EBF1 (Applied Biosystems, Assay ID: Mm00432948_m1), Cyclin D1 (Applied Biosystems, Assay ID: Mm00432359_m1), Cyclin D2 (Applied Biosystems, Assay ID: Mm00438070_m1). Using cDNA template (from 18 ng total RNA) and the TaqMan Universal PCR MM (Applied Biosystems), the PCR reactions were set up for the detection and quantification of mRNA. GAPDH was used as the endogenous housekeeping gene and the control or untreated samples as the calibrator. The parameters for the PCR reactions thermal profile were Activation 50°C for 2 min, 95°C for 10 min, 95°C for 15 s, and 60°C for 1 min for 40 cycles. The quantitative real-time (qRT)-PCR was carried out using the 7900 HT system (Applied Biosystems) with 384-well block. For relative mRNA quantification, we used the Comparative CT method. The ΔCT values between the test and housekeeping genes and the fold difference (2−ΔΔCT) in the expressions for all the samples were determined and plotted as a bar or line graph in Sigma Plot.
Statistics and data analysis
All data were analyzed with SigmaPlot version 12.5, using one way analyses of variance and Dunnetts t test where applicable, for the determination of differences between control and treatment groups. For immunoblotting, samples were treated and ran in triplicates as shown on the blots. Image J (1.48v) software (NIH download from website: http://rsb.info.nih.gov/ij/) was used to obtain band intensities for treatment and control samples. For the qRT-PCR studies, samples were treated in triplicate and each replicate was run in triplicate to obtain 9 data points per sample. The ΔCT and 2−ΔΔCT was used to plot the fold change in induction between control and treatment samples.
RESULTS
MMA+3 Inhibits STAT5 Phosphorylation at Lower Dose than As+3 in Mouse pre-B Cells
Based on our previous work, we know that MMA+3 inhibits pre-B cell formation in mouse BM in vivo and in vitro (Ezeh et al., 2014). The activation of the transcriptional factor, STAT5, is required for pre-B cell development in mouse BM. Therefore, we determined whether STAT5 phosphorylation is inhibited by As+3 and MMA+3 exposure using multiparameter flow cytometry. Primary mouse BM cells were treated in vitro with 5, 50, and 500 nM As+3 or MMA+3 for 24 h. CD45R (B220) and CD127 were used as surface markers to identify the low abundance pre-B cell population in the BM (Matthias et al., 2005, Figure 1A). STAT5 signaling was detected by the presence of intracellular p-STA5. STAT5 phosphorylation was detected in both IL-7 treated and nontreated BM pre-B cells, suggesting that either STAT5 phosphorylation is constitutively present in these pre-B cells or that there are cellular sources of IL-7 or other cytokines that signal STAT5. STAT5 phosphorylation decreased significantly in pre-B cells exposed to 50 and 500 nM MMA+3 and at 500 nM As+3 (Figure 1B). Thus, these results show that in primary BM pre-B cells STAT5 signaling is inhibited by MMA+3at 50 nM in vitro, which is more potent than As+3
FIG. 1.
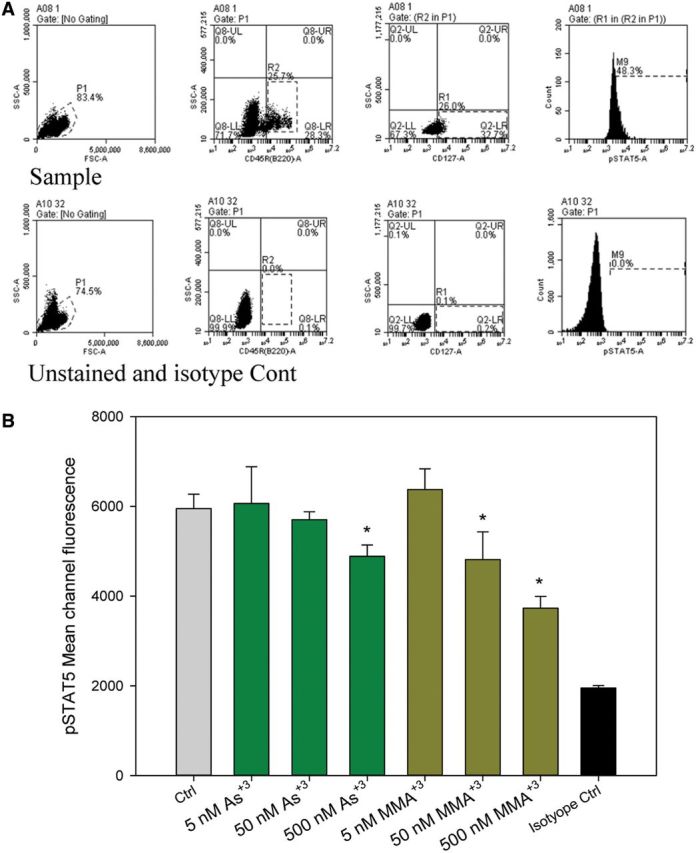
Inhibition of STAT5 phosphorylation by As+3 and MMA+3 in mouse pre-B cells in vitro detected using multiparameter flow cytometry. A, Gating of CD127 and CD45R stained BM cells and isotope control for pSTAT5 intracellular staining. B, Phospho-STAT5 signaling Mean Channel Fluorescence was measured in mouse BM pre-B cells identified by CD45R (B220) and CD127 surface markers. Error bars are ± SD. *Significantly different compared with control.
Comparative Effects of As+3 and MMA+3 on 2E8 Cell Growth
Mouse 2E8 cells are a cloned and differentiation-limited pre-B cell line that maintains its responsiveness to IL-7 (Ishihara et al., 1991). To determine the suitability of 2E8 for the proposed IL-7 signaling studies, we first determined the IL-7 dependence of the cells grown in culture. As shown in Figure 2A, 2E8 cells were found to have an absolute growth factor dependence on IL-7. When IL-7 was removed from the media, 2E8 failed to grow and began to lose cell viability measured using AO/PI, 4 days after seeding cultures.
FIG. 2.
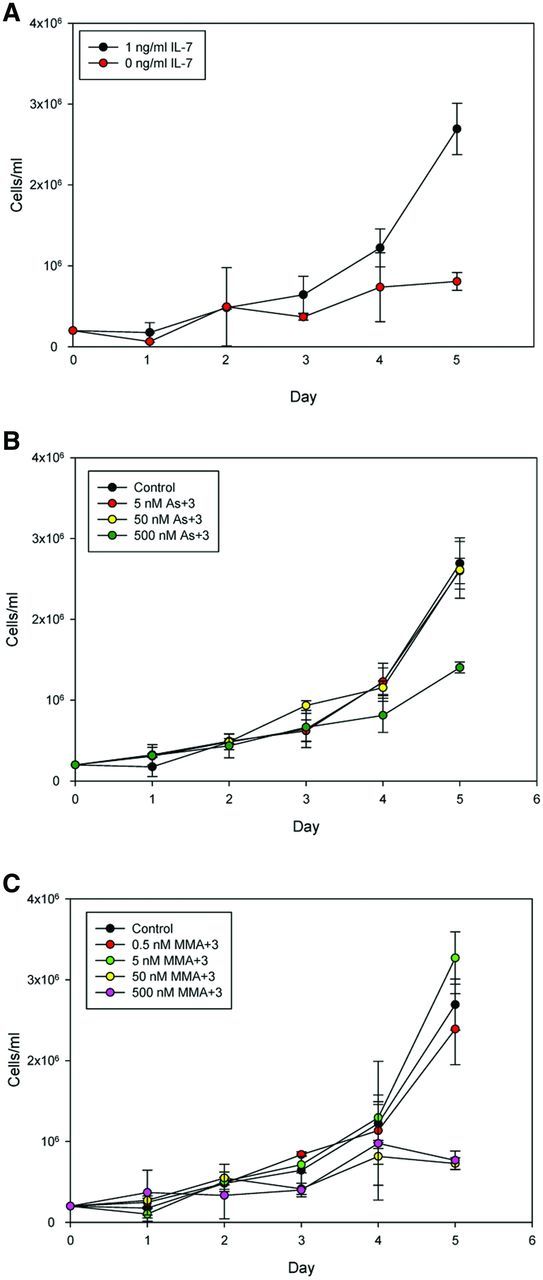
Five-day cell proliferation (cells/ml) and viability (%) by automated cell counter and trypan blue exclusion. Cell culture in medium with or without 1 ng/ml IL-7 (A); in medium containing IL-7 and different concentrations of As+3 (B); in medium containing IL-7 and different concentrations of MMA+3 (C). Error bars are ± SD.
As+3 did not modify the growth of 2E8 cells in the presence of IL-7 until cytotoxic concentrations (500 nM) were used (Figure 2B). However, MMA+3 was found to inhibit cell growth at concentrations as low as 50 nM on days 3–5 (Figure 2C). The effects of MMA+3 were not due to an increase in cytotoxicity on day 3. We therefore postulated that MMA+3 was interfering with IL-7 signaling.
MMA+3 Decreases STAT5 Activation in 2E8 Cells, but Does not Inhibit JAK1 Activation
IL-7 signals cell growth and differentiation through a STAT5 pathway involving phosphorylation of tyrosine 694 (Clark et al., 2014). We found that STAT5 activation was dependent on the presence of IL-7, as the levels of STAT5 phosphorylation were almost undetectable when cells were incubated overnight (approximately 24 h) in the absence of IL-7 (Figure 3A and C). STAT5 signaling might be downregulated in noncycling cells. To examine the effects of As+3 and MMA+3 on STAT5 activation, we analyzed the phosphorylation of STAT5 via Western blots. MMA+3 inhibited IL-7-induced STAT5 phosphorylation in a concentration-dependent manner (50–500 nM), as determined by the density of band images obtained in triplicate (Figure 3C and D). STAT5 signaling was IL-7 dependent and 50 nM MMA+3 was unable to modify STAT5 phosphorylation in the absence of IL-7. In contrast to MMA+3, As+3 had no effect on pSTAT in the concentration range of 5–500 nM (Figure 3A and B). These results clearly demonstrate that the effects of arsenic on IL-7 signaling in vitro is associated with MMA+3 and not As+3.
FIG. 3.

Effect of As+3 and MMA+3 on STAT5 activation in 2E8 cells (by Western). A, blot of pSTAT5 and total STAT5 in As+3 treated 2E8 cells. B, pSTAT5 band intensity to STAT5 band intensity ratio in (A). C, blot of pSTAT5 and total STAT5 in MMA+3 treated 2E8 cells. D, pSTAT5 band intensity to STAT5 band intensity ratio in (C).Cells were incubated overnight (approximately 24 h),in medium without IL-7, but containing 0–500 nM As+3 or MMA+3, then washed twice and treated with 0 or 10 ng/ml IL-7 and incubated for 30 min Error bars are ± SD. *Significantly different compared with control.
Activation of JAK1 and JAK3, the immediate upstream activator of STAT5, was also examined under identical conditions used to evaluate the effects of MMA+3 and As+3 on STAT5 activation. 500 nM As+3 only inhibited JAK3 phosphorylation, while 500 nM MMA+3 inhibited both JAK1 and JAK3 phosphorylation (Figs. 4 and 5). MMA+3 at 50 nM also suppressed JAK3 phosphorylation while As+3 had no effect at this dose (Figs. 4A and C and 5 A and C). Therefore, these results clearly demonstrated that MMA+3 has a stronger suppressive effects on JAK1 and JAK3 phosphorylation, the upstream signal for STAT5 activation, comparing to As+3 in vitro.
FIG. 4.

Effect of As+3 on JAK1 and JAK3 activation in 2E8 cells. A, pJAK1, JAK1, pJAK3, and JAK3 blots. B, pJAK1 to JAK1 band intensity ratio. C, pJAK3 to JAK3 band intensity ratio. Cells were incubated overnight (approximately 24 h) in medium without IL-7, but containing 0–500 nM As+3, then washed twice and treated with 0 or 10 ng/ml IL-7 and incubated for 30 min. Error bars are ± SD. *Significantly different compared with control.
FIG. 5.

Effect of MMA+3 on JAK1 and JAK3 activation in 2E8 cells. A, pJAK1, JAK1, pJAK3, and JAK3 blots. B, pJAK1 to JAK1 band intensity ratio. C, pJAK3 to JAK3 band intensity ratio. Cells were incubated overnight (approximately 24 h) in medium without IL-7, but containing 0–500 nM MMA+3, then washed twice and treated with 0 or 10 ng/ml IL-7 and incubated for 30 min. Error bars are ± SD. *Significantly different compared with control.
MMA+3 Suppressed IL-7 Receptor Expression on Cell Surface
Because JAK1 and JAK3 activation was inhibited by MMA+3, IL-7 receptor was very likely to be affected. Therefore, we stained 2E8 cells treated with As+3 at 5–500 nM and MMA+3 at 0.5–500 nM in vitro. IL-7 receptor expression is significantly inhibited by 500 nM MMA+3 not As+3 at both 4 and 18 h (Figures 6A and B), suggesting that the suppression of JAK1, JAK3, and STAT5 activation may be the results of the absence of IL-7 receptor expression on cell surface after exposure to MMA+3. 50 nM MMA+3 also inhibited IL-7 receptor expression, which correlates with the suppression of STAT5 and JAK3 activation with the same dose.
FIG. 6.
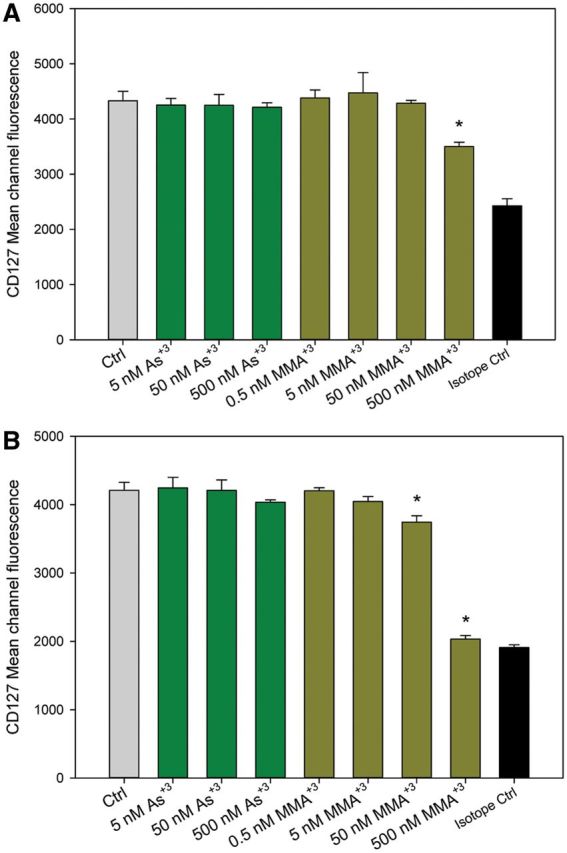
Effect of As+3 and MMA+3 on IL-7 R cell surface expression in 2E8 cells. 2E8 cells were exposed to 5–500 nM As+3 or 0.5–500 nM MMA+3 for 4 and 18 h, IL-7 R expression was measured by CD127 mean channel fluorescence. A, 4 h; B, 18 h. Error bars are ± SD. *Significantly different compared with control.
MMA+3 Inhibits Multiple Genes Associated with STAT5 Activation and B-Cell Development
E2A, EBF, and PAX5 are three major factors in B cell development (Liu et al., 2014; Rothenberg and Taghon 2005). There is also evidence indicating that STAT5 inhibition alters the expression of cell cycle genes such as Cyclin D1 (de Groot et al., 2000). We determined the mRNA expression of E2A, EBF, PAX5, Cyclin D1, and Cyclin D2 genes using qPCR to see if As+3 or MMA+3 induced pSTAT5 inhibition will lead to alterations in these genes. PAX5 and E2A gene expression were inhibited by MMA+3 in a concentration-dependent manner there was a trend towards inhibition in the range of 50–200 nM (Figure 7A). Early B cell factor gene, EBF1 and Cyclin D1 were found to be suppressed by both removal of IL-7 and 200 nM MMA+3 exposure, indicating that these genes are highly regulated by pSTAT5 in the IL-7 signaling pathway. Interestingly, 500 nM As+3 significantly increased the expression of EBF1, indicating that As+3 induced STAT5 phosphorylation inhibition may occur through a different mechanism. In Figure 7B, we demonstrate the Ebf1 protein expression by 2E8 cells was very IL-7-dependent, and it was inhibited by MMA+3 in the 50–100 nm range. Interesting Ebf1 protein expression was increased at 500 nM As+3, which is consistent with the mRNA results shown in Figure 7A.
FIG. 7.

STAT5 transcriptionally regulated mRNA and protein induction after exposure to As+3 and MMA+3. A, Fold change in p-STAT5 responsive B cell development factors (EBF1, E2A, PAX5) and cell cycle gene (CYCD1, CYCD2) expression in As+3 or MMA+3-treated cells (by qPCR). B, Effect of As+3 or MMA+3 on Ebf1 total protein (by Western). Cells were incubated overnight (approximately 24 h) in medium without IL-7, but containing 0–500 nM As+3, or 0–200 nM MMA+3, then washed twice and treated with 0 or 10 ng/ml IL-7 and incubated for 30 min. Error bars for fold change are ± SD.
DISCUSSION
Numerous diseases including cancers, vascular diseases, lung diseases, and diabetes have been linked to environmental exposures to As+3 in drinking water and food (Argos et al., 2010, 2014; Chen et al., 2003, 2011; Navas-Acien et al., 2005, 2006). Although numerous mechanisms of As+3 toxicity in cells have been proposed, most studies have used extremely high levels of As+3 for in vivo and in vitro studies. At micromolar concentrations, As+3 is well known to produce oxidative stress that impacts many cellular pathways (Flora et al., 2011; Kitchin and Conolly, 2010). Previous studies have examined DNA damage and repair pathways that are associated with the genotoxicity of As+3 (Cooper et al., 2013; King et al., 2012). However, there have been few studies that have examined nongenotoxic pathways associated with cell signaling.
Immune and inflammatory diseases could play a role in many chronic diseases associated with arsenic exposures. Prenatal arsenic exposures have been linked to immune suppression and altered lymphoid cell development (Ahmed et al., 2012; Nadeau et al., 2014). We have previously observed selective targeting of murine BM lymphoid progenitors by extremely low doses of As+3 in vivo and MMA+3 in vitro administered at environmentally relevant exposure levels (Ezeh et al., 2014). MMA+3 is formed through a series of metabolic reactions via the action of arsenite-3-methyltransferase (AS3MT), which is expressed in the liver and a few other extrahepatic tissues (Aposhian et al., 2000). AS3MT is not found at appreciable levels in lymphoid cells. Thus, for in vitro studies, it is necessary to add MMA+3 directly to cell cultures to observe its effects on lymphoid cell function. It is important to understand the effects of MMA+3 on lymphoid cells because they are very sensitive to low exposure levels. Also, we know that AS3MT is polymorphic in humans and has been causally linked to many arsenic-induced environmental diseases (Pierce et al., 2012).
This study assessed the role of As+3 and one of its key metabolites, MMA+3, on lymphoid progenitor cell activation. Beginning from the Pre-pro B to the large B cell stage, lymphoid progenitors express the IL-7 receptor alpha (IL-7Rα, CD127) and respond to IL-7 (Clark et al., 2014; Peschon, et al., 1994). IL-7 is a cytokine growth factor produced by the BM stromal cells (Fry and Mackall, 2001) and it is associated with proliferation, survival, and differentiation of B and T cells (Corfe et al., 2012). Humoral immune responses, which we have shown are extremely sensitive to in vivo As+3 exposure (Ezeh et al., 2014), utilize the ability of the lymphoid progenitors to expand following antigen challenge. Inhibition of B cell differentiation and proliferation by environmental pollutants can negatively impact the humoral immune responses, leading to decreased immunity and increased susceptibility to disease.
The IL-7 R consists of the IL-7Rα (CD127) which is specific for IL-7 ligand, and the γc chain (CD134) usually shared by other cytokines. Early B cells express CD127 for IL-7 signaling. Upon IL-7 binding to CD127 in a B cell, a cascade of phosphorylation initially begins with the receptor-associated Janus Kinases 1 and 3 (JAK1 and JAK3), and results in the phosphorylation of intracellular Signal Transduction and Activator of Transcription 5 (STAT5) molecules on tyrosine residues Y694 and Y699. These phosphorylated STAT5 molecules dimerize and translocate to the nucleus to drive the transcription of genes responsible for commitment maintenance, further differentiation, proliferation, and survival of the B lineage cells. It has been shown that deletion of total STAT5 protein (STAT5a/5 b) in cultures of fetal liver cells inhibited IL-7-induced lymphoid expansion (Dai et al., 2007). Although STAT5 activation could occur with several other cytokines, it is predominantly activated by IL-7 (Snow et al., 2002). STAT5 is highly activated by IL-7 in our cloned pre-B 2E8 cell system. Downstream of STAT5, the transcription factor, early B factor (Ebf1) and cyclin D1, in concert with E2A upregulate PAX5 gene expression (O’Riordan and Grosschedl, 1999). Due to the observed minimal levels of PAX5 in pre-pro B cells (Nut et al., 1999; Rolink et al., 1999), it has been suggested that pre-pro B cells Inhibition of STAT5 phosphorylation and PAX5 gene expressions could lead to a diversion from the B lineage commitment and unbalanced immune responses. Our studies show that Enf1 expression in 2 E* cells is very dependent on IL-7 signaling, and that Ebf1 protein expression induced by IL-7 is inhibited at low levels of MMA+3 (50–100 nm).
This studies show that MMA+3 inhibits phosphorylation of STAT5 at low (50 nM) concentrations that are not cytotoxic to primary pre-B cells and 2E8 cells. MMA+3 also inhibited JAK1 and JAK3 activation, and suppressed IL-7 Rα (CD127) expression on cell surface. Although the activation of STAT5 is known to be dependent on both JAK1 and JAK3 phosphorylation, it is suggested that JAK3 is not a primary activator of STAT5, but functions as a stabilizer of pJAK1 (Haan et al., 2011). In this study, MMA+3 at 50 nM only inhibited JAK3 phosphorylation, resulting in a slight suppression of STAT5 activation, while 50 nM MMA+3 inhibited both JAK1 and JAK3 activation, leading to a more significant suppression of STAT5 (Figs. 4 and 5). Also, the possibility that MMA+3 may directly inhibit STAT5 phosphorylation by acting on sites recently shown for direct inhibitors of STAT5 activation cannot be excluded (Behbod et al., 2003; Page et al., 2012).
The different responses of STAT5 and some STAT5 regulated genes such as PAX5 to As+3 and MMA+3 could be due to a number of reasons including differences in the cysteine residue content of the peptides, and the presence or absence of other thiol-containing amino acids in these proteins. As+3 is known to bind peptides that contain 3 (C3) and 4 (C4) cysteine residues whereas MMA+3 is capable of binding peptides containing 2 (C2) cysteine residues (Zhao et al., 2012; Zhou et al., 2011). Thus, MMA+3 has a different spectrum of action than does As+3.
In summary, our studies demonstrate important differences between the immunotoxicity of MMA+3 and As+3 on mouse pre-B cells. We demonstrate that MMA+3 can inhibit pre-B cell development through alteration of IL-7 signaling and STAT5 pathways that may interfere with B cell development, proliferation, or survival. MMA+3 acts at several stages including inhibition of IL-7 R(α) expression, activation of Jaks, and direct inhibition of STAT5 activation. The specific stage of pro-pre-B or pre-B cells affected, and the specific pathways altered by MMA+3 that interact with IL-7 and STA5 signaling are yet to be determined, but they likely involve other signaling pathways acting through direct and indirect mechanisms (Boller and Grosschedl, 2014). Finally, because MMA+3 B cell formation and IL-7 signaling at extremely low concentrations of arsenic that are environmentally relevant, the importance of nongenotoxic pathways in the action of arsenic is demonstrated.
FUNDING
This work was funded by a grant from the National Institute of Environmental Health Sciences of National Institute of Health (R01-ES019968)
ACKNOWLEDGMENTS
Monomethylarsonous acid (MMA+3) was obtained from Dr Terry Monks and Dr Todd Camenisch at the Southwest Environmental Health Sciences Center P30-ES006694, Synthetic Core, which was funded by the University of Arizona Superfund Program (P50-ES04940).
REFERENCES
- Ahmed S., Ahsan K. B., Kippler M., Mily A., Wagatsuma Y., Hoque A. M., Ngom P. T., El Arifeen S., Raqib R., et al. (2012). In utero arsenic exposure is associated with impaired thymic function in newborns possibly via oxidative stress and apoptosis. Toxicol. Sci. 129, 305–314. [DOI] [PubMed] [Google Scholar]
- Aposhian V. H., Gurzau E. S., Le X. C., Guazau A., Healy S. M., Lu X., Ma M., Yip L., Zakharyan R. A., Maiorino R. M., et al. (2000). Occurrence of monomethylarsonous acid in urine of humans exposed to inorganic arsenic. Chem. Res. Toxicol. 13, 693–697. [DOI] [PubMed] [Google Scholar]
- Argos M., Kalra T., Rathouz P. J., Chen Y., Pierce B., Parvez F., Islam T., Ahmed A., Rakibuz-Zaman M., Hasan R., Sarwar G., et al. (2010). Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): A prospective cohort study. Lancet. 376, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos M., Parvez F., Rahman M., Rakibuz-Zaman M., Ahmed A., Hore S. K., Islam T., Chen Y., Pierce B. L., Slavkovich V., et al. (2014). Arsenic and lung disease mortality in Bangladeshi adults. Epidemiology. 25, 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behbod F., Nagy Z. S., Stepkowski S. M., Karras J., Johnson C.R., Jarvis W. D., Kirken R. A. (2003). Specific inhibition of Stat5a/b promotes apoptosis of IL-2-responsive primary and tumor-derived lymphoid cells. J. Immunol. 171, 3919–3927. [DOI] [PubMed] [Google Scholar]
- Boller S., Grosschedl R. (2014). The regulatory network of B-cell differentiation: A focused view of early B-cell factor 1 function. Immunol. Rev. 261, 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. C., Guo Y. L., Su H. J., Hsueh Y., Smith T. J., Ayan L. M., Lee M-S., Chao S., Lee J. Y., et al. (2003). Arsenic methylation and skin cancer risk in southwestern Taiwan. J. Occup. Env. Med. 45, 241–248. [DOI] [PubMed] [Google Scholar]
- Chen Y., Graziano J. H., Parvez F., Liu M., Slavkovich V., Kalra T., Argos M., Islam T., Ahmed A., Rakibuz-Zaman M., et al. (2011). Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: Prospective cohort study. BMJ. 342, d2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. R., Mandal M., Ochiai K., Singh H. (2014). Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signaling. Nat. Rev. Immunol. 14, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper K. L., King B. S., Sandoval M. M., Liu K. J., Hudson L. G. (2013). Reduction of arsenite-enhanced ultraviolet radiation-induced DNA damage by supplemental zinc. Toxicol. Appl. Pharmacol. 269, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corfe S. A., Paige C. J. (2012). The many roles of IL-7 in B cell development; mediator of survival, proliferation and differentiation. Semin. Immunol. 24,198–208. [DOI] [PubMed] [Google Scholar]
- Dai X., Chen Y., Di L., Podd A., Li G., Bunting K.D., Hennighausen L., Wen R., Wang D. (2007). STAT5 is essential for Early B cell Development but not for B cell maturation and function. J. Immunol. 179, 1068–1079. [DOI] [PubMed] [Google Scholar]
- de Groot R. P., Raaijmakers J. A., Lammers J. W., Koenderman L. (2000). STAT5-dependent cyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol. Cell Biol. Res. Commun. 3, 299–305. [DOI] [PubMed] [Google Scholar]
- Ezeh P. C., Lauer F. T., MacKenzie D., McClain S., Liu K. J., Hudson L. G., Gandolfi A. J., Burchiel S. W. (2014). Arsenite selectively inhibits mouse bone marrow lymphoid progenitor cell development in vivo and in vitro and suppresses humoral immunity in vivo. PLoS One. 9, e93920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora S. J. (2011). Arsenic-induced oxidative stress and its reversibility. Free Radic. Biol. Med. 51, 257–281. [DOI] [PubMed] [Google Scholar]
- Fry T. J., Mackall C. L. (2001). Interleukin-7: Master regulator of peripheral T cell homeostasis? Trends Immunol. 22, 564–571. [DOI] [PubMed] [Google Scholar]
- Haan C., Rolvering C., Raulf F., Kapp M., Drückes P., Thoma G., Behrmann I., Zerwes H. G. (2011). Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem. Biol. 18, 314–323. [DOI] [PubMed] [Google Scholar]
- Ishihara K., Medina K., Hayashi S., Pietrangeli C., Namen A. E., Miyake K., Kincade P. W. (1991). Stromal-cell and cytokine-dependent lymphocyte clones which span the pre-B- to B-cell transition. Dev. Immunol. 1, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K., Lai A. Y., Hsu C. L., Kondo M. (2005). IL-7 receptor signaling is necessary for stage transition in adult B cell development through up-regulation of EBF. J. Exp. Med. 201, 1197–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B. S., Cooper K. L., Liu K. J., Hudson L. G. (2012). Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J Biol. Chem. 287, 39824–39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchin K. T., Conolly R. (2010). Arsenic-induced carcinogenesis–Oxidative stress as a possible mode of action and future research needs for more biologically based risk assessment. Review. Chem. Res. Toxicol. 23, 327–335. [DOI] [PubMed] [Google Scholar]
- Kozul C. D., Ely K. H., Enelow R. I., Hamilton J. W. (2009). Low-dose arsenic compromises the immune system response to influenza A infection in vivo. Environ. Health Perspect. 117, 1441–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Lauer F. T., Liu K. J., Hudson L.G., Burchiel S. W. (2010). Low-dose synergistic immunosuppression of T-dependent antibody responses by polycyclic aromatic hydrocarbons and arsenic in C57BL/6J murine spleen cells. Tox. Appl. Pharmacol. 245, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. J., Cimmino L., Jude J. G., Hu Y., Witkowsk M. T., McKenzie M. D., Kartal-Kaess M., Best S. A., Tuohey L., Liao Y., et al. (2014). Pax5 loss imposes a reversible differentiation block in B-progenitor acute lymphoblastic leukemia. Genes Dev. 28, 1337–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias P., Rolink A. G. (2005). Transcriptional networks in developing and mature B cells. Nat. Rev. Immunol. 5, 497–508. [DOI] [PubMed] [Google Scholar]
- Nadeau K. C., Li Z., Farzan S., Koestler D., Robbins D., Fei D. L., Malipatlolla M., Maecker H., Enelow R., Korrick S., et al. (2014). In utero arsenic exposure and fetal immune repertoire in a US pregnancy cohort. Clin. Immunol. 155, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas-Acien A., Sharrett A. R., Silbergeld E. K., Schwartz B. S., Nachman K. E., Burke T. A., Guallar E. (2005). Arsenic exposure and cardiovascular disease: A systematic review of the epidemiologic evidence. Am. J. Epidemiol. 162, 1037–1049. [DOI] [PubMed] [Google Scholar]
- Navas-Acien A., Silbergeld E. K., Streeter R. A., Clark J. M., Burke T. A., Guallar E. (2006). Arsenic exposure and type 2 diabetes: A systematic review of the experimental and epidemiological evidence. Environ. Health Perspect. 114, 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt S. L., Heavey B., Rolink A.G., Busslinger M. (1999). Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401, 556–562. [DOI] [PubMed] [Google Scholar]
- O’Riordan M., Grosschedl R. (1999). Coordinate Regulation of B cell differentiation by transcription factors EBF and E2A. Immunity 11, 21–31. [DOI] [PubMed] [Google Scholar]
- Oya-Ohta Y., Kaise T., Ochi T. (1996). Induction of chromosomal aberrations in cultured human fibroblasts by inorganic and organic arsenic compounds and different roles of glutathione in such induction. Mutat. Res. 357, 123–129. [DOI] [PubMed] [Google Scholar]
- Page B. D., Khoury H., Laister R. C., Fletcher S., Vellozo M., Manzoli A., Yue P., Turkson J., Minden M. D., Gunning P. T. (2012). Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J. Med. Chem. 55, 1047–1055. [DOI] [PubMed] [Google Scholar]
- Peschon J. J., Morrissey P. J., Grabstein K. H., Ramsdell F. J., Maraskovsky E., Gliniak B. C., Park L. S., Zeigler S. F., Williams D. E., Ware C.B., et al. (1994). Early lymphocyte expansion is severely impaired in interleukin-7 receptor-deficient mice. J. Exp. Med. 180, 1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce B. L., Kibriya M. G., Tong L., Jasmine F., Argos M., Roy S., Paul-Brutus R., Rakibuz-Zaman M., Parvez F., Ahmed A., et al. (2012). Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh. PLOS Genet. 8, e1002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X. J., Liu W., Li Y. N., Sun X., Hai C. X., Hudson L. G., Liu K. J. (2012). Poly (ADP-ribose) polymerase-1 inhibition by arsenite promotes the survival of cells with unrepaired DNA lesions induced by UV exposure. Toxicol. Sci. 127, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink A. G., Nutt S. L., Melchers F., Busslinger M. (1999). Long-term in vivo reconstitution of T-cell development by Pax5-deficient B-cell progenitors. Nature 401, 603–606. [DOI] [PubMed] [Google Scholar]
- Rothenberg E. V., Taghon T. (2005). Molecular genetics of T cell development. Annu. Rev. Immunol. 23, 601–649. [DOI] [PubMed] [Google Scholar]
- Snow J. W., Abraham N., Ma M. C., Abbey N. W., Herndier B., Goldsmith M. A. (2002). STAT5 promotes multilineage hematolymphoid development in vivo through effects on early hematopoietic progenitor cells. Blood 99, 95−101. [DOI] [PubMed] [Google Scholar]
- Styblo M., Del Razo L. M., Vega L., Germolec D. R., LeCluyse E. L., Hamilton G. A., Reed W., Wang C., Cullen W. R., Thomas D. J. (2000). Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 74, 289–299. [DOI] [PubMed] [Google Scholar]
- Wang F., Zhou X., Liu W., Sun X., Chen C., Hudson L. G., Liu K. J. (2013). Arsenite- induced ROS/RNS generation causes zinc loss and inhibits the activity of poly(ADP-ribose) polymerase-1. Free Radic. Biol. Med. 61, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D., Liu W., Cooper K., Qin X. -J., de Souza Bergo P. L., Hudson L. G., Liu K. J. (2009). Inhibition of poly(ADP-ribose)polymerase-1 by arsenite interferes with repair of oxidative DNA Damage. J. Biol. Chem. 284, 6809–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Chen S., Jia L., Shu S., Zhu P., Liu Y. (2012). Selectivity of arsenite interaction with zinc finger proteins. Metallomics 4, 988–994. [DOI] [PubMed] [Google Scholar]
- Zhou X., Sun X., Cooper K. L., Wang F., Liu K. J., Hudson L. G. (2011). Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J. Biol. Chem. 286, 22855–22863. [DOI] [PMC free article] [PubMed] [Google Scholar]